Unlocking the molecular mechanisms of antipsychotics – a new frontier for discovery
DOI: https://doi.org/10.4414/smw.2016.14314
Heather
Bowling, Emanuela
Santini
Summary
Despite the use of antipsychotics to treat schizophrenia for the last several decades, little was understood about their molecular mechanisms of action. In this review, we discuss recent studies that have helped elucidate mechanisms of action of antipsychotics and their potential interplay with genetic, metabolomic, proteomic, and other cellular process-related discoveries in schizophrenia pathology. We also highlight genes that have been identified in multiple studies in both schizophrenia patients and in antipsychotic action that are related to glucose and cellular metabolism, the cytoskeleton, protein synthesis, cell adhesion and synaptic activity. Though some questions of antipsychotic mechanisms of action, such as primary versus off-target effects, remain, the recent gains in understanding how to treat schizophrenia at the molecular level are promising. We propose that these recent insights provide a new and more complete landscape for drug discovery and patient biomarker development.
Antipsychotics – a history
Antipsychotics were discovered in the late 1950s in an effort to improve current anesthetic agents at the time. Though these new agents were ineffective as anesthetics, they robustly attenuated psychotic symptoms in patients, and thus became rapidly popular for their clinical application.
In the 1960s and 70s, researchers determined that this first class of agents, or “typical” antipsychotics, acted primarily on dopamine D2 and related receptors (DRD2). The pharmacological profile of first generation antipsychotics concurred, together with other experimental observations, to the hypothesis that schizophrenia was caused by abnormal dopamine signaling [1–3]. Subsequent studies have revealed irregularities in synaptic function across a broad range of brain regions and in different neurotransmitters in schizophrenia patients, which have led to a clearer, but still incomplete, understanding of the disorder and the drugs that treat it.
With between 0.4% and 1% of the population suffering from schizophrenia in their lifetime [4, 5] and over 3.1 million people being treated with antipsychotics in the United States of America in 2011 alone [6], it remains an important goal to establish the underlying mechanisms of the disorder and its treatments to improve the quality of care and life for schizophrenia patients.
Although antipsychotics were efficacious for positive psychotic symptoms, such as hallucinations, delusions, and paranoia, they remained less effective for the negative (e.g. anhedonia and improper facial affect) and cognitive symptoms associated with schizophrenia [7]. In addition, the first generation of antipsychotics, including haloperidol, were associated with debilitating extrapyramidal side effects following long-term treatment including tremors and Parkinsonism.
In an effort to improve the treatment of the negative and cognitive symptoms and to decrease the side effect profile, “second generation” or “atypical” antipsychotics were introduced in the late 1980s and early 1990s. The atypical antipsychotics continued to target DRD2, but they also targeted other receptor groups such as the serotonin 5HT2a receptors, which are more widely expressed in the hippocampus and other brain regions associated with learning and memory. Atypical antipsychotics were quickly lauded for their improved efficacy and attenuated side effects, and they became the standard of care very quickly. The claims of increased efficacy with fewer side effects of the atypical antipsychotic drugs were increasingly questioned as clinical studies emerged suggesting that, while indeed they did not induce as many extrapyramidal side effects, they were associated with weight gain and obesity in patients [8]. In addition, both typical and atypical antipsychotics presented similar challenges to basic scientists attempting to study cognition in animal models of schizophrenia as, with the exception of clozapine, they induced catalepsy, or “a condition of diminished responsiveness usually characterized by a trancelike state and constantly maintained immobility” [9, 10]. Catalepsy rendered learning and memory studies difficult to interpret, and subsequently interfered in attempts to understand fully antipsychotic actions on cognition.
The question of improved clinical efficacy in treating schizophrenia of atypical versus typical antipsychotics drew further attention in 2005, when multicenter trials revealed no overall amelioration of schizophrenia (both positive/negative symptoms) or side effects between the two groups, with important differences in the types of side effects affected [11]. Extrapyramidal side effects and tremor were more common with typical antipsychotics, while obesity and subsequent diabetes were more common with atypical antipsychotics. Clozapine, which had been previously thought to be favorable for treatment-resistant schizophrenia patients [12], was deemed a potentially promising alternative because of the occurrence of fewer extrapyramidal side effects [13]. However, clozapine usage has also been associated with rare, but severe toxicity, including an increased rate of agranulocytosis, adverse cardiovascular events, seizures, and potentially increased fatality in elderly patients, leading to multiple Boxed Warnings in the US prescribing information. Aripiprazole, another atypical antipsychotic approved in 2002, a partial DRD2 agonist and 5HT2a antagonist, also showed promise, and many also believed its efficacy profile offered improvement for those who could not tolerate the side effects of the other antipsychotics [14, 15]. However, it did not yield a significant improvement in schizophrenia symptoms over risperidone, with conflicting reports on efficacy compared with haloperidol based on treatment duration [15, 16]. Therefore, despite the initial enthusiasm for atypical antipsychotics, further studies revealed that they did not improve efficacy compared with typical antipsychotics, were associated with concerning, albeit different, side effects, and therefore the search for better schizophrenia treatments was renewed.
Because the bulk of new antipsychotics were derivatives of the original discovery, new development efforts searched for a new direction to improve efficacy against both positive and negative symptoms. Many new attempts in the 1990s through the past decade have centered on the hypothesis that schizophrenia results from disrupted glutamatergic signaling (reviewed in [17]). Two such compounds, bitopertin, a glycine transporter type 1 inhibitor that increases glycine in the synaptic cleft, which co-activates N-methyl-D-aspartate receptors (NMDAR) with glutamate, and pomaglumetad, a metabotropic glutamate receptor group II agonist were tested in clinical trials; however, both resulted in phase III trial failure, as they did not achieve their primary endpoints [18—20]. With these disappointments in the past few years, there is a need to refocus on understanding the pathophysiology of schizophrenia and the mechanism of action of current antipsychotic drugs. To that end, this review summarizes several recent studies that further elucidate the molecular targets of antipsychotic activity and how they might interface with schizophrenia pathology.
Antipsychotic signaling – what we know now
Although initial studies focused on antipsychotic action at the receptor level, questions remained about the action of antipsychotics at the molecular and signaling levels. Because DRD2 are G-protein coupled receptors Gi/o type receptors, the primary characterized downstream signaling pathways are adenylyl cyclase-cyclic AMP (cAMP) – protein kinase A (PKA) (G-protein dependent cascade) and β-arrestin2 – phosphatase 2A-Akt (G-protein independent cascade).
The majority of antipsychotics block DRD2 receptors and increase cAMP production by removing the tonic inhibition exerted by Gi/o proteins on adenylyl cyclase activity, which depends on the action of the neuromodulator adenosine. The enhanced adenylyl cyclase activity increases cAMP levels, activating PKA and increasing PKA-dependent phosphorylation of DARPP-32 (dopamine- and cAMP-regulated phosphoprotein of molecular weight 32000) [21–24]. An important functional consequence of the opposite role of DRD2 and adenosine 2A receptors (A2AR) on cAMP production is that genetic or pharmacological blockade of A2AR prevents the ability of DRD2 antagonists (haloperidol, eticlopride, etc.) to increase the cAMP-PKA-DARPP-32 signaling pathway [25, 26]. Antipsychotics acting as DRD2 antagonists also inhibit the assembly and activation of the β-arrestin2 and phosphatase 2A complex. When assembled, this complex dephosphorylates and deactivates Akt (=protein kinase B), and therefore its blockade leads to increased Akt signaling [27, 28].
Despite evidence that PKA and Akt signaling pathways are modulated by multiple first and second generation antipsychotics, the extent of conservation of these mechanisms varies between different antipsychotic drugs. Masri and colleagues noted that the antipsychotics haloperidol, risperidone, ziprasidone clozapine, olanzapine, and aripiprazole all prevented the association of β-arrestin2 to the DRD2 induced by administration of a DRD2R agonist (quinpirole) in a cell culture-based assay [29]. In contrast, the accumulation of cAMP varied between these drugs, suggesting there may be slight variations in the PKA signaling pathway between antipsychotic classes [29]. Because prevention of β-arrestin2/phosphatase2A recruitment leads to the phosphorylation and activation of Akt [28–30], Akt became a strong focus for convergent antipsychotic signaling mechanisms.
The conserved activation of Akt signaling nicely complimented previous work demonstrating that typical antipsychotics lead to phosphorylation of glycogen synthase kinase 3β (GSK3β), altering β-catenin signaling [28, 31, 32]. Interestingly, activation of Akt-GSK3β signaling has also been demonstrated for the newer generation of atypical antipsychotics, such as aripiprazole [33]. Akt-GSK3β action additionally occurs in the different context of mood stabilizers, such as lithium, which also targets DRD2 and GSK3β directly [30]. Therefore, Akt signaling remains the most common and robust downstream signaling effector of the different classes of antipsychotics acting on DRD2.
Moreover, activation of Akt is upstream of multiple signaling cascades involved in transcription, protein synthesis, metabolism and other cellular processes. Therefore, the implication of Akt signaling in the mechanism of action in antipsychotics raised the question of whether other cellular processes downstream of Akt might also be involved in antipsychotic action. The Akt-mTORC1 (mechanistic target of rapamycin complex 1)-protein synthesis pathway was of particular interest as previous studies have shown that long-term antipsychotic treatment alters the proteome in human schizophrenia patients [34]. Recently, Bowling and colleagues demonstrated that acute antipsychotic treatment induces the Akt-mTORC1-protein synthesis pathway resulting in immediate changes in protein synthesis [35]. The two main downstream effectors of mTOR are both upstream of protein synthesis and are 4E-binding protein (4E-BP) and p70 ribosomal S6 kinase 1 (S6K1). Interestingly, other studies have demonstrated that ribosomal protein S6 (Rps6), which has multiple phosphorylation sites that are specific and nonspecific targets of S6K1, can be phosphorylated downstream of PKA signaling as well [21, 36], suggesting some level of convergence on ribosomal signaling following antipsychotic treatment.
In addition to the mTORC1 pathway, investigation has also demonstrated an antipsychotic-mediated activation of the Akt-FOXO (forkhead box O) pathway [37] suggesting that other downstream Akt effectors may be involved in the antipsychotic signaling as well. Overall, these data indicate a critical role for PKA and Akt signaling in the mechanism of action for antipsychotics (fig. 1).
Animal models
In the past, the most commonly utilized animal models of schizophrenia were centered on the idea that dopamine dysfunction was implicated in this disorder. Indeed, as mentioned earlier in this review, many of the effective antipsychotic drugs were designed as antagonists of dopamine receptors and dopamine agonists may induce symptoms that resemble psychosis. However, the recent discoveries about the pathophysiology of schizophrenia determine the emergence of novel animal models, mimicking some of the clinically relevant human phenotypes. To date, there are four major classes of animal models used to investigate schizophrenia pathology and the efficacy of antipsychotics: genetic models, pharmacological models, neurodevelopmental models, and lesion models [38]. The bulk of publications and effort have focused on genetic and pharmacological models as a means of exploring antipsychotic efficacy and mechanism of action.
