Current treatment of dyslipidaemia: PCSK9 inhibitors and statin intolerance
DOI: https://doi.org/10.4414/smw.2016.14333
Konstantinos
Koskinas, Matthias
Wilhelm, Stephan
Windecker
Summary
Statins are the cornerstone of the management of dyslipidaemias and prevention of cardiovascular disease. Although statins are, overall, safe and well tolerated, adverse events can occur and constitute an important barrier to maintaining long-term adherence to statin treatment. In patients who cannot tolerate statins, alternative treatments include switch to another statin, intermittent-dosage regimens and non-statin lipid-lowering medications. Nonetheless, a high proportion of statin-intolerant patients are unable to achieve recommended low-density lipoprotein (LDL) cholesterol goals, thereby resulting in substantial residual cardiovascular risk. Proprotein convertase subtilisin/kexin type 9 (PCSK9) is a protease implicated in LDL receptor degradation and plays a central role in cholesterol metabolism. In recent studies, PCSK9 inhibition by means of monoclonal antibodies achieved LDL cholesterol reductions of 50% to 70% across various patient populations and background lipid-lowering therapies, while maintaining a favourable safety profile. The efficacy and safety of the monoclonal antibodies alirocumab and evolocumab were confirmed in statin-intolerant patients, indicating that PCSK9 inhibitors represent an attractive treatment option in this challenging clinical setting. PCSK9 inhibitors recently received regulatory approval for clinical use and may be considered in properly selected patients according to current consensus documents, including patients with statin intolerance. In this review we summarise current evidence regarding diagnostic evaluation of statin-related adverse events, particularly statin-associated muscle symptoms, and we discuss current recommendations on the management of statin-intolerant patients. In view of emerging evidence of the efficacy and safety of PCSK9 inhibitors, we further discuss the role of monoclonal PCSK9 antibodies in the management of statin-intolerant hypercholesterolaemic patients.
Introduction
The efficacy of statins for reduction of low-density lipoprotein cholesterol (LDL-C) and prevention of cardiovascular events is well established [1, 2]. Statins are the most widely prescribed class of drugs worldwide and are generally safe and well tolerated; however, statin intolerance is not uncommon in real-world practice [3]. Skeletal muscle-related events are the most frequent adverse event associated with statin treatment, followed by asymptomatic liver enzyme elevation and other, less frequent, side effects [4]. While adverse events are in principle fully reversible upon statin withdrawal or dose reduction, they constitute an important barrier to maintaining long-term compliance with statin treatment [5]. Furthermore, a substantial proportion of patients – both statin-tolerant and, to a greater extent, statin-intolerant – are unable to achieve recommended LDL-C goals despite alternative treatment strategies including switch to another statin, intermittent-dosage statin regimens, and/or non-statin lipid-lowering medications. Thereby, residual cardiovascular risk is particularly prevalent among high-risk, secondary prevention patients.
Proprotein convertase subtilisin/kexin type 9 (PCSK9) is a secreted protein that enhances degradation of the LDL receptor and thereby plays a central role in LDL-C metabolism [6]. Consequently, inhibition of PCSK9 has emerged as a promising new target in lowering plasma LDL-C. In recent studies, monoclonal antibodies that functionally inhibit PCSK9 achieved LDL-C reductions of 50% to 70% across various patient populations and background lipid-lowering therapies while maintaining a favourable safety profile [7, 8]. PCSK9 inhibitors have specifically been tested in statin-intolerant patients and resulted in greater reductions of LDL-C levels and fewer adverse effects compared with other non-statin medications [9, 10], thus representing an attractive treatment option in this clinical setting.
In this review we summarise evidence regarding the incidence, risk factors and diagnostic evaluation of statin-related adverse events, particularly statin-associated myopathy, and we discuss current recommendations on the management of statin-intolerant patients. In view of emerging evidence of the efficacy and safety of PCSK9 inhibitors, we further discuss the role of monoclonal PCSK9 antibodies for management of statin-intolerant hypercholesterolaemic patients.
Statin intolerance: incidence and clinical impact
Statins represent the cornerstone of the management of dyslipidaemias and prevention of cardiovascular disease [1, 2]. The Cholesterol Treatment Trialists’ (CTT) collaboration, an individual-patient pooled dataset including >170 000 patients, demonstrated a reduction in the annual rate of major vascular events by 22% with each LDL-C reduction og 1 mmol/l and thereby provided unequivocal evidence of substantial cardiovascular benefit across the spectrum of patient risk categories [11, 12]. Although statins are generally safe and well tolerated, a number of adverse effects have been attributed to them. These include muscle-related symptoms, liver enzyme elevation and a slightly elevated risk of new-onset diabetes [13], as well as other potential side effects with less-established evidence of causality such as gastrointestinal discomfort, insomnia and neurocognitive symptoms [14]. The absolute excess risk of these adverse effects of statins is very small and is outweighed by well-established beneficial effects on major cardiovascular events.
Statin-associated muscle symptoms represent the leading manifestation of statin intolerance. Skeletal muscle-related adverse events range from symptoms with normal creatine kinase (CK) levels (“myalgia”) to a combination of symptoms with minor to moderate CK elevation [<10× the upper limit of the normal range (ULN)] or more marked CK elevation >10× ULN (“myositis”), to extremely rare cases of rhabdomyolysis (CK elevation >40× ULN associated with renal impairment and/or myoglobinuria) [5]. Accurate estimation of the incidence of statin-associated muscle symptoms is hindered by the vast heterogeneity of definitions across clinical studies and regulatory reports. Randomised trials found similarly low rates of myalgia in patients treated with statin or placebo [15, 16]. In contrast, observational studies reported statin-associated muscle symptoms in 7–11% [17, 18] up to 29% in a single study [19], most commonly associated with normal or slightly elevated CK concentrations. More marked CK elevations (>10× ULN) were observed in 1 per 1000 to 1 per 10 000 patients per year [20], depending on the statin type and dosage, and presence of risk factors. In this context, it is important to consider that the exclusion of patients who are at highest risk of developing side effects from most randomised trials likely resulted in an underestimation of the true rate of adverse events. On the other hand, muscle symptoms are a nonspecific complaint (table 1), such that frequently reported muscle symptoms in statin-treated patients may not be attributable to statins in observational registries [20].
Statin-associated myopathy is largely clinically benign, in that symptoms as well as CK elevations are reversible upon withdrawal of the statin in the vast majority of affected patients. Persistent muscle symptoms during statin treatment are accompanied by characteristic histological patterns of muscle damage [21], and there is some evidence that statins may induce long-standing ultrastructural muscular changes, even in the absence of symptoms [22]. In extremely rare cases, an autoimmune myopathy develops in patients treated with statins; this disorder results in progressive weakness that must be controlled with immunosuppressive therapy [23]. While in principle benign and reversible, statin-induced muscle symptoms may compromise physical activity – an important component of cardiovascular prevention. Importantly, these side effects have substantial impact on drug adherence and consequently on cardiovascular risk reduction, as they frequently result in discontinuation or suboptimal dosing of statins [5]. Discontinuation rates amount to 75% in patients who develop muscle-related symptoms [24]; the occurrence of side effects is in fact the main reason for nonadherence to statin therapy [25]. Notably, low adherence to statin therapy is associated with a 25% increase in all-cause mortality [26] and a 15% increase in cardiovascular risk [27] among secondary prevention patients.
Risk factors that increase the likelihood of statin-induced myopathy include demographic features (advanced age, frailty, Asian descent); genetic factors; and patient co-morbidities [3, 5] (table 2). Moreover, differences in physicochemical properties of each statin, and their metabolism and dosage influence the risk of developing myopathy. For any given statin, the risk of developing muscle symptoms is higher with increasing doses. Similarly, growing numbers of patients are expected to receive treatment with high-dose statins in the future as the importance of adequately controlled blood lipid levels for prevention of cardiovascular disease is increasingly appreciated [28, 29], and as genetic (familial) forms of hypercholesterolaemia are increasingly recognised [30]. All statins can cause muscle symptoms, but the risk appears to vary between different statins and appears to be lowest with fluvastatin and pravastatin [31]. In-vitro studies suggested greater direct muscle toxicity with lipophilic statins (simvastatin, lovastatin), whereas more hydrophilic statins (pravastatin, rosuvastatin) may be less toxic due to lower penetration into muscle cells [32]; however, individual patient differences are likely more important than statin differences in affecting statin uptake by skeletal muscle tissue. Pharmacokinetic interactions with concomitant medications represent a well-established risk factor of statin-induced myopathy. The possibility of drug interactions is higher for statins mainly metabolised by the CYP3A4 enzyme system (simvastatin, atorvastatin, lovastatin) than with rosuvastatin and pravastatin, which are not extensively metabolized by CYP3A4. Medications with the potential for drug-drug interactions that increase statin exposure and thereby raise the risk of myopathy include potent CYP3A4 inhibitors (itraconazole, ketoconazole, macrolides), human immunodeficiency virus (HIV) protease inhibitors, immunosuppressive drugs (ciclosporin), and commonly prescribed cardiovascular medications (amiodarone, diltiazem, verapamil).
Regarding hepatic involvement, asymptomatic liver enzyme elevation is observed in 0.1–3.0% of treated patients and is a dose-dependent class effect for statins [33]. The condition is considered clinically benign and is not accompanied by histological alterations in the liver. Clinically relevant hepatic impairment is very infrequent and requires exclusion of secondary causes or possible drug-drug interactions. Of note, elevated enzyme levels tend to normalise despite continuation of therapy, which is likely related to statin-mediated reduction in hepatic steatosis [34]. It is, in fact, believed that liver enzyme elevation may represent adaptation of the liver to lower LDL-C levels rather than true hepatic toxicity [35].