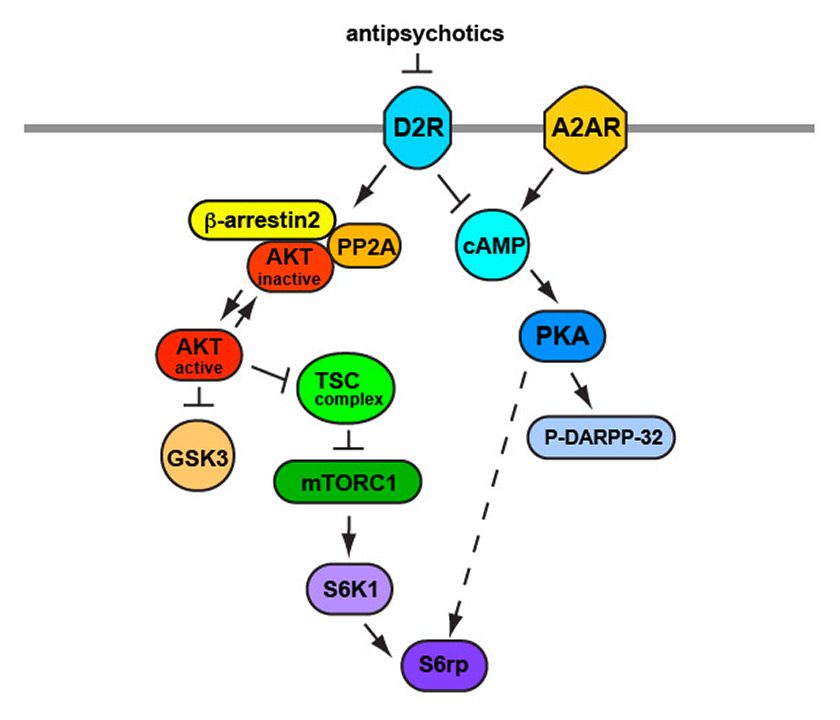
Figure 1
Schematic representing the action of antipsychotics on intracellular signaling cascade.
Inhibition of D2Rs by antipsychotic results in increased cAMP levels produced by removing the tonic inhibition on adenylyl cyclase activity (see text for more details). This leads to activation of PKA and phosphorylation of DARPP-32. Moreover, antipsychotics, by antagonizing D2Rs, inhibit the activation of β-arrestin2/PP2A complex resulting in increased AKT activity. Both AKT and PKA lead to activation of mTORC1 and S6 ribosomal proteins, which regulated protein synthesis.
AKT ≡ protein kinase B; cAMP = cyclic AMP; D2R = dopamine 2 receptor; DARPP-32 = dopamine- and cAMP-regulated phosphoprotein of molecular weight 32 000; mTORC1 = mechanistic target of rapamycin complex 1; PKA = protein kinase A; PP2A = phosphatase 2A; S6K1 = p70 ribosomal S6 kinase 1; S6rp = S6 ribosomal protein; TSC = tuberous sclerosis complex
Genetic models
Genetic animal models of human disorders are based on the idea that manipulation of the susceptibility genes account for the risk of illness and, together with environmental factors, for phenotypic variation.
Schizophrenia is a highly heritable neuropsychiatric disorder that probably involves multiple genes [39]. Targeted manipulations of schizophrenia-associated genes may provide unique advantages in understanding the genetic contribution to the pathophysiology of the disorder and the disease-related endophenotypes. This approach is important to gain knowledge on the molecular pathways, neuronal circuits and behaviors affected in schizophrenia. Moreover, the genetic models allow investigation of interactions between susceptibility genes, the relations between genes and environment, and the effects of genetic manipulation on disease development [40].
We shall describe two such models based on the association between schizophrenia and the candidate susceptibility genes, neuregulin 1 (NRG1) and disrupted-in-schizophrenia 1 (DISC1).
Neuregulin 1 (NRG1)
The genetic association between NRG1 and, to lesser extent, its receptor erbB4, and schizophrenia has been supported in most genetic and meta-analysis studies of various populations [41, 42] including genome-wide association studies [43–45]. Animal models lacking different portions of the Nrg1 gene have been engineered and studied in order to understand the different array of molecular and behavioral phenotypes correlated with the particular deletion. In these models, all the deletions in the Nrg1 gene are in heterozygosis since the homozygote deletions are lethal [41]. Overall, these mice display deficits in prepulse and latent inhibition (PPI and LI, respectively), anxiety, alteration in motor activity and abnormal social behavior [46–49]. Surprisingly, mice with heterozygote deletion of Nrg1 gene have an intact spatial and working memory. The behavioral deficits are also accompanied by changes in neurotransmitter and synaptic receptors, such as decreased expression of NMDA receptors and increased expression of serotonin 2A receptors and serotonin transporter [49]. Importantly, some of the behavioral phenotypes displayed by these mice could be corrected by treatment with the antipsychotic drug, clozapine [41, 46]. Also, transgenic mice with overexpression of the Nrg1 type 1 isoform show deficits in acoustic startle and PPI, hyperactivity and age-dependent disruption of short-term memory [50, 51]. These studies suggest that alteration of Nrg1 gene dosage (deletion or overexpression) generates behavioral phenotypes consistent with clinical symptoms of neuropsychiatric disorders.
Moreover, given the important role of Nrg1 in oligodendrocyte development, Roy and colleagues developed transgenic mice expressing a dominant negative form of the gene exclusively in oligodendrocytes [42]. They demonstrated that blocking NRG1-erbB signaling in oligodendrocytes resulted in changes in the number and morphology of oligodendrocyte as well as decreased myelin thickness and reduced conduction velocity in the central nervous system axons. Moreover, these animals displayed anxiety, hypoactivity and deficit in social behavior. The behavioral and morphological phenotypes were accompanied by increased levels of dopamine receptors and dopamine transporter. This study indicates that NRG1-erbB signaling is important in oligodendrocyte development in vivo and the resulting alteration in brain myelinization is correlated with the onset of behaviors consistent with human neuropsychiatric disorders.
Disrupted-in-schizophenia (DISC1)
Disrupted-in-schizophenia (DISC1) was initially identified as a gene affected by a translocation mutation that segregated with psychiatric disorders, such as schizophrenia and depression, in a Scottish family [53]. Subsequent studies suggested that variation in DISC1 may play a role in schizophrenia and other psychiatric disorders in normal individuals [54–56].
Mouse models with heterozygote Disc1 deletion were generated to model the translocation mutation discovered in patients [57, 58]. Behaviorally, these mice show higher impulsivity, while locomotor activity and PPI are not different from wild-type littermates. However, the mice display a specific pattern of cognitive impairments, with working memory being severely affected. These cognitive deficits are accompanied by a specific neuronal pathology in the hippocampus, particularly in the neurogenesis process. These studies demonstrated that deletion of Disc1 give rise to phenotypes resembling human neuropsychiatric disorders.
Other models with inducible expression of the human DISC1 gene in specific brain regions, such as cerebral cortex, hippocampus and striatum [59] were produced in accordance with the idea that a mutant truncated DISC1 protein with dominant-negative effects is generated as result of the human translocation mutation. These mice showed an enlargement of the ventricles but no other gross anatomical abnormalities in the brain. Behaviorally, the mice displayed hyperactivity, and deficits in social behavior and spatial memory. Similarly, mice expressing a dominant-negative form of DISC1 [60] present enlargement of the brain ventricles and behavioral abnormalities, including hyperactivity, PPI and affective behaviors. Importantly, these studies suggest that the expression of a truncated non-functional DISC1 protein is associated to anatomical and behavioral changes mimicking human neuropsychiatric disorders.
Animal models with reversible and inducible induction of the C-terminal part of Disc1 [61] were produced on the basis of the demonstration that during development Disc1 is highly expressed in the brain. In these mice, early postnatal Disc1 expression affects synaptic transmission and morphology of neurons in the dentate gyrus. Moreover, this is accompanied by depressive-like behaviors, abnormalities in social behaviors and impairments in working memory. This study suggests that alteration of Disc1 during development may determine the appearance of schizophrenia-like behaviors in the adulthood.
Additionally, RNA interference was utilized to study the function of Disc1 during development. Small hairpin RNA (shRNA)-mediated downregulation of Disc1 during neurogenesis led to morphological and cytological changes in the dentate gyrus, for instance the appearance of neurons with ectopic apical and basal dendrites. Moreover, the neurons showed increased excitability, alteration in the neurogenesis process and aberrant localization of the new-born neurons [62]. These studies underscore the importance of DISC1 expression during early development.
Pharmacology models
While the most popular models for the negative symptoms of schizophrenia are genetic, the most commonly used models for positive symptoms are pharmacological. The two most commonly used paradigms unsurprisingly correspond to the two popularly held neurotransmitter theories of schizophrenia: glutamate and dopamine. For one paradigm, a “schizophrenia-like” state is induced by manipulating NMDA receptors with either phencyclidine or MK-801 and in the other dopamine release is triggered in large quantities by increasing dopamine release with amphetamine or related agents.
MK801/phencyclidine
In the 1950s, phencyclidine was developed as an anesthetic and quickly gained notoriety for causing psychotic-like states in healthy patients. Clinicians noted the similarities between the psychosis induced by phencyclidine and that of schizophrenia patients, and sought to better understand the pathology of psychosis by administering phencyclidine to schizophrenia patients. The administration of phencyclidine led to an exacerbation of symptoms in both stabilized and acute patients, which did not occur with lysergic acid diethylamide (LSD) or other pharmacological agents (reviewed in [63]). These data contributed to the formation of the glutamate hypothesis of schizophrenia and quickly the injection of phencyclidine or another NMDAR antagonist into rodent models to simulate schizophrenia-like behavior. Treatment with this class of inhibitor leads to increased locomotion and altered abnormal sensory-motor gating, and may mimic some of the negative symptoms of schizophrenia such as anhedonia, abnormal social behavior, and decreased cognitive performance on learning tasks (reviewed in [64]). There have been conflicting reports on the ability of typical and atypical antipsychotics to rescue these induced phenotypes [65–67], as well as of newer drugs targeting glycine transporters that increase glutamatergic signaling and glutamate receptor agonists [68–71].
Amphetamine
Amphetamine increases the amount of dopamine in the synaptic cleft and the activity of dopamine receptors. Because the dopamine hypothesis of schizophrenia suggested increased dopamine signaling, amphetamines and related stimulants were a natural early choice for modeling schizophrenia in rodents. In the amphetamine-based studies, mice were given a dose of amphetamine, which increased locomotor activity, agitation, altered sensory-motor gating and was thought to induce psychotic-like symptoms and behavior including increased locomotion, altered sensory motor gating, and disordered attentional processing (reviewed in [72–76]). Either preceding or following the induction of this altered state where rodents are believed to exhibit “schizophrenia-like” symptoms, animals are given antipsychotics to prevent or correct these behaviors. To date, typical and atypical antipsychotics have been shown to reverse or prevent many of these “positive symptom-like” behavioral deficits; however this is somewhat dependent on the treatment paradigm ([75], reviewed in [38]). The benefit of this model is the induction of “psychosis-related behavior.” However, other studies have contradicted the effect of antipsychotics on these induced “schizophrenia like states,” making them harder to interpret. In addition, the kinetics of the two pharmacological agents – the induction of the symptoms and, separately, the treatment – increase the margin of error and introduce potential differences in pharmacokinetics, pharmacodynamics, signaling changes, and metabolism between the two phases of drug treatment. These potential interactions should be considered when interpreting the data from these models. Many studies have also use wild-type mice to study the mechanisms of action of antipsychotics on circuits at molecular, electrophysiological and behavioral levels, and to predict side effect profiles, which are discussed throughout this review.