|
Table 1: Differential diagnosis of muscle symptoms or creatine kinase elevation. |
| Hypo-/hyperthyroidism
Excessive physical activity
Trauma
Viral infection
Vitamin-D deficiency
Rheumatic disorders (polymyositis, systemic lupus erythematosus)
Peripheral arterial disease
Medications (glucocorticoids, antipsychotics, immunosuppressant, gemfibrozil)
Substance abuse (opioids, cocaine, amphetamines)
Metabolic/infectious myopathies
Neuropathies |
|
Table 2: Risk factors and precipitating factors of statin-associated myopathy. Adapted with permission from [5]. |
|
Demographic factors
Age >80 years
Female gender
Low body mass index
Asian descent |
|
Comorbidities
Acute infection
Hypothyroidism (untreated or undertreated)
Impaired renal/hepatic function
Biliary tree obstruction
Organ transplant recipients
Severe trauma
Human immunodeficiency virus infection
Diabetes mellitus
Vitamin D deficiency
Surgery with high metabolic demands |
|
Relevant history
History of creatine kinase elevation
History of pre-existing muscle/joint/tendon pain
Inflammatory or metabolic neuromuscular defects
Previous statin-induced myopathy
Previous myotoxicity by non-stain lipid-lowering medications |
|
Other risk factors
Genetic factors (polymorphisms in genes encoding CYP450 isoenzymes)
Dietary effects (grapefruit juice)
Excess alcohol use
Drug abuse (cocaine, amphetamines, heroin) |
Diagnostic assessment of statin-associated muscle symptoms
Definitive diagnosis of statin-associated myopathy is difficult because of the subjective, nonspecific symptoms and the absence of a gold-standard diagnostic test. A combination of clinical and laboratory findings can help to assess the probability that muscle complaints are indeed caused by a statin. The localisation of symptoms, magnitude of CK elevation and temporal association with statin initiation, discontinuation or re-challenge need to be carefully reviewed. Statin-induced muscular complaints are typically symmetrical and proximal, and generally affect large muscle groups including the thighs, buttocks, calves and back muscles [5]. These symptoms usually occur early (within 4–6 weeks) after starting statin therapy or increasing the dose [36], although delayed occurrence (even after years of treatment) is possible. Symptoms may be triggered by an increase in statin dose, initiation of interacting medications, or presence of precipitating factors (e.g., alcohol excess, drug abuse, major surgery) (table 2), and are more frequent in physically active individuals. Recently, a risk score was proposed, incorporating the aforementioned clinical and laboratory variables, to facilitate differential diagnosis and define the probability that muscle complaints and/or CK elevation are attributed to statins [37].
Management of statin-intolerant patients
Initial evaluation of stain-treated patients who present with muscle symptoms should focus on exclusion of alternative causes and identification of possible precipitating factors. Along these lines, glucocorticoids, antipsychotic (risperidone, haloperidol), immunosuppressant or antiviral agents (HIV protease inhibitors) and lipid-modifying drugs (gemfibrozil), as well as alcohol or drug abuse (opioids, cocaine) are potential causes of muscle-related symptoms irrespective of statin use. Once secondary causes or triggering factors are excluded, the need for continued statin therapy should be re-evaluated on the basis of individual patient risk. For patients at high or very high cardiovascular risk (e.g., those with known cardiovascular disease, familial hypercholesterolaemia or diabetes mellitus), the benefits of statin therapy need to be weighed against the burden of muscle symptoms. As general measures, patients need to be counselled regarding the risk of side effects and the high probability that these can be dealt with successfully, and the role of dietary or other lifestyle measures for cardiovascular risk reduction needs to be emphasised [5].
A treatment algorithm for patients with statin-associated muscle symptoms was proposed in a recent consensus statement by the European Atherosclerosis Society [5] (fig. 1). Withdrawal of statin therapy usually leads to resolution of symptoms and, along with symptom recurrence upon re-challenge, can help determine a causal link between the drug and the adverse event. In patients with initial CK elevation <4× ULN, statin cessation with a 2–4 week washout period is recommended; persistence of symptoms practically excludes the causal role of the statin regimen. If symptoms improve, treatment with a lower dose of the same statin or with an alternative statin should be considered. Doses can be up-titrated to achieve LDL-C goals or reduce LDL-C to the maximal extent with minimal muscle complaints. More efficacious statins with a long half-life (rosuvastatin, atorvastatin, pitavastatin), usually starting at a lower dose, or intermittent (non-daily) regimens are recommended. In the case of higher initial CK elevation (>4× ULN), a low-dose or alternate day regimen of a potent statin is recommended after a prolonged (6-week) washout resulting in symptom improvement and normalization of CK levels (fig. 1). In the extremely rare event of rhabdomyolysis, statins should not be reintroduced.
In the proposed algorithm, the primary goal is achievement of LDL-C targets with maximally tolerated statin doses. However, combination with non-statin therapies is recommended in patients at high risk for cardiovascular disease with LDL-C levels above respective targets despite maximally tolerated statin therapy. Ezetimibe reduces LDL-C by 15–20%, has few side effects [38] and has been shown to reduce cardiovascular events when added to statin therapy [39]. Other non-statin lipid-lowering medications include bile acid sequestrants and fenofibrate (which, unlike gemfibrozil, does not increase the risk of rhabdomyolysis when added to a statin). Niacin is no longer prescribed in Europe because of reports of excessive adverse effects without cardiovascular benefit [40]. Although complementary therapy with ubiquinone (coenzyme Q10) has been suggested to improve statin tolerability, this approach has not proven effective in randomised controlled trials [41] and is currently not recommended [5]. It is important to note that statins are believed to confer cardiovascular benefits by means of pleotropic effects beyond their LDL-C–lowering potential (e.g., favourable effects on atherosclerotic plaque composition [42, 42], anti-inflammatory effects [44], immunomodulatory properties [45], or direct effects of the vascular endothelium). Whether these properties are also afforded by non-statin lipid-lowering medications is unclear and remains to be determined. Along these lines, recent evidence demonstrated the incremental value of ezetimibe when added to a statin for regression of coronary atherosclerosis [46] and reduction of blood levels of C-reactive protein [47].