Disadvantages and advantages of animal models
Although animal models have been useful in probing mechanistic questions of antipsychotics and side effects, they have had limited benefit in predicting efficacy. There are many complex reasons that the lack of translation from animal models to the patient population may occur. This lack of efficacy prediction could be the result of over-extrapolation of the behaviors, as mice are less cognitively complex as humans, or it could be related to an over interpretation of efficacy. In addition the human schizophrenia population is diverse with no single unifying environmental or genetic known cause, which could lead to subgroups that respond differently to drugs, whereas mouse models are comparatively a very homogenous population. These differences between animal models and humans complicate both preclinical studies and clinical trial design and interpretation; however, when used with caution, animal models can be informative of mechanism and side effect profiles. Many researchers have also noted that part of the issue with designing better animal models is that the pathology in humans is so poorly understood and unbiased metrics are lacking for research and clinical trials [77]. In summary, there are many benefits to using different schizophrenia animal models to understand the mechanism of action for antipsychotics, but the field should remain cautious in directly extrapolating animal model data to human patients without further confirmatory studies.
Antipsychotics and “omics”: additional insights from transcriptomic and proteomic studies
While these recent molecular signaling cascade studies provide important insights into the pathways that are immediately affected, how these molecular shifts lead to antipsychotic action at the cellular, circuit and behavioral level remains unclear. Previous clinical research has shown that though antipsychotic action can take up to 3 weeks to reach fully efficacy, some symptom improvement is noted within the first 24 hours of antipsychotic treatment [78, 79]. To better understand how the fast-acting molecular changes ultimately incite prolonged antipsychotic efficacy, transcriptomics and proteomics studies have been employed to offer insight into the establishment of long-term changes in neuronal processes (summarized in table 1).
Interestingly, transcriptomic and proteomic studies in both human patients and rodents have highlighted a series of convergent targets suggesting that specific cellular processes and transcripts may play a role in antipsychotic action (table 1). One such cellular process, energy metabolism, has been proposed to be a target of antipsychotic action by both acute and chronic antipsychotic studies in both humans and rodents. Changes in the levels of metabolism-related transcripts and proteins, such as DISC1, optic atrophy 1 (OPA1), glutamate dehydrogenase 1 (GLUD1), and AKT1 were regulated by acute and chronic antipsychotic treatment [34, 35, 80–82]. In a 2009 study, Ji and colleagues examined the proteome of cortical synaptoneurosomes of rats treated with antipsychotics for 34 days and noted altered expression of proteins involved in glycolysis and glucose metabolism, further suggesting altered mitochondrial and energy metabolism in the brain following prolonged antipsychotic treatment [83].
The role of metabolic changes was also underscored in a recent study performed on peripheral blood mononuclear cells of schizophrenia patients sampled before and after 6 to 8 weeks of antipsychotic treatment [84]. Gene expression profiling revealed that antipsychotic treatment led to changes in proteins involved in different forms of metabolism, including carbohydrate, amino acid and nucleic acid metabolism. In addition, they demonstrated aberrant transcript abundance of AKT1 and DISC1 in mononuclear patient cells as compared with healthy volunteers before antipsychotic treatment. Interestingly, antipsychotic treatment normalized AKT1 but not DISC1 transcript levels, suggesting that AKT1 may be a stronger antipsychotic target in peripheral tissues.
In the same study, antipsychotics also restored physiological levels of ribosomal proteins and a transfer RNA (tRNA) synthesizing protein in schizophrenia patients [84]. Consistently, another study also indicated an antipsychotic-driven change in the level of the RNA helicase Ddx5,atranslation-related molecule that has been implicated in protein synthesis initiation [85]. Translational proteins in the form of ribosomal proteins were additionally implicated in a study in mice treated with antipsychotics for 2 weeks, as treatment increased the mRNA expression of two ribosomal proteins [86]. Other studies have also noted the upregulation of mTOR and Rps6 transcripts in rodents following antipsychotic treatment [87], which is especially remarkable as ribosomal proteins are thought to be mTOR-specific targets [88], and underscoring the evidence of the role of mTOR signaling in antipsychotic action. The upregulation of proteins involved in protein synthesis, such as ribosomal protein and tRNAs, suggest another interesting cellular process that is targeted by antipsychotics.
Another piece of evidence supporting the role of changes in protein synthesis (and potentially transcription as well) is data by Bowling et al. [35] that suggest that antipsychotics induce mTORC1 signaling and subsequent changes in ribosomal proteins within the first few hours of treatment and that this effect is maintained for at least 48 hours. Following the initial increase in protein synthesis related proteins such as ribosomal, chaperone and tRNA-related proteins, there was a later phase of increased cytoskeletal proteins. Interestingly, many of these candidate proteins overlapped with the previous reports in rodents and patients who had undergone prolonged antipsychotic treatment, suggesting that cytoskeletal protein expression may be induced within a day of treatment and maintained. One such previously reported cytoskeletal protein was microtubule-associated protein 2 (MAP2). It has been shown that Map2 transcripts and protein levels were increased in the rodent cortex after prolonged (3-week) treatment with antipsychotics [82, 89]. Additional cytoskeletal proteins other than Map2 were identified as upregulated following antipsychotic treatment in rodents and rodent neural progenitor cells (NPCs), including dynamin 1 (Dnm1) and glial fibrillary acidic protein (Gfap) [81, 82]. In summary, transcriptomic and proteomic studies support a role of cytoskeletal proteins following long-term antipsychotic treatment, especially for Map2.
In addition to the identified changes in proteins relating to the cytoskeleton, several large-scale studies also showed alterations in abundance of cell-adhesion and synaptic-activity proteins. Chan et al. [34] examined cortices of human schizophrenia patients treated with antipsychotics and compared their proteomes with those of human control subjects. They identified multiple altered proteins, most notably neural cell adhesion molecule 1 (NCAM1) and synaptosomal-associated protein, 91kDa (SNAP91), which are involved in cell adhesion and clathrin-dependent presynaptic vesicle assembly. Synaptic activity proteins Snap91 and synapsin I (Syn1) were also increased in proteomic rodent brain and neuron in culture-based antipsychotic studies [35, 82]. The changes in the abundance of proteins associated with metabolism, protein synthesis, cytoskeleton, cell adhesion and synaptic activity across multiple independent studies (table 1) suggest critical roles for these cellular processes in the action of antipsychotics.
|
Table 1:Cellular processes that are affected in schizophrenia patients and targeted by antipsychotics. Cellular processes with at two or more genes and multiple publications selecting their involvement in both schizophrenia pathology and antipsychotic action. |
|
Cellular process class
|
Reported genes and
cellular processes
|
Gene/RNA/protein
|
Schizophrenia pathology?
|
Antipsychotic action?
|
| Metabolism
Evidence
Kim et al. 2009 [103],
Prabakaran et al. 2004 [95],
Beaulieu et al. 2004 [30],
Emamian et al, 2004 [31],
Ahmed et al. 2012 [81],
Ma et al. 2009 [82],
Chan et al. 2011 [34],
Rosenfeld et al. 2011 [90],
Engman et al. 2011 [91],
Brennand et al. 2015 [94],
English et al. 2011 [96],
Mas et al. 2015 [126],
Kumarasinghe et al. 2013 [84]
|
AMPK, GLUD1, DISC1, AKT1, MTOR, OPA1,
|
Gene, RNA, protein
Patients and animal models |
Yes |
Yes |
|
Glucose metabolism, ATP synthesis, response to insulin signaling, amino acids, ATP/AMP ratio, glycolysis – gluconeogenesis pathway disruptions, increased reactive oxygen species
|
Human genetic screen, proteomic data in human patient iPSC NPCs, human metabolomic and proteomic screens
|
Microarray in human patient blood following treatment, Proteomic data in cultured rodent neurons following treatment, AMPK and mTOR signaling transcripts unbalanced in blood from patients with EPS symptoms
|
| Protein synthesis and processing
Evidence
Valjent et al. 2011 [21],
Bonito-Oliva et al. 2013 [36],
Bowling et al. 2014 [35],
Mas et al. 2015 [85],
Mas et al. 2015 [104],
Mas et al. 2015 [126],
Kumarasinghe et al. 2013 [84],
Thomas et al. 2003 [86]
|
mTOR, RPS6, EEF1A2, ribosomal proteins, CCT, RAPTOR, DDX5
|
Gene, RNA, protein
Patients and animal models |
Yes |
Yes
|
|
Translation initiation, translation elongation, protein folding
|
Transcripts reduced in patients compared to control subjects in blood cells, raptor SNP part of risk group for EPS in patients
|
Microarrays in human patient blood following treatment, one suggests difference between patients with and without EPS, proteomic data in cultured rodent neurons following treatment, microarray data in mouse brain following treatment
|
| Cytoskeleton
Evidence
Law et al. 2004 [89],
Ma et al. 2009 [82],
Ahmed et al. 2012 [81],
Shelton et al. 2015 [107],
Rosoklija et al. 2005 [108],
Jones et al. 2002 [109],
Arnold et al. 1991 [110],
Clark et al. 2006 [111],
Pennington et al. 2007 [112],
Millar et al. 2000 [53],
Chiba et al. 2006 [80]
|
MAP2, DISC1, DNM1
|
Gene, RNA, protein
Patients and animal models |
Yes |
Yes |
|
Microtubule dynamics, dendritic branching, spine dynamics
|
Human genetic screen, microarray and proteomic data in human iPSC NPCs, protein levels in human patient brains
|
Proteomic data in cultured rodent neurons following treatment, increase in transcripts in rodent brains following antipsychotic treatment
|
| Cell adhesion Goes et al. 2015 [116],
Atz et al. 2007 [119]
Tanaka et al. 2007 [120]
Vawter et al. 2001 [121],
Poltorak et al. 1997 [122],
Sullivan et al. 2007 [123],
Ayalew et al. 2012 [124],
Bowling et al. 2014 [35],
Brennand et al. 2015 [94],
Chan et al. 2011 [34]
|
NCAM1, CNTN4, NRXN1
|
Gene, RNA, protein
Patients and animal models |
Yes |
Yes |
|
Cell adhesion molecules, synaptic stability
|
Human genetic screen, microarray and proteomic data in human patient iPSC NPCS
|
Proteomic data in cultured rodent neurons and human patient brains following treatment
|
| Synaptic Activity Tam et al. 2010 [114],
Yu et al. 2012 [115],
Dolgin et al. 2014 [125],
Ma et al. 2009 [82],
Chan et al. 2011 [34],
Brennand et al. 2015 [94],
Bowling et al. 2014 [35],
Chiba et al. 2006 [80],
Millar et al. 2000 [53]
|
DISC1, SNAP91, SYN1, BIN1
|
Gene, RNA, protein
Patients and animal models |
Yes |
Yes |
|
Synaptic transmission, catecholamine synthesis and release, postsynaptic synaptic scaffolding, vesicle recycling
|
Human genetic screen, microarray and proteomic data in human patient iPSC NPCs
|
Proteomic data in cultured rodent neurons and rodent brains following treatment
|
| AMPK = 5' AMP-activated protein kinase; EPS = extrapyramidal symptoms; iPSC = induced pluripotent stem cell; mTOR = mechanistic target of rapamycin; NPC = neural progenitor cell; SNP = single nucleotide polymorphism |
Mechanisms underlying disease versus treatment
The wealth of new information on the molecular basis of antipsychotic action provided a more complete picture as to how antipsychotics modulate cellular outputs and processes. However, the question remained of how antipsychotics could be addressing underlying schizophrenia pathology. Several studies in humans, human cell culture systems and animal models have been performed to elucidate some of the potential mechanisms underlying schizophrenia pathology. Many of the studies highlight deficits in regulation in the genes and proteins targeted by schizophrenia that we have summarized in table 2.