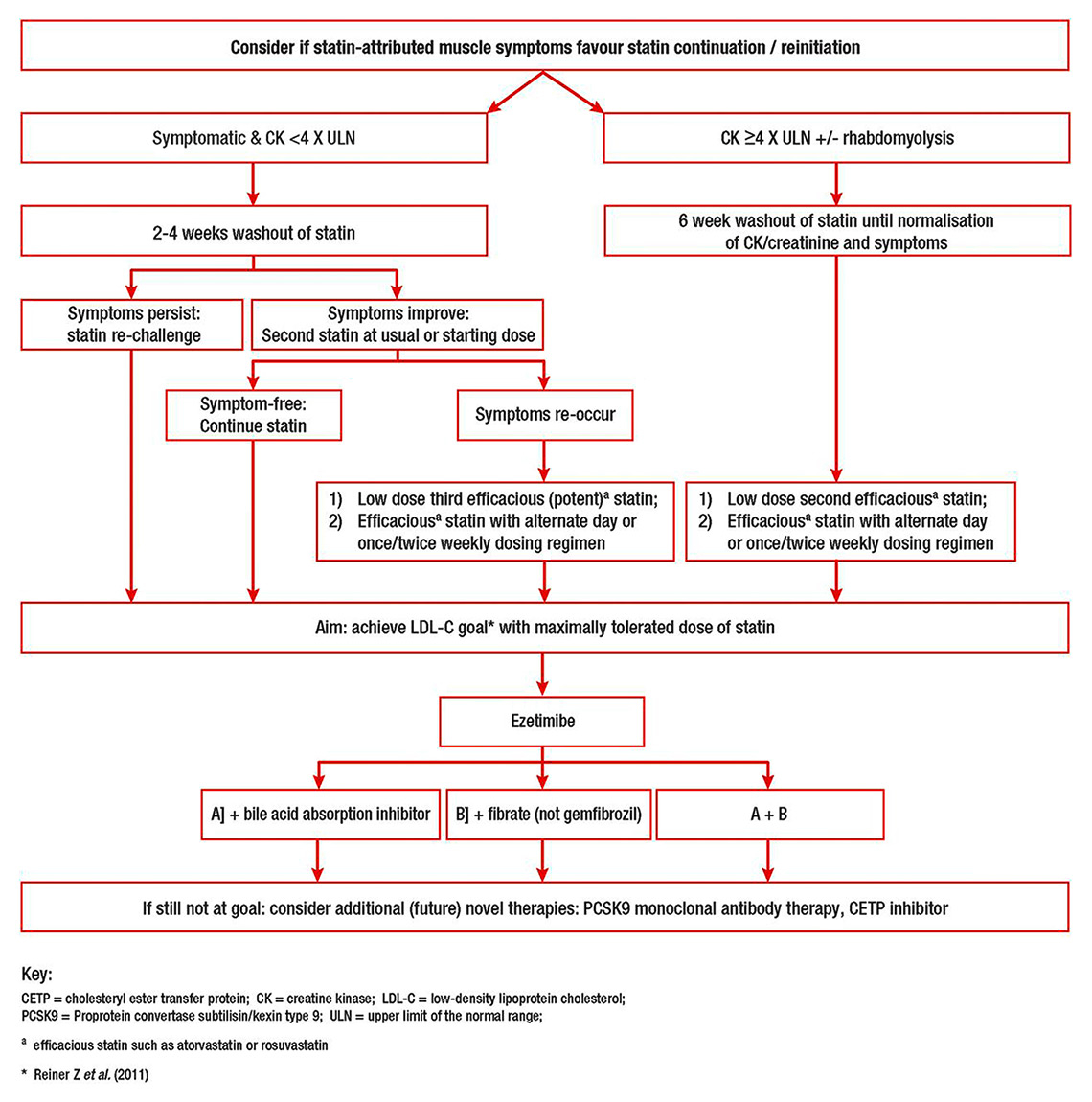
Figure 1
Proposed algorithm for management of patients with statin-associated muscle symptoms. Adapted with permission from [5].
CETP = cholesteryl ester transfer protein; CK = creatine kinase; LDL-C = low-density lipoprotein cholesterol; PCSK9 = proprotein convertase subtilisin/kexin type 9; ULN = upper limit of the normal range
a efficacious stain such as atorvastatin or rosuvastatin
* Reiner et al. 2011 [28]
Regarding liver enzyme elevation, the Swiss Atherosclerosis Association recommends continued statin treatment in the event of aspartate transaminase and alanine transaminase levels <3× ULN and repeated checking after 4–6 weeks. If enzyme levels are >3× ULN, statin discontinuation or dose reduction is recommended, followed by a repeat check after 4–6 weeks and treatment resumption once enzymes return to normal.