|
Table 2:Targets associated with both schizophrenia pathology and antipsychotic action. The following genes have been implicated in at least three studies in both schizophrenia pathology and the mechanism of action of antipsychotics at the genetic, transcript and/or protein level. |
|
Gene name
|
Identified at the genomic/transcript/ protein/signaling level?
|
Human or rodent studies?
|
Implicated in schizophrenia?
|
Implicated in antipsychotic action?
|
References
|
|
AKT1
|
Gene, signaling |
Human
Rodent |
SNPs correlate with schizophrenia risk, reduced AKT1 protein levels in patient brains, one of the SNPs in a cluster that may predict patient EPS risk |
Increased signaling in response to acute treatment in rodent neurons, part of DISC1 regulation of neuronal development |
Bajestan et al. 2006 [98],
Emamian et al. 2004 [31],
Kim et al. 2009 [103],
Mas et al. 2015 [104],
Beaulieu et al. 2004 [30],
Bowling et al. 2014 [35],
Schwab et al. 2005 [97] |
|
BIN1
|
Gene, proteome |
Human
Rodent |
Rare insertion variants in human patients, altered in human patient iPSC NPCs |
Upregulated in rat brain proteome following chronic antipsychotic treatment |
Tam et al. 2010[14],
Ma et al. 2009 [82],
Brennand et al. 2015 [94] |
|
DDX5
|
Transcript, proteome |
Human
Rodent |
Measured as altered in human patient iPSC NPCs |
Transcripts differentially regulated in EPS vs non-EPS mice treated with antipsychotics, altered in rodent neurons at the proteome level following antipsychotic treatment |
Mas et al. 2015 [85],
Brennand et al. 2015 [94],
Bowling et al. 2014 [35] |
|
DNM1
|
Proteome |
Human
Rodent |
Increased in patient brains (proteome) |
Increased at proteome level following antipsychotic treatment in rodents |
Ma et al. 2009 [82],
Prabakaran et al. 2004 [95],
Clark et al. 2006 [111],
Pennington et al. 2007 [112] |
|
GLUD1
|
Transcript, Proteome, Enzymatic activity |
Human
Rodent |
Increased glutamate dehydrogenase activity in human patient prefrontal cortex, differentially regulated in human patient iPSC NPCs |
Altered in response to antipsychotic treatment in rodents and in patients. |
Burbaeva et al. 2003 [92],
Ma et al. 2009 [82],
Brennand et al. 2015 [94],
Bowling et al. 2014 [35]
Chan et al. 2011 [34] |
|
GSK3B
|
Gene, signaling |
Human
Rodent |
SNPs are a risk factor for schizophrenia |
Signaling changes following antipsychotic treatment |
Emamian et al. 2004 [31],
Beaulieu et al. 2011 [30],
Chen et al. 2015 [106],
Li et al. 2007 [32] |
|
MAP2
|
Transcript, proteome |
Human
Rodent |
Differential immunostaining in human patients (usually decreased) |
Increased at the transcript and protein level following antipsychotic treatment |
Jones et al. 2002 [109],
Rosoklija et al. 2005 [108],
Shelton et al. 2015 [107],
Law et al. 2004 [89],
Bowling et al. 2014 [35],
Ma et al. 2009 [82] |
|
MTOR
(overall pathway)
|
Gene, transcript, signaling |
Human
Rodent |
SNP of mTORC1 member Raptor indicated as risk factor for EPS in patients in concert with other SNPs |
Signaling increased following acute antipsychotic treatment, altered at the transcript level in patient blood following antipsychotic treatment with transcript differences between EPS and non-EPS patients in blood |
Mas et al. 2015 [85],
Mas et al. 2015 [104],
Mas et al. 2015 [126],
Bowling et al. 2014 [35],
Korostynski et al. 2013 [ |
|
NCAM1
|
Gene, transcript, proteome |
Human
Rodent |
SNPs are a risk factor for schizophrenia, change in abundance in cerebral spinal fluid in patients, changes in abundance in patient serum |
Changes at transcript and proteomic level following antipsychotic treatment |
Atz et al. 2007 [119],
Sullivan et al. 2007 [123],
Ayalew et al, 2012 [124],
Tanaka et al. 2007 [120],
Vawter et al. 2001 [121],
Poltorak et al. 1997 [2011],
Chan et al. 2011 [34] |
|
OPA1
|
Proteome, signaling |
Human
Rodent |
Reduced protein in patient prefrontal cortex, altered abundance in human patient iPSC NPCs |
Increased in human patient brains following treatment with antipsychotics |
Engmann et al. 2011 [91],
Chan et al. 2011 [34],
Brennand et al. 2015 [94] |
|
RPS6
|
Proteome, signaling |
Human
Rodent |
No |
Multiple pathways suggested upstream of RPS6 in response to acute antipsychotic treatment , differential phosphorylation associated with increased risk in EPS in mice |
Mas et al. 2015 [85],
Bowling et al. 2014[35],
Valjent et al. 2011 [21],
Korostynski et al. 2013 [87]
(website accessed Aug 2015) |
|
SNAP91
|
Gene, proteome |
Human
Rodent |
Identified in a GWAS and correlated with associated risk of schizophrenia |
Increased at proteome level following long term antipsychotic treatment in human patients and rodents |
Dolgin 2014 [125],
Ma et al. 2009 [82],
Chan et al. 2011 [34] |
|
SYN1
|
Gene, transcript, proteome |
Human
Rodent |
SNP is risk factor for schizophrenia, altered at the transcript level compared in human patient iPSC NPCs |
Increased with antipsychotic treatment in rodents |
Yu et al. 2012 [115],
Ma et al. 2009 [82],
Bowling et al. 2014 [35],
Brennand et al. 2015 [94] |
| AKT1 = a serine/threonine-protein kinase ; DISC1 = disrupted in schizophrenia 1; EPS = extrapyramidal symptoms; GWAS = genome-wode association study; iPSC = induced pluripotent stem cell; mTORC1 = mechanistic target of rapamycin complex 1; NPC = neural progenitor cell; RPS& = ribosomal protein S6; SNP = single nucleotide polymorphism |
There are multiple lines of evidence suggesting a disruption in metabolism that nicely compliment the data on antipsychotic mechanism of action. For instance, genetic studies of mitochondrial proteins have noted decreased expression of OPA1 [90, 91] that correlate to increased schizophrenia risk. In addition, increased glutamine dehydrogenase activity (including that of both GLUD1 and GLUD2) has been noted in the prefrontal cortex of schizophrenia patients compared with healthy controls [92]. GLUD1 is basally more active than GLUD2 [93], and likely contributes towards this increased activity [92].
Mitochondria-associated oxidative stress and damage were also evident in NPCs derived from human patient induced pluripotent stem cells (hiPSCs) with increased oxidative stress, decreased mitochondrial size, and altered cellular distribution [94]. Mitochondrial dysfunction, altered metabolism and oxidative stress were again noted in a separate metabolomic and proteomic study in human prefrontal cortex tissue [95]. Metabolic dysfunction was also observed to be a common thread in multiple studies of human schizophrenia patient tissue [96]. Given that antipsychotics have been shown to alter transcription and protein abundance of several of the specific genes indicated in mitochondrial and metabolism defects, the cellular processes regulating metabolism appears to be a site of interplay between known schizophrenia pathology and the action of antipsychotics.
In addition to metabolism changes, genes encoding for signaling molecules involved in metabolism and other key cellular processes such as AKT1, DISC1, GSK3B and genes encoding for proteins regulating the mTOR pathway have been implicated in schizophrenia risk and pathology. Although not all populations appear to have a correlation between variants in the AKT1 gene and schizophrenia, several studies across the world have reported AKT1as a risk gene for schizophrenia [31, 97–101]. Schizophrenia has also been associated with genetic disruptions such as in the DISC1 gene identified in smaller cohort of patients [102]. DISC1 protein, which is involved in the regulation of mitochondrial and cytoskeletal function, has also been shown to interact with Akt-mTOR signaling in mice [103], further suggesting a convergence of signaling in schizophrenia pathology. Indeed, a single nucleotide polymorphism (SNP) in RAPTOR, encoding for the protein interacting with mTOR in mTORC1, has also been found to be predictive of adverse effects in patients who develop extrapyramidal symptoms (EPS) [104]. Protein synthesis and ribosomal proteins, which are also downstream of Akt-mTORC1, have additionally been reported to be downregulated in olfactory cells of schizophrenia patients [105]. GSK3β, another downstream effector of AKT1, also has variations associated with increased risk for schizophrenia in the Han Chinese population [106], suggesting another point of interaction of the signaling cascades implicated in both pathology and antipsychotic action. Because these signaling molecules have been heavily implicated in multiple cellular processes including metabolism, cytoskeleton, protein synthesis and transcription, they also represent an important point of convergence for pathology and mechanism of action of antipsychotics at the protein and signaling levels.
The next category of cellular processes that is a target of antipsychotic action is the cytoskeleton. MAP2, which was upregulated following antipsychotic exposure in multiple studies, has been overwhelmingly reported to be downregulated in the brains of human schizophrenia patient [107–110]. DNM1, another cytoskeleton protein, was shown to be increased in patient brains at the proteomic level [95, 111, 112] and was also targeted by antipsychotics [35, 82]. These data firmly support a role of cytoskeletal proteins in both schizophrenia and in its treatment.