PCSK9 inhibitors: overview of efficacy and safety
PCSK9 is a serine protease that promotes degradation of the LDL receptor, thereby resulting in reduced hepatic uptake and increased circulating levels of LDL-C [6]. In 2003, mutations in the PCSK9 gene were identified as one of the causes of familial hypercholesterolaemia [48]. Gain-of-function mutations in PCSK9 are associated with elevated LDL-C levels and premature cardiovascular disease, whereas loss-of-function mutations in PCSK9 result in reduced LDL-C levels and lower cardiovascular event rates [49]. Following these observations, intensive research has focused on the development of therapeutic approaches to inhibition of PCSK9 function as a means of lowering atherogenic lipoprotein levels; these approaches include antibodies, antisense oligonucleotides and small interfering RNAs targeting PCSK9 synthesis. Currently, monoclonal antibodies are the most advanced and clinically documented approach to PCSK9 inhibition. In several published phase II and phase III studies, PCSK9 inhibitors were able to decrease LDL-C by 50–70% across a wide range of patient populations and background therapies, including patients who were unable to tolerate statin therapy, patients with heterozygous and homozygous familial hypercholesterolaemia, or patients unable to reach LDL-C targets despite lipid-lowering medications [7, 8]. In these studies, LDL-C reductions were consistent and dose-dependent, and were not affected by age, gender, or baseline LDL-C concentrations.
Current evidence on PCSK9 inhibitors is reviewed in detail elsewhere [50]. Here we briefly discuss the largest published phase III studies of the two most extensively studied antibodies, alirocumab (Regeneron/Sanofi) and evolocumab (Amgen). The randomised, placebo-controlled ODYSSEY LONG-TERM trial evaluated the efficacy and safety of treatment with alirocumab for 78 weeks in 2341 patients with established coronary heart disease (CHD) or CHD equivalent with LDL-C levels ≥70 mg/dl (≥1.8 mmol/l) on maximum tolerated statin dose or other lipid-lowering therapy [51]. Alirocumab resulted in a reduction of LDL-C levels by 61% at 24 weeks and by 52% at 78 weeks. Reductions in non-HDL cholesterol of 52%, apolipoprotein B of 54%, and lipoprotein(a) of 26% were also observed. The goal of an LDL-C level <1.8 mmol/l was met by 79% of patients in the alirocumab group compared with 8% of patients in the placebo group. In a predefined exploratory analysis, treatment with alirocumab was associated with a 48% lower rate of major adverse cardiovascular events compared with placebo (hazard ratio [HR] 0.52; 95% confidence interval [CI] 0.31–0.90; p = 0.02) [51].
Evolocumab was studied in two open-label, randomised trials (OSLER 1 and OSLER 2) [52]. A total of 4465 patients were randomised in a 2:1 ratio to receive either evolocumab (140 mg every 2 weeks or 420 mg monthly) plus standard therapy or standard therapy alone. After 12 weeks of treatment, evolocumab reduced LDL-C by 61% to a median level of 48 mg/dl; this reduction was maintained throughout 48 weeks of follow-up. In a post-hoc analysis, treatment with evolocumab was associated with a significantly lower rate of cardiovascular events at 1 year compared with standard therapy (HR 0.47; 95% CI 0.28–0.78; p = 0.003) [52]. In a pooled meta-analysis including 17 trials with 13 083 patients randomised to PCSK9 inhibitors (alirocumab or evolocumab), ezetimibe or placebo, PCSK9 inhibitors reduced LDL-C by 57% relative to placebo and 36% relative to ezetimibe, and were associated with a 57% reduction in all-cause mortality (odds ratio 0.43, 95% CI 0.22–0.82; p = 0.01) [53].
Adding to the marked efficacy in reducing levels of LDL-C and other atherogenic lipoproteins, the safety and tolerability of the two most extensively studied antibodies appears to be promising in up to 2 years of follow-up. In the OSLER trials, adverse events occurred with similar frequency in the two groups, with the exception of neurocognitive events which were more frequent in the evolocumab group, and did not vary according to the achieved level of LDL-C [52]. Similarly, alirocumab was tolerated well without a significant increase in adverse effects attributable to the study drug in the ODYSSEY LONG TERM trial [51]. The finding of a two-fold higher incidence of neurocognitive adverse events with PCSK9 inhibitors than with placebo in a pooled meta-analysis [53] requires further investigation in larger patient cohorts with longer follow-up, and is currently being tested in a dedicated study of evolocumab (the EBBINGHAUS study; ClinicalTrials.gov Identifier: NCT02207634).
Studies investigating PCSK9 inhibitors in statin-intolerant patients
In published studies testing PCSK9 inhibitors, large LDL-C reductions were associated with very low rates of muscle symptoms, thus reinforcing the concept that statins and not LDL-C lowering per se are implicated in the development of muscle-related symptoms. Both alirocumab and evolocumab have been assessed specifically in formally statin-intolerant patients.