Because of the hypotheses that schizophrenia is caused by alterations in neurotransmitter signal transduction and synaptic activity, proteins that affect synaptic activity, stability and function have long been an area of intense investigation in schizophrenia. Recently, it has been shown that evoked synaptic release of catecholamines (dopamine, epinephrine and norepinephrine) from hiPSCs was different in schizophrenia and control patients [113]. Not only did the schizophrenia patient-derived neurons exhibit increased catecholamine release, but also proportionally more neurons had proteins to synthesize catecholamines. This suggests that there may be increased catecholamine synthesis and improper increased release in human patients under specific evoked conditions. In addition, they also discovered variations in genes that encode proteins involved in synaptic endocytosis, release and synaptic transmission such as bridging integrator 1 (BIN1) [114], SYN1 [115], contactin 4 (CNTN4) [116, 117] as well as single nucleotide polymorphisms in cell adhesion molecule NCAM1[119, 123] and exonic deletions in Neurexin 1 (NRXN1) [118]. Moreover, the changes in the cell adhesion molecule NCAM1 in human patient cerebral spinal fluid and serum [119–124] are important because NCAM1 is also a known target of antipsychotic action. Though more investigation is needed, changes in synaptic release proteins, cell-cell interactions and synaptic properties have been reported in patients and by other investigators following antipsychotic treatment [34, 35, 82, 94, 115, 125], therefore, the idea that antipsychotics may correct schizophrenia pathology by decreasing the effects of overly secreted catecholamines and altering the synaptic landscape by changing cell adhesion molecules is potentially promising and merits further investigation.
Primary versus off-target effects?
Given the new insights gained in the potential molecular mechanisms of action of antipsychotics, one important question still remains: how many of these molecular changes are related to the efficacy of treatment versus the appearance of undesirable side effects, such as extrapyramidal symptoms (EPS) and potential neurotoxicity? Though this is an emerging area of research, some studies suggest that this relationship may be complex. Recently, it was discovered that the presence of four SNPs in gene encoding for proteins regulating the Akt-mTORC1 pathway in schizophrenia patients (including AKT1 and mTORC1 member, RAPTOR) predicted an increased risk for EPS after 15 days of treatment [104]. Patients who did not experience EPS had altered abundance of mRNA transcripts related to protein folding in their peripheral blood. In contrast, patients with EPS had changes in transcripts related to mTOR and 5' AMP-activated protein kinase (AMPK), suggesting a different role of mTOR balance and energy signaling in the onset of EPS [126]. Although these data were not confirmed at the protein or signaling level, taken together, they suggest that changes in the Akt-mTOR pathway may predict the likelihood of EPS [126]. These findings provide evidence that there may discreet signaling differences between EPS and non-EPS patients that could be characterized for biomarkers.
To fully understand the relationship between the Akt-mTORC1 pathway and extrapyramidal symptoms, studies in animal models are required. Indeed, the relationship between mTOR signaling and EPS was investigated in the brains of two rodent strains with different sensitivities to EPS, and it was discovered that the strain with increased sensitivity to EPS had reduced phosphorylation in the mTORC1-dependent Rps6 site, and increased phosphorylation in the site that has also been shown to be downstream of MEK-ERK and RSK signaling [126–128].
Another preliminary unpublished study discussed by Chao and Klann [129], found that disruptions in the mTORC1 pathway did not prevent the onset of catalepsy following acute (within 2 hours of) antipsychotic treatment. Because catalepsy in animal models has been reported to predict EPS liability in humans [130], these data further suggest that the normal activity of the mTORC1 pathway may not be critical to the onset of EPS following antipsychotic treatment, but disruptions in mTOR signaling may influence EPS liability. Indeed, multiple studies have suggested that molecular signaling pathways other than mTOR contribute to antipsychotic-induced catalepsy, and thus, potentially EPS.
Many of these pathways are associated with PKA that is a key regulator of dopamine signaling in the striatum [22]. PKA-cFos signaling and delta FosB have both been reported to be upregulated following antipsychotic treatment and catalepsy induction [131–133]. Another downstream effector of PKA, Darpp32 has also been implicated in the induction of catalepsy as DARPP32 knockout mice have reduced catalepsy following antipsychotic treatment [134]. Muscarinic receptors have additionally been suggested to play a role in catalepsy induction as acetylcholine muscarinic receptor 4 (M4) knockout mice have diminished catalepsy as well [135]. These studies support a role for signaling pathways other than mTORC1 in the onset of catalepsy, which may be a predictor for EPS in human patients.
These findings clearly demonstrate a role of multiple signaling pathways in the induction of extrapyramidal symptoms following antipsychotic treatment. Although some pathways, such as those in the PKA intracellular signaling cascade suggest a convergent role in EPS, other findings such as the potential role of atypical mTORC1 signaling remain under characterized. However, the upregulation of PKA and aberrant or underactive mTOR signaling may play a role in EPS in patients and may indicate a potential area for future biomarkers of EPS.
Biomarkers for patient stratification
The potentially predictive markers for the induction of EPS in humans and rodents signify possibilities for biomarkers that could stratify the patient population. As there is no known unifying genetic or environmental cause of schizophrenia, stratification could both streamline clinical trials and improve care for patients, as those at risk of developing EPS could be identified even if it is after beginning treatment but before the development of full EPS. Though both PKA and Akt-mTORC1 signaling may be promising for developing biological and unbiased means of patient stratification and EPS biomarkers, they still require more investigation. In addition, the fact that mTORC1 and translation-related proteins and signaling have been positively correlated with efficacy of antipsychotics, and dysregulation with their side effects, may suggest that careful titration of these pathways is supremely important in separating efficacy from undesirable off-target drug actions. In summary, the idea of distinguishing which of these newly identified molecular candidates is important for efficacy versus side effects (and if this role is temporally dependent) will be an important focus for future research.
Biomarkers for efficacy
Another hurdle that identifying molecular markers for antipsychotic action could help overcome is that of unbiased markers of efficacy. To date, the majority of clinical trials have relied on rating scales and not unbiased biological metrics to assess antipsychotic efficacy; however, these scales can be problematic, difficult to interpret, limited by rater reliability and their reporting can introduce bias [136, 137]. Therefore, an unbiased biologically based metric performed either through blood tests, positron emission tomography, or magnetic resonance imaging or through peripheral tissue would be beneficial in assessing the efficacy of treatment for further trial efforts in most psychiatric and neuroscience based indications.
The identification of key molecular pathways and players in efficacious treatment of schizophrenia would not only open a new avenue for drug discovery, but would also provide a launching pad for identifying markers of efficacy that could be used to better design clinical trials. Given that AKT1 and RPS25 transcripts have been shown to be rescued in blood cells following 6 to 8 weeks of antipsychotic treatment [84], blood-based efficacy markers do seem possible. Though we have mentioned some key genes, proteins and cell signaling cascades that could prove an informed starting point, more careful examination of their relationship with antipsychotic efficacy in specific temporal intervals would be required to fully establish any of them as true biomarkers. Although many novel targets associated with antipsychotic efficacy have emerged following recent research, it is important that they be put into the context of known genetic mutations and proteomic differences associated with schizophrenia patients (table 2).
Conclusion
Given the wealth of new data in the past decade, there is now a clearer framework for the mechanisms of action of antipsychotics and how they could interact with schizophrenia pathology (tables 1–2, fig. 2). Although studies need to be performed to establish primary versus off-target effects, this is far more possible than it was a decade ago. Future efforts should focus primarily on (a) elucidating the role of these targets in efficacy vs side effects (may not be direct, but dose or temporally related) (b) identifying biomarkers as a means of tracking efficacy in an unbiased manner for more expedient and clear clinical trials and (c) drug discovery efforts for drugs with more improvements and fewer side effects. With these advances in our understanding, there is renewed optimism for future drug discovery and biomarker efforts.
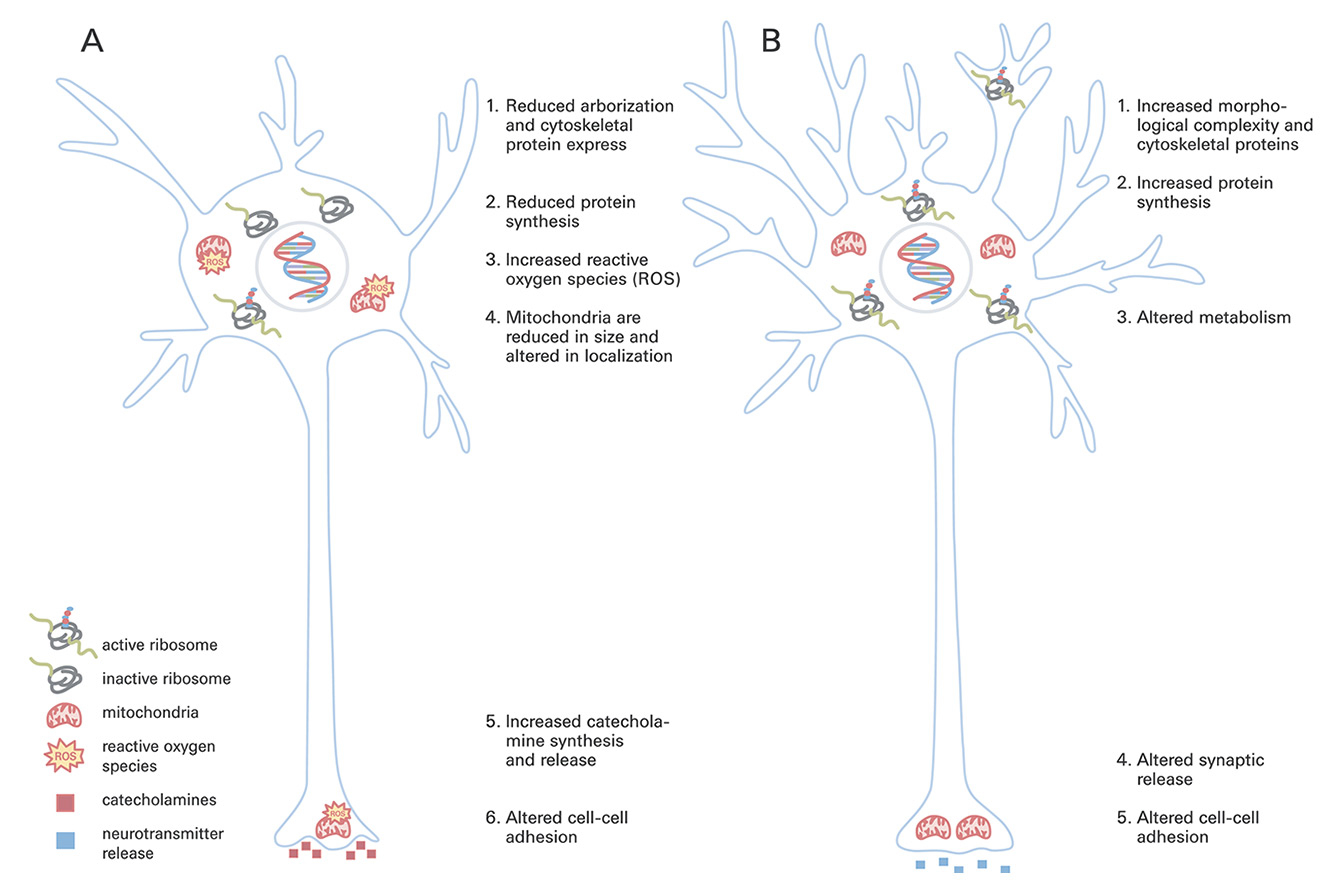
Figure 2
Schematic representation of reported changes to neurons in schizophrenia (A) and following antipsychotic treatment (B). Organelles are drawn to scale.
Acknowledgements:NJ Arnhols is acknowledged for her contribution. H. B. was supported by NIH grants NS034007 and NS047384. E.S. was supported by NIH grant NS087112. We also acknowledge that we could not include all aspects of antipsychotics and schizophrenia in this review and apologize to those whose excellent work we did not discuss.
Authors’ contribution: Equal contributors.
Disclosure statement: The authors have no known conflicts of interest. All outside support has been from the NIH, with no commercial money provided.
Correspondence: Heather Bowling, PhD, Center for Neural Science, New York University, USA-New York, NY, hlb248[at]nyu.edu
References
1 van Rossum JM. The significance of dopaminereceptor blockade for the action of neuroleptic drugs. Archives Internationales de Pharmacodynamie et de Therapie 1966;160:492–4.
2 Creese I, Burt DR, Snyder SH. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science. 1976;192(4238):481–3.
3 Snyder SH. The dopamine hypothesis of schizophrenia: focus on the dopamine receptor. Am J Psychiatry. 1976;133(2):197–202.
4 Regier DA, Narrow WE, Rae DS, Manderscheid RW, Locke BZ, Goodwin FK. The de facto US mental and addictive disorders service system. Epidemiologic catchment area prospective 1-year prevalence rates of disorders and services. Arch Gen Psychiatry. 1993:50(2):85–94.
5 Saha S, Chant D, Welham J, McGrath J. A systematic review of the prevalence of schizophrenia. PLoS Med. 2005;2(5):e141.
6 The Use of Medicines in the United States: Review of 2011. IMS Institute for Healthcare Informatics. 2012
7 Erhart SM, Marder SR, Carpenter WT. Treatment of schizophrenia negative symptoms: future prospects. Schizophr Bull. 2006;32(2):234–7.
8 Rummel-Kluge C, Komossa K, Schwarz S, Hunger H, Schmid F, Lobos CA, Kissling W, Davis JM, Leucht S. Head-to-head comparisons of metabolic side effects of second generation antipsychotics in the treatment of schizophrenia: a systematic review and meta-analysis. Schizophr Res. 2010;123(2-3):225–33.
9 Marchese G, Casti P, Ruiu S, Saba P, Sanna A, Casu G, Pani L. Haloperidol, but not clozapine, produces dramatic catalepsy in delta9-THC-treated rats: possible clinical implications. Br J Pharmacol. 2003;140(3):520–6.
10 http://medical-dictionary.thefreedictionary.com/catalepsy
11 Lewis S, Lieberman J. CATIE and CUtLASS: can we handle the truth? Br J Psychiatry. 2008;192(3):161–3.
12 Kane J, Honigfeld G, Singer J, Meltzer H. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789–96.
13 Miller CH, Mohr F, Umbricht D, Woerner M, Fleischhacker WW, Lieberman JA. The prevalence of acute extrapyramidal signs and symptoms in patients treated with clozapine, risperidone, and conventional antipsychotics. J Clin Psychiatry. 1998;59(2):69–75.
14 Marder SR1, McQuade RD, Stock E, Kaplita S, Marcus R, Safferman AZ, et al. Aripiprazole in the treatment of schizophrenia: safety and tolerability in short-term, placebo-controlled trials. Schizophr Res. 2003;61(2-3):123–36.
15 Citrome L. A review of aripiprazole in the treatment of patients with schizophrenia or bipolar I disorder. Neuropsychiatr Dis Treat. 2006:2(4):427–43.
16 McCue RE, Waheed R, Urcuyo L, Orendain G, Joseph MD, Charles R, et al. Comparative effectiveness of second-generation antipsychotics and haloperidol in acute schizophrenia. Br J Psychiatry. 2006;189:433–40. Replaced Boettger
17 Moghaddam B, Javitt D. From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology. 2012;37(1):4–15.
18 Roche Media Release April 14, 2014.
19 Alberati D, Moreau JL, Lengyel J, Hauser N, Mory R, Borroni E, et al. Glycine reuptake inhibitor RG1678: a pharmacologic characterization of an investigational agent for the treatment of schizophrenia. Neuropharmacology. 2012;62(2):1152–61.
20 “Lilly Stops Phase III Development of Pomaglumetad Methionil For the Treatment of Schizophrenia Based on Efficacy Results” Eli Lilly Media Release. August 29, 2012.
21 Valjent E, Bertran-Gonzalez J, Bowling H, Lopez S, Santini E, Matamales M, et al. Haloperidol regulates the state of phosphorylation of ribosomal protein S6 via activation of PKA and phosphorylation of DARPP-32. Neuropsychopharmacology. 2011;36(12):2561–70.
22 Borgkvist A, Fisone G. Psychoactive drugs and regulation of the cAMP/PKA/DARPP-32 cascade in striatal medium spiny neurons. Neurosci Biobehav Rev. 2007;31(1):79–88.
23 Blasi G, Napolitano F, Ursini G, Taurisano P, Romano R, Caforio G, et al. DRD2/AKT1 interaction on D2 c-AMP independent signaling, attentional processing, and response to olanzapine treatment in schizophrenia. Proc Natl Acad Sci U S A. 2011;108(3):1158–63.
24 Håkansson K, Galdi S, Hendrick J, Snyder G, Greengard P, Fisone G. Regulation of phosphorylation of the GluR1 AMPA receptor by dopamine D2 receptors. J Neurochem. 2006;96(2):482–8.
25 Svenningsson P, Lindskog M, Ledent C, Parmentier M, Greengard P, Fredholm BB, Fisone G. Regulation of the phosphorylation of the dopamine- and cAMP-regulated phosphoprotein of 32 kDa in vivo by dopamine D1, dopamine D2, and adenosine A2A receptors. Proc Natl Acad Sci U S A. 2000;15;97(4):1856–60.
26 Bertran-Gonzalez J, Håkansson K, Borgkvist A, Irinopoulou T, Brami-Cherrier K, Usiello A, et al. Histone H3 phosphorylation is under the opposite tonic control of dopamine D2 and adenosine A2A receptors in striatopallidal neurons. Neuropsychopharmacology. 2009;34(7):1710–20.
27 Girgis RR, Javitch JA, Lieberman JA. Antipsychotic drug mechanisms: links between therapeutic effects, metabolic side effects and the insulin signaling pathway. Mol Psychiatry. 2008;13(10):918–29.
28 Beaulieu JM, Del’guidice T, Sotnikova TD, Lemasson M, Gainetdinov RR. Beyond cAMP: The Regulation of Akt and GSK3 by Dopamine Receptors. Front Mol Neurosci. 2011;4:38.
29 Masri B, Salahpour A, Didriksen M, Ghisi V, Beaulieu JM, Gainetdinov RR, Caron MG. Antagonism of dopamine D2 receptor/beta-arrestin 2 interaction is a common property of clinically effective antipsychotics. Proc Natl Acad Sci U S A. 2008;105(36):13656–61.
30 Beaulieu JM, Sotnikova TD, Yao WD, Kockeritz L, Woodgett JR, Gainetdinov RR, Caron MG. Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci U S A. 2004;101(14):5099–104.
31 Emamian ES, Hall D, Birnbaum MJ, Karayiorgou M, Gogos JA. Convergent evidence for impaired AKT1-GSK3beta signaling in schizophrenia. Nat Genet. 2004;36(2):131–7.
32 Li X, Rosborough KM, Friedman AB, Zhu W, Roth KA. Regulation of mouse brain glycogen synthase kinase-3 by atypical antipsychotics. Int J Neuropsychopharmacol. 2007;10(1):7–19.
33 Pan B, Chen J, Lian J, Huang XF, Deng C. Unique Effects of Acute Aripiprazole Treatment on the Dopamine D2 Receptor Downstream cAMP-PKA and Akt-GSK3β Signalling Pathways in Rats. PLoS One. 2015;10(7):e0132722.
34 Chan MK, Tsang TM, Harris LW, Guest PC, Holmes E, Bahn S. Evidence for disease and antipsychotic medication effects in post-mortem brain from schizophrenia patients. Mol Psychiatry. 2011;16(12):1189–202.
35 Bowling H, Zhang G, Bhattacharya A, Pérez-Cuesta LM, Deinhardt K, Hoeffer CA, Neubert TA, Gan WB, Klann E, Chao MV. Antipsychotics activate mTORC1-dependent translation to enhance neuronal morphological complexity. Sci Signal. 2014;7(308):ra4.
36 Bonito-Oliva A, Pallottino S, Bertran-Gonzalez J, Girault JA, Valjent E, Fisone G. Haloperidol promotes mTORC1-dependent phosphorylation of ribosomal protein S6 via dopamine- and cAMP-regulated phosphoprotein of 32 kDa and inhibition of protein phosphatase-1. Neuropharmacology. 2013;72:197–203.
37 Weeks KR, Dwyer DS, Aamodt EJ. Antipsychotic drugs activate the C. elegans akt pathway via the DAF-2 insulin/IGF-1 receptor. ACS Chem Neurosci. 2010;1(6):463–73.
38 Jones CA, Watson DJG, Fone KCF. Animal models of schizophrenia. Br J Pharmacol. 2011;164:1162–94.
39 Kendler KS, MacLean CJ, O’Neill FA, Burke J, Murphy B, Duke F, et al. Evidence for a schizophrenia vulnerability locus on chromosome 8p in the Irish Study of High-Density Schizophrenia Families. Am J Psychiatry. 1996;153:1534–40.
40 Arguello PA, Gogos JA. Modeling madness in mice: one piece at a time. Neuron. 2006;52:179–96.
41 Stefansson H, et al. Neuregulin 1 and susceptibility to schizophrenia. Am J Hum Genet. 2002;71:877–92.
42 Munafò MR, Thiselton DL, Clark TG, Flint J. Association of the NRG1 gene and schizophrenia: a meta-analysis. Mol Psychiatry. 2006;11:539–46.
43 Mei L, Xiong W-C. Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat Rev Neurosci. 2008;9:437–52.
44 Sullivan PF, Lin D, Tzeng J-Y, van den Oord E, Perkins D, Stroup TS, et al. Genomewide association for schizophrenia in the CATIE study: results of stage 1. Mol Psychiatry. 2008;13:570–84.
45 Agim ZS, Esendal M, Briollais L, Uyan O, Meschian M, Martinez LAM, et al. Discovery, validation and characterization of Erbb4 and Nrg1 haplotypes using data from three genome-wide association studies of schizophrenia. Hu VW, ed. PLoS ONE 2013;8:e53042.
46 Rimer M, Barrett DW, Maldonado MA, Vock VM, Gonzalez-Lima F. Neuregulin-1 immunoglobulin-like domain mutant mice: clozapine sensitivity and impaired latent inhibition. Neuroreport. 2005;16:271–5.
47 O’Tuathaigh CM, O’Sullivan GJ, Kinsella A, Harvey RP, Tighe O, Croke DT, Waddington JL. Sexually dimorphic changes in the exploratory and habituation profiles of heterozygous neuregulin-1 knockout mice. Neuroreport. 2006;17:79–83.
48 Karl T, Duffy L, Scimone A, Harvey RP, Schofield PR. Altered motor activity, exploration and anxiety in heterozygous neuregulin 1 mutant mice: implications for understanding schizophrenia. Genes Brain Behav. 2007;6:677–87.