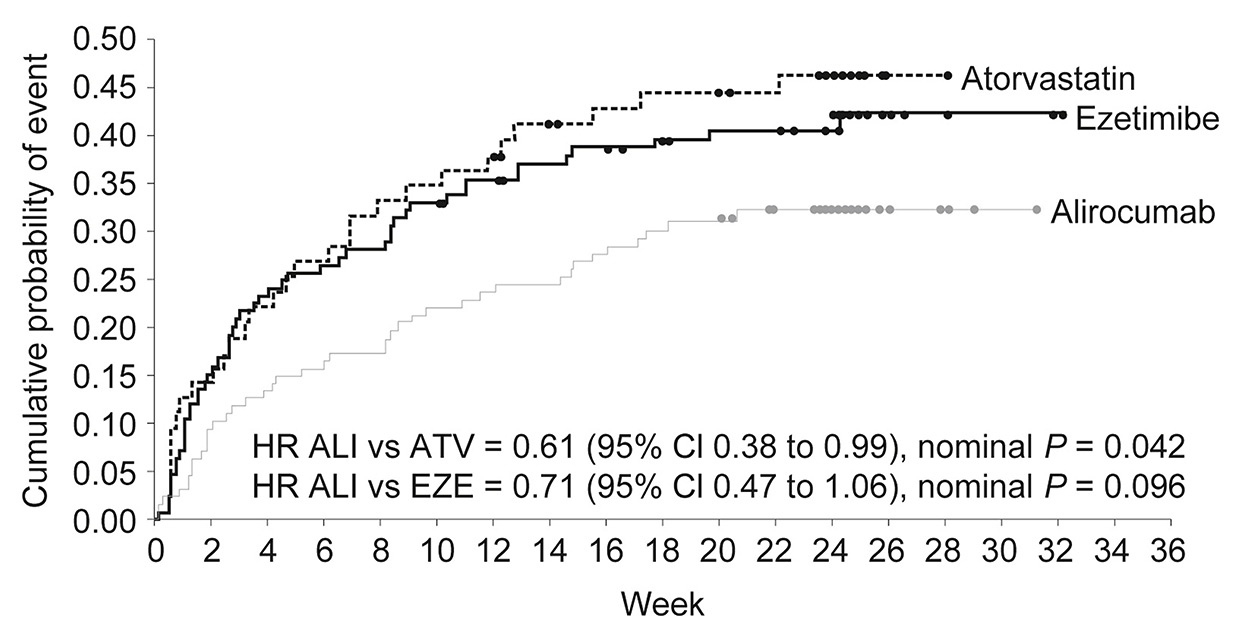
Figure 2
Kaplan-Meier estimates for time to first skeletal muscle-related adverse event across treatment groups in the ODYSSEY ALTERNATIVE randomised trial. Adapted with permission from [10].
ALI = alirocumab; ATV =atorvastatin; CI = confidence interval; EZE = ezetimibe; HR = hazard ratio.
The GAUSS-2 randomised trial compared the efficacy and safety of evolocumab versus ezetimibe in 307 hypercholesterolaemic patients unable to tolerate effective statin doses [9]. Patients had baseline LDL-C levels above their NCEP ATP-III risk category goal, i.e., ≥100 mg/dl (2.6 mmol/l) with diagnosed CHD or risk equivalent; ≥130 mg/dl (3.4 mmol/l) with two or more risk factors; ≥160 mg/dl (4.1 mmol/l) with a single risk factor; or ≥190 mg/dL (4.9 mmol/l) without CHD and without risk factors. Evidence of intolerance to at least two statins was an inclusion criterion. Patients were randomised to evolocumab 140 mg every 2 weeks or 420 mg monthly plus oral placebo, or to subcutaneous placebo plus ezetimibe. Mean percent LDL-C reductions from baseline to 12 weeks were 56% with evolocumab 140 mg every 2 weeks and 55% with 420 mg once monthly, corresponding to treatment differences of 37% and 39%, respectively, compared with ezetimibe. Attainment of LDL-C targets within 12 weeks was more frequent in patients treated with evolocumab than with ezetimibe (76% vs 5.5%). Muscle-related adverse events occurred in 12% of evolocumab-treated patients and 23% of ezetimibe-treated patients, and rates of discontinuation due to musculoskeletal side effects were 5% in the evolocumab compared with 6% in the ezetimibe group [9].
In the ODYSSEY ALTERNATIVE trial, alirocumab was evaluated in 314 patients with statin intolerance [10]. Unlike GAUSS-2, this trial had a more extended follow-up of 24 weeks and included a statin rechallenge arm in an attempt to confirm intolerance. Patients were randomised to alirocumab 75 mg every 2 weeks, ezetimibe 10 mg daily, or atorvastatin 20 mg daily. At 24 weeks, reduction in LDL-C was 45% with alirocumab as compared with 15% in ezetimibe-treated patients. Treatment-related adverse events did not differ between groups. Rates of skeletal muscle-related events were lower in the alirocumab than in the atorvastatin group (HR 0.61, 95% CI 0.38–0.99; p = 0.04), with a similar trend for the comparison between alirocumab and ezetimibe (HR 0.71, 95% CI 0.47–1.06; p = 0.096) [10] (fig. 2).
PCSK9 inhibitors in statin-intolerant patients: current status and future perspectives
Statin-associated adverse events, particularly myopathy, affect a non-negligible proportion of treated patients, and the global burden of statin intolerance is magnified by the large numbers of patients receiving statins worldwide. While proposed management strategies including alternative statin– and non-statin–based regimens are effective at least in part, there remains an unmet need for improved lipid lowering, particularly among true statin-intolerant patients who are at high cardiovascular risk and cannot reach established LDL-C targets. According to current evidence in populations selected on the basis of documented statin-induced myopathy [9, 10], LDL-C reduction was more pronounced and muscle adverse events were less frequent following treatment with PCSK9 inhibitors than with ezetimibe, the most widely used non-statin drug in this clinical setting. These studies provided evidence of marked efficacy combined with favourable tolerability, thereby placing PCSK9 monoclonal antibodies as a promising therapy for treatment of high-risk statin-intolerant patients with persistently elevated blood lipid levels.
Alirocumab and evolocumab were recently approved for clinical use by the US Food and Drug Administration as well as the European Medicines Agency (EMA). According to the EMA approval [54, 55], these antibodies are indicated in adults with primary hypercholesterolaemia or mixed dyslipidaemia, as an adjunct to diet: (i) in combination with a statin, or statin with other lipid-lowering therapies in patients unable to reach LDL-C goals with the maximum tolerated dose of a statin; or (ii) alone or in combination with other lipid-lowering therapies in patients who are statin-intolerant. In Switzerland, evolocumab was approved by the Swiss Agency for Therapeutic Products, Swissmedic, in February 2016 [56].
In addition to regulatory approval, current scientific documents indicate that PCSK9 monoclonal antibodies may be considered for properly selected patients, including those with statin intolerance. In a consensus statement on statin-associated muscle symptoms by the European Atherosclerosis Society [5], PCSK9 inhibitors were included in the relevant treatment algorithm (fig. 1) and were reserved for patients with LDL-C above target levels despite maximally tolerated statin doses and addition of non-statin medications (ezetimibe, fibrates or bile acid absorption inhibitors). The aforementioned approach is relatively conservative with regard to the use of PCSK9 inhibitors, but needs to be interpreted in view of the fact that the consensus paper [5] was published prior to publication of the two largest studies to date attesting to the efficacy and safety of PCSK9 monoclonal antibodies [51, 52]. Subsequently, recommendations were issued by the US National Lipid Association, indicating that PCSK9 inhibitors may be considered in selected high- or very high-risk patients who are statin-intolerant and require substantial additional cholesterol reduction despite the use of other lipid lowering therapies [57]. An updated guideline document by the European Society of Cardiology on the management of dyslipidaemias will be published in 2016 and is expected also to provide recommendations with regard to the use of PCSK9 inhibitors in statin-intolerant patients.
Collectively, patients who are at high cardiovascular risk but cannot achieve LDL-C goals as a result of statin intolerance appear to be particularly well suited for treatment with PCSK9 inhibitors. Currently, more evidence is expected from ongoing trials focusing on the long-term safety and efficacy of alirocumab (the ODYSSEY Outcomes trial; NCT01663402), evolocumab (the FOURIER trial; NCT01764633) and the monoclonal antibody bococizumab (Pfizer) (SPIRE 1 trial; NCT01975376 and SPIRE 2 trial; NCT01975389). These studies will primarily test the impact of PCSK9 monoclonal antibodies on long-term cardiovascular outcomes and will thereby determine whether the documented LDL-C reduction and promising effects on mid-term clinical outcomes by means of pharmacological PCKS9 inhibition [53, 58] translate into definitive evidence of long-term cardiovascular benefit. Of note, there have been concerns that the high production cost and currently high market price may become appreciable barriers to wide-spread use of these effective medications [59]. As in the case of statins [60], robust evidence of clinical benefit with these agents will be required for definitive affordability assessment and cost-effectiveness analyses. Whether PCSK9 inhibitors prove broadly effective for cardiovascular event reduction, and whether healthcare savings due to the afforded clinical benefit might counterbalance treatment cost remains to be determined. The expected new insights from ongoing studies will significantly influence future recommendations as well as real-world penetration of these promising therapies overall – and specifically in the challenging setting of statin-intolerant patients.
References
1 Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr, Kastelein JJ, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–207.
2 Ridker PM, Cannon CP, Morrow D, Rifai N, Rose LM, McCabe CH, et al.; Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 (PROVE IT-TIMI 22) Investigators. C-reactive protein levels and outcomes after statin therapy. N Engl J Med. 2005;352:20–8.
3 Chatzizisis YS, Koskinas KC, Misirli G, Vaklavas C, Hatzitolios A, Giannoglou GD. Risk factors and drug interactions predisposing to statin-induced myopathy: implications for risk assessment, prevention and treatment. Drug Saf. 2010;33:171–87.
4 Armitage J. The safety of statins in clinical practice. Lancet. 2007;370:1781–90.
5 Stroes ES, Thompson PD, Corsini A, Vladutiu GD, Raal FJ, Ray KK, et al. Statin-associated muscle symptoms: impact on statin therapy. European Atherosclerosis Society consensus panel statement on assessment, aetiology and management. Eur Heart J. 2015;36:1012–22.
6 Bergeron N, Phan BA, Ding Y, Fong A, Krauss RM. Proprotein convertase subtilisin/kexin type 9 inhibition: a new therapeutic mechanism for reducing cardiovascular disease risk. Circulation. 2015;132:1648–66.
7 Giugliano RP, Sabatine MS. Are PCSK9 Inhibitors the Next Breakthrough in the Cardiovascular Field? J Am Coll Cardiol. 2015;65:2638–51.
8 Shimada YJ, Cannon CP. PCSK9 (Proprotein convertase subtilisin/kexin type 9) inhibitors: past, present, and the future. Eur Heart J. 2015;36:2415–24.
9 Stroes E, Colquhoun D, Sullivan D, Civeira F, Rosenson RS, Watts GF, et al.; GAUSS-2 Investigators. Anti-PCSK9 antibody effectively lowers cholesterol in patients with statin intolerance: the GAUSS-2 randomized, placebo-controlled phase 3 clinical trial of evolocumab. J Am Coll Cardiol. 2014;63:2541–8.
10 Efficacy and safety of alirocumab vs ezetimibe in statin-intolerant patients, with a statin rechallenge arm: The ODYSSEY ALTERNATIVE randomized trial. J Clin Lipidol. 2015;9:758–69.
11 Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, et al., Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–78.
12 Cholesterol Treatment Trialists’ (CTT) Collaboration, Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, Bhala N, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670–81.
13 Sattar N, Preiss D, Murray HM, Welsh P, Buckley BM, de Craen AJ, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet. 2010;375:735–42.
14 Macedo AF, Taylor FC, Casas JP, Adler A, Prieto-Merino D, Ebrahim S. Unintended effects of statins from observational studies in the general population: systematic review and meta-analysis. BMC Med. 2014;12:51.
15 Kashani A, Phillips CO, Foody JM, Wang Y, Mangalmurti S, Ko DT, Krumholz HM. Risks associated with statin therapy: a systematic overview of randomized clinical trials. Circulation. 2006;114:2788–97.
16 Heart Protection Study Collaborative Group.MRC/BHFHeart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomized placebo-controlled trial. Lancet. 2002;360:7–22.
17 Bruckert E, Hayem G, Dejager S, Yau C, Begaud B. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients – the PRIMO study. Cardiovasc Drugs Ther. 2005;19:403–14.
18 Zhang H, Plutzky J, Skentzos S, Morrison F, Mar P, Shubina M, Turchin A. Discontinuation of statins in routine care settings: A cohort study. Ann Intern Med. 2013;158:526–34.
19 Cohen JD, Brinton EA, Ito MK, Jacobson TA. Understanding statin use in America and gaps in patient education (USAGE): an internet-based survey of 10,138 current and former statin users. J Clin Lipidol. 2012;6:208–15.
20 Law M, Rudnicka AR. Statin safety: a systematic review. Am J Cardiol. 2006;97(8A):52C–60C.
21 Mohaupt MG, Karas RH, Babiychuk EB, Sanchez-Freire V, Monastyrskaya K, Iyer L, et al. Association between statin-associated myopathy and skeletal muscle damage. CMAJ. 2009;181:E11–8.
22 Draeger A, Monastyrskaya K, Mohaupt M, Hoppeler H, Savolainen H, Allemann C, Babiychuk EB. Statin therapy induces ultrastructural damage in skeletal muscle in patients without myalgia. J Pathol. 2006;210:94–102.
23 Mammen AL. Statin-associated autoimmune myopathy. N Engl J Med. 2016;374:664–9.
24 Chodick G, Shalev V, Gerber Y, Heymann AD, Silber H, Simah V, Kokia E. Longterm persistence with statin treatment in a not-for-profit health maintenance organization: a population-based retrospective cohort study in Israel. Clin Ther. 2008;30:2167–79.
25 Cohen JD, Brinton EA, Ito MK, Jacobson TA. Understanding statin use in America and gaps in patient education (USAGE): an internet-based survey of 10,138 current and former statin users. J Clin Lipidol. 2012;6:208–15.
26 Jackevicius CA,MamdaniM,Tu JV. Adherence with statin therapy in elderly patients with and without acute coronary syndromes. JAMA. 2002;288:462–7.
27 Chowdhury R, Khan H, Heydon E, Shroufi A, Fahimi S, Moore C, et al. Adherence to cardiovascular therapy: a meta-analysis of prevalence and clinical consequences. Eur Heart J. 2013;34:2940–8.
28 Reiner Z, Catapano AL, De Backer G, Graham I, Taskinen MR, Wiklund O, et al. ESC/EAS Guidelines for the management of dyslipidaemias: the Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Eur Heart J. 2011;32:1769–818.
29 Lloyd-Jones DM, Morris PB, Ballantyne CM, Birtcher KK, Daly DD Jr, DePalma SM, et al. 2016 ACC Expert Consensus Decision Pathway on the Role of Non-Statin Therapies for LDL-Cholesterol Lowering in the Management of Atherosclerotic Cardiovascular Disease Risk: A Report of the American College of Cardiology Task Force on Clinical Expert Consensus Documents. J Am Coll Cardiol. 2016 Mar 28. pii: S0735-1097(16)32398–1.
30 de Ferranti SD, Rodday AM, Mendelson MM, Wong JB, Leslie LK, Sheldrick RC. Prevalence of Familial Hypercholesterolemia in the 1999 to 2012 United States National Health and Nutrition Examination Surveys (NHANES). Circulation. 2016;133:1067–72.
31 Bruckert E, Hayem G, Dejager S, Yau C, Bégaud B. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients – the PRIMO study. Cardiovasc Drugs Ther. 2005;19:403–14.
32 Gadbut AP, Caruso AP, Galper JB. Differential sensitivity of C2-C12 striated muscle cells to lovastatin and pravastatin. J Mol Cell Cardiol. 1995;27:2397–402.
33 Athyros VG, Tziomalos K, Gossios TD, Griva T, Anagnostis P, Kargiotis K, et al.; GREACE Study Collaborative Group. Safety and efficacy of long-term statin treatment for cardiovascular events in patients with coronary heart disease and abnormal liver tests in the Greek Atorvastatin and Coronary Heart Disease Evaluation (GREACE) Study: a post-hoc analysis. Lancet. 2010;376:1916–22.
34 Navarro VJ, Senior JR. Drug-related hepatotoxicity. N Engl J Med. 2006;354:731–9.
35 Bader T. The myth of statin-induced hepatotoxicity. Am