49 Dean B, Karl T, Pavey G, Boer S, Duffy L, Scarr E. Increased levels of serotonin 2A receptors and serotonin transporter in the CNS of neuregulin 1 hypomorphic/mutant mice. Schizophr Res. 2008;99:341–9.
50 Deakin IH, Law AJ, Oliver PL, Schwab MH, Nave KA, Harrison PJ, Bannerman DM. Behavioural characterization of neuregulin 1 type I overexpressing transgenic mice. Neuroreport. 2009;20:1523–8.
51 Deakin IH, Nissen W, Law AJ, Lane T, Kanso R, Schwab MH, et al. Transgenic overexpression of the type I isoform of neuregulin 1 affects working memory and hippocampal oscillations but not long-term potentiation. Cereb Cortex. 2012;22:1520–9.
52 Roy K, Murtie JC, El-Khodor BF, Edgar N, Sardi SP, Hooks BM, et al. Loss of erbB signaling in oligodendrocytes alters myelin and dopaminergic function, a potential mechanism for neuropsychiatric disorders. Proc Natl Acad Sci USA. 2007;104:8131–6.
53 Millar JK, Wilson-Annan JC, Anderson S, Christie S, Taylor MS, Semple CA, et al. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet. 2000;9:1415–23.
54 Hennah W, Varilo T, Kestilä M, Paunio T, Arajärvi R, Haukka J, et al. Haplotype transmission analysis provides evidence of association for DISC1 to schizophrenia and suggests sex-dependent effects. Hum Mol Genet. 2003;12:3151–9.
55 Callicott JH, Straub RE, Pezawas L, Egan MF, Mattay VS, Hariri AR, et al. Variation in DISC1 affects hippocampal structure and function and increases risk for schizophrenia. Proc Natl Acad Sci USA. 2005;102:8627–32.
56 Cannon TD, Hennah W, van Erp TGM, Thompson PM, Lonnqvist J, Huttunen M, et al. Association of DISC1/TRAX haplotypes with schizophrenia, reduced prefrontal gray matter, and impaired short- and long-term memory. Arch Gen Psychiatry. 2005;62:1205–13.
57 Koike H, Arguello PA, Kvajo M, Karayiorgou M, Gogos JA. Disc1 is mutated in the 129S6/SvEv strain and modulates working memory in mice. Proc Natl Acad Sci USA. 2006;103:3693–7.
58 Kuroda K, et al. Behavioral alterations associated with targeted disruption of exons 2 and 3 of the Disc1 gene in the mouse. Hum Mol Genet. 2011;20:4666–83.
59 Pletnikov MV, Ayhan Y, Nikolskaia O, Xu Y, Ovanesov MV, Huang H, et al. Inducible expression of mutant human DISC1 in mice is associated with brain and behavioral abnormalities reminiscent of schizophrenia. Mol Psychiatry. 2008;13:173–86–115.
60 Hikida T, Jaaro-Peled H, Seshadri S, Oishi K, Hookway C, Kong S, et al. Dominant-negative DISC1 transgenic mice display schizophrenia-associated phenotypes detected by measures translatable to humans. Proc Natl Acad Sci USA. 2007;104:14501–6.
61 Li W, Zhou Y, Jentsch JD, Brown RAM, Tian X, Ehninger D, et al. Specific developmental disruption of disrupted-in-schizophrenia-1 function results in schizophrenia-related phenotypes in mice. Proc Natl Acad Sci. 2007;104:18280–5.
62 Duan X, Chang JH, Ge S, Faulkner RL, Kim JY, Kitabatake Y, et al. Disrupted-In-Schizophrenia 1 regulates integration of newly generated neurons in the adult brain. Cell. 2007;130:1146–58.
63 Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148(10):1301–8.
64 Mouri A, Noda Y, Enomoto T, Nabeshima T. Phencyclidine animal models of schizophrenia: approaches from abnormality of glutamatergic neurotransmission and neurodevelopment. Neurochem Int. 2007;51(2-4):173–84.
65 Hoffman DC, Donovan H, Cassella JV. The effects of haloperidol and clozapine on the disruption of sensorimotor gating induced by the noncompetitive glutamate antagonist MK-801. Psychopharmacology (Berl). 1993;111(3):339–44.
66 Abdul-Monim Z, Reynolds GP, Neill JC. The effect of atypical and classical antipsychotics on sub-chronic PCP-induced cognitive deficits in a reversal-learning paradigm. Behav Brain Res. 2006;169(2):263–73.
67 Steinpreis RE, Anders KA, Branda EM, Kruschel CK. The effects of atypical antipsychotics and phencyclidine (PCP) on rotorod performance. Pharmacol Biochem Behav. 1999;63(3):387–94.
68 Profaci CP, Krolikowski KA, Olszewski RT, Neale JH. Group II mGluR agonist LY354740 and NAAG peptidase inhibitor effects on prepulse inhibition in PCP and D-amphetamine models of schizophrenia. Psychopharmacology (Berl). 2011;216(2):235–43.
69 Schlumberger C, Schäfer D, Barberi C, Morè L, Nagel J, Pietraszek M, et al. Effects of a metabotropic glutamate receptor group II agonist LY354740 in animal models of positive schizophrenia symptoms and cognition. Behav Pharmacol. 20(1):56–66.
70 Harada K, Nakato K, Yarimizu J, Yamazaki M, Morita M, Takahashi S, et al. A novel glycine transporter-1 (GlyT1) inhibitor, ASP2535 (4-[3-isopropyl-5-(6-phenyl-3-pyridyl)-4H-1,2,4-triazol-4-yl]-2,1,3-benzoxadiazole), improves cognition in animal models of cognitive impairment in schizophrenia and Alzheimer's disease. Eur J Pharmacol. 2012;685(1-3):59–69.
71 Liu Y, Guo L, Duan H, Zhang L, Jiang N, Zhen X, Shen J. Discovery of 4-benzoylpiperidine and 3-(piperidin-4-yl)benzo[d]isoxazole derivatives as potential and selective GlyT1 inhibitors. RSC Adv. 2015;5:40964–77.
72 Marcotte ER, Pearson DM, Srivastava LK. Animal models of schizophrenia: a critical review. J Psychiatry Neurosci. 2001;26(5):395–410.
73 McKinney WT, Moran EC. Animal models of schizophrenia. Am J Psychiatry. 1981;138(4):478–83.
74 Rebec GV, Bashore TR. Critical issues in assessing the behavioral effects of amphetamine. Neurosci Biobehav Rev. 1984;8(1):153–9.
75 Andersen MP, Pouzet B. Effects of acute versus chronic treatment with typical or atypical antipsychotics on d-amphetamine-induced sensorimotor gating deficits in rats. Psychopharmacology (Berl). 2001;156(2-3):291–304.
76 Weiner I, Lubow RE, Feldon J. Abolition of the expression but not the acquisition of latent inhibition by chronic amphetamine in rats. Psychopharmacology (Berl). 1984;83(2):194–9.
77 Improving the Utility and Translation of Animal Models for Nervous System Disorders: Workshop Summary.Institute of Medicine (US) Forum on Neuroscience and Nervous System Disorders. Washington (DC): National Academies Press (US); 2013.
78 Kapur S, Arenovich T, Agid O, Zipursky R, Lindborg S, Jones B. Evidence for onset of antipsychotic effects within the first 24 hours of treatment. Am J Psychiatry. 2005;162:939–46 CrossRefMedlineWeb of ScienceGoogle Scholar
79 Wright P, Lindborg SR, Birkett M, Meehan K, Jones B, Alaka K, et al. Intramuscular olanzapine and intramuscular haloperidol in acute schizophrenia: Antipsychotic efficacy and extrapyramidal safety during the first 24 hours of treatment. Can J Psychiatry. 2003;48:716–21.
80 Chiba S, Hashimoto R, Hattori S, Yohda M, Lipska B, Weinberger DR, Kunugi H. Effect of antipsychotic drugs on DISC1 and dysbindin expression in mouse frontal cortex and hippocampus. J Neural Transm (Vienna). 2006;113(9):1337–46.
81 Ahmed EU, Ahmed S, Ukai W, Matsumoto I, Kemp A, McGregor IS, Kashem MA. Antipsychotic induced alteration of growth and proteome of rat neural stem cells. Neurochem Res. 2012;37(8):1649–59.
82 Ma D, Chan MK, Lockstone HE, Pietsch SR, Jones DN, Cilia J, et al. Antipsychotic treatment alters protein expression associated with presynaptic function and nervous system development in rat frontal cortex. J Proteome Res. 2009;8(7):3284–97.
83 Ji B, Zhang Z, Zhang M, Zhu H, Zhou K, Yang J, et al. Differential expression profiling of the synaptosome proteome in a rat model of antipsychotic resistance. Brain Res. 2009;1295:170–8.
84 Kumarasinghe N, Beveridge NJ, Gardiner E, Scott RJ, Yasawardene S, Perera A, et al. Gene expression profiling in treatment-naive schizophrenia patients identifies abnormalities in biological pathways involving AKT1 that are corrected by antipsychotic medication. Int J Neuropsychopharmacol. 2013;16(7):1483–503.
85 Mas S, Gassó P, Boloc D, Rodriguez N, Marmol F, Sanchez J, et al. Network analysis of gene expression in mice provides new evidence of involvement of the mTOR pathway in antipsychotic-induced extrapyramidal symptoms. Pharmacogenomics J. 2015: in press (a).
86 Thomas EA, George RC, Danielson PE, Nelson PA, Warren AJ, Lo D, Sutcliffe JG. Antipsychotic drug treatment alters expression of mRNAs encoding lipid metabolism-related proteins. Mol Psychiatry. 2003;8(12):983–93.
87 Korostynski M, Piechota M, Dzbek J, Mlynarski W, Szklarczyk K, Ziolkowska B, Przewlocki R. Novel drug-regulated transcriptional networks in brain reveal pharmacological properties of psychotropic drugs. BMC Genomics. 2013;14:606.
88 Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature. 2012;485(7396):109–13.
89 Law AJ, Hutchinson LJ, Burnet PW, Harrison PJ. Antipsychotics increase microtubule-associated protein 2 mRNA but not spinophilin mRNA in rat hippocampus and cortex. J Neurosci Res. 2004;76(3):376–82.
90 Rosenfeld M, Brenner-Lavie H, Ari SG, Kavushansky A, Ben-Shachar D. Perturbation in mitochondrial network dynamics and in complex I dependent cellular respiration in schizophrenia. Biol Psychiatry. 2011;69(10):980–8.
91 Engmann O, Hortobágyi T, Pidsley R, Troakes C, Bernstein HG, Kreutz MR, et al. Schizophrenia is associated with dysregulation of a Cdk5 activator that regulates synaptic protein expression and cognition. Brain. 2011;134(Pt 8):2408–21.
92 Burbaeva GSh, Boksha IS, Turishcheva MS, Vorobyeva EA, Savushkina OK, Tereshkina EB. Glutamine synthetase and glutamate dehydrogenase in the prefrontal cortex of patients with schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27(4):675–80.
93 Spanaki C, Zaganas I, Kounoupa Z, Plaitakis A. The complex regulation of human glud1 and glud2 glutamate dehydrogenases and its implications in nerve tissue biology. Neurochem Int. 2012;61(4):470–81.
94 Brennand K, Savas JN, Kim Y, Tran N, Simone A, Hashimoto-Torii K, et al. Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol Psychiatry. 2015;20(3):361–8.
95 Prabakaran S, Swatton JE, Ryan MM, Huffaker SJ, Huang JT, Griffin JL, et al. Mitochondrial dysfunction in schizophrenia: evidence for compromised brain metabolism and oxidative stress. Mol Psychiatry. 2004;9(7):684–97.
96 English JA, Pennington K, Dunn MJ, Cotter DR. The neuroproteomics of schizophrenia. Biol Psychiatry. 2011;69(2):163–72.
97 Schwab SG, Hoefgen B, Hanses C, Hassenbach MB, Albus M, Lerer B, et al. Further evidence for association of variants in the AKT1 gene with schizophrenia in a sample of European sib-pair families. Biol Psychiatry. 2005;58(6):446–50.
98 Bajestan SN, Sabouri AH, Nakamura M, Takashima H, Keikhaee MR, Behdani F, et al. Association of AKT1 haplotype with the risk of schizophrenia in Iranian population. Am J Med Genet B Neuropsychiatr Genet. 2006;141B(4):383–6.
99 Ikeda M, Iwata N, Suzuki T, Kitajima T, Yamanouchi Y, Kinoshita Y, et al. Association of AKT1 with schizophrenia confirmed in a Japanese population. Biol Psychiatry. 2004;56(9):698–700.
100 Xu MQ1, Xing QH, Zheng YL, Li S, Gao JJ, He G, et al. Association of AKT1 gene polymorphisms with risk of schizophrenia and with response to antipsychotics in the Chinese population. J Clin Psychiatry. 2007;68(9):1358–67.
101 Thiselton DL1, Vladimirov VI, Kuo PH, McClay J, Wormley B, Fanous A, et al. AKT1 is associated with schizophrenia across multiple symptom dimensions in the Irish study of high density schizophrenia families. Biol Psychiatry. 2008;63(5):449–57.
102 Millar JK, Wilson-Annan JC, Anderson S, Christie S, Taylor MS, Semple CA, et al. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet. 2000;9(9):1415–23.
103 Kim JY, Duan X, Liu CY, Jang MH, Guo JU, Pow-anpongkul N, et al. DISC1 regulates new neuron development in the adult brain via modulation of AKT-mTOR signaling through KIAA1212. Neuron. 2009;63(6):761–73.
104 Mas S, Gassó P, Ritter MA, Malagelada C, Bernardo M, Lafuente A. Pharmacogenetic predictor of extrapyramidal symptoms induced by antipsychotics: multilocus interaction in the mTOR pathway. Eur Neuropsychopharmacol. 2015;25(1):51–9 b.
105 English JA, Fan Y, Föcking M, Lopez LM, Hryniewiecka M, Wynne K, et al. Reduced protein synthesis in schizophrenia patient-derived olfactory cells. Transl Psychiatry. 2015;5:e663.
106 Chen J, Wang M, Waheed Khan RA, He K, Wang Q, et al. The GSK3B gene confers risk for both major depressive disorder and schizophrenia in the Han Chinese population. J Affect Disord. 2015;185:149–55.
107 Shelton MA, Newman JT, Gu H, Sampson AR, Fish KN, MacDonald ML, Moyer CE, DiBitetto JV, Dorph-Petersen KA, Penzes P, Lewis DA, Sweet RA. Loss of Microtubule-Associated Protein 2 Immunoreactivity Linked to Dendritic Spine Loss in Schizophrenia. Biol Psychiatry. 2015;78(6):374–85.
108 Rosoklija G, Keilp JG, Toomayan G, Mancevski B, Haroutunian V, Liu D, et al. Altered subicular MAP2 immunoreactivity in schizophrenia. Prilozi. 2005;26(2):13–34.
109 Jones LB, Johnson N, Byne W. Alterations in MAP2 immunocytochemistry in areas 9 and 32 of schizophrenic prefrontal cortex. Psychiatry Res. 2002;114(3):137–48.
110 Arnold SE, Lee VM, Gur RE, Trojanowski JQ. Abnormal expression of two microtubule-associated proteins (MAP2 and MAP5) in specific subfields of the hippocampal formation in schizophrenia. Proc Natl Acad Sci U S A. 1991;88(23):10850–4.
111 Clark D, Dedova I, Cordwell S, Matsumoto I. A proteome analysis of the anterior cingulate cortex gray matter in schizophrenia. Mol Psychiatry. 2006;11(5):459–70, 423.
112 Pennington K, Beasley CL, Dicker P, Fagan A, English J, Pariante CM, et al. Prominent synaptic and metabolic abnormalities revealed by proteomic analysis of the dorsolateral prefrontal cortex in schizophrenia and bipolar disorder. Mol Psychiatry. 2008;13(12):1102–17.
113 Hook V, Brennand KJ, Kim Y, Toneff T, Funkelstein L, Lee KC, Ziegler M, Gage FH. Human iPSC neurons display activity-dependent neurotransmitter secretion: aberrant catecholamine levels in schizophrenia neurons. Stem Cell Reports. 2014: 3(4):531-8.
114 Tam GW, van de Lagemaat LN, Redon R, Strathdee KE, Croning MD, Malloy MP, et al. Confirmed rare copy number variants implicate novel genes in schizophrenia. Biochem Soc Trans. 2010;38(2):445–51.
115 Yu GI, Kim SK, Park HJ, Kim JW, Chung JH, Shin DH. The C allele of synonymous SNP (rs1142636, Asn170Asn) in SYN1 is a risk factor for the susceptibility of Korean female schizophrenia. Synapse. 2012;66(11):979–83.
116 Goes FS, McGrath J, Avramopoulos D, Wolyniec P, Pirooznia M, Ruczinski I, et al. Genome-wide association study of schizophrenia in Ashkenazi Jews. Am J Med Genet B Neuropsychiatr Genet. 2015: in press.
117 Pak C, Danko T, Zhang Y, Aoto J, Anderson G, Maxeiner S, et al. Human Neuropsychiatric Disease Modeling using Conditional Deletion Reveals Synaptic Transmission Defects Caused by Heterozygous Mutations in NRXN1. Cell Stem Cell. 2015;17(3):316–28.
118 Todarello G, Feng N, Kolachana BS, Li C, Vakkalanka R, Bertolino A, et al. Incomplete penetrance of NRXN1 deletions in families with schizophrenia. Schizophr Res. 2014;155(1-3):1–7.
119 Atz ME, Rollins B, Vawter MP. NCAM1 association study of bipolar disorder and schizophrenia: polymorphisms and alternatively spliced isoforms lead to similarities and differences. Psychiatr Genet. 2007;17(2):55–67.
120 Tanaka Y, Yoshida S, Shimada Y, Ueda H, Asai K. Alteration in serum neural cell adhesion molecule in patients of schizophrenia. Hum Psychopharmacol. 2007;22(2):97–102.
121 Vawter MP, Usen N, Thatcher L, Ladenheim B, Zhang P, VanderPutten DM, et al. Characterization of human cleaved N-CAM and association with schizophrenia. Exp Neurol. 2001;172(1):29–46.
122 Poltorak M, Wright R, Hemperly JJ, Torrey EF, Issa F, Wyatt RJ, Freed WJ. Monozygotic twins discordant for schizophrenia are discordant for N-CAM and L1 in CSF. Brain Res. 1997;751(1):152–4.
123 Sullivan PF, Keefe RS, Lange LA, Lange EM, Stroup TS, Lieberman J, Maness PF. NCAM1 and neurocognition in schizophrenia. Biol Psychiatry. 2007;61(7):902–10.
124 Ayalew M, Le-Niculescu H, Levey DF, Jain N, Changala B, Patel SD, et al. Convergent functional genomics of schizophrenia: from comprehensive understanding to genetic risk prediction. Mol Psychiatry. 2012;17(9):887–905.
125 Dolgin E. Massive schizophrenia genomics study offers new drug directions. Nat Rev Drug Discov. 2014;13(9):641–2.
126 Mas S, Gassó P, Parellada E, Bernardo M, Lafuente A. Network analysis of gene expression in peripheral blood identifies mTOR and NF-κB pathways involved in antipsychotic-induced extrapyramidal symptoms. Pharmacogenomics J. 2015;15(5):452–60 c.
127 Roux PP, Shahbazian D, Vu H, Holz MK, Cohen MS, Taunton J, et al. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J Biol Chem. 2007;282(19):14056–64.
128 Steelman LS, Chappell WH, Abrams SL, Kempf RC, Long J, Laidler P, et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging (Albany NY). 2011;3(3):192–222. (replaces Costa-mattioli)
129 Moses V. Chao, Eric Klann, and Annalisa M. VanHook. A conversation about a Research Article published in the 14 January 2014 issue of Science Signaling. Podcast Science Signaling. 2014: http://stke.sciencemag.org/content/suppl/2014/01/10/7.308.pc2.DC1.
130 Hoffman DC, Donovan H. Catalepsy as a rodent model for detecting antipsychotic drugs with extrapyramidal side effect liability. Psychopharmacology (Berl). 1995;120(2):128–33.
131 Adams MR, Brandon EP, Chartoff EH, Idzerda RL, Dorsa DM, McKnight GS. Loss of haloperidol induced gene expression and catalepsy in protein kinase A-deficient mice. Proc Natl Acad Sci U S A. 1997;94(22):12157–61.
132 Atkins JB, Chlan-Fourney J, Nye HE, Hiroi N, Carlezon WA Jr, Nestler EJ. Region-specific induction of deltaFosB by repeated administration of typical versus atypical antipsychotic drugs. Synapse. 1999;33(2):118–28.
133 MacGibbon GA, Lawlor PA, Bravo R, Dragunow M. Clozapine and haloperidol produce a differential pattern of immediate early gene expression in rat caudate-putamen, nucleus accumbens, lateral septum and islands of Calleja. Brain Res Mol Brain Res. 1994;23(1-2):21–32.
134 Bateup HS, Santini E, Shen W, Birnbaum S, Valjent E, Surmeier DJ, et al. Distinct subclasses of medium spiny neurons differentially regulate striatal motor behaviors. Proc Natl Acad Sci U S A. 2010;107(33):14845–50.
135 Fink-Jensen A, Schmidt LS, Dencker D, Schülein C, Wess J, Wörtwein G, Woldbye DP. Antipsychotic-induced catalepsy is attenuated in mice lacking the M4 muscarinic acetylcholine receptor. Eur J Pharmacol. 2011;656(1-3):39–44.
136 Marshall M, Lockwood A, Bradley C, Adams C, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomised controlled trials of treatments for schizophrenia. Br J Psychiatry. 2000;176:249–52.
137 Leucht S, Heres S, Hamann J, Kane JM. Methodological issues in current antipsychotic drug trials. Schizophr Bull. 2008;34(2):275–85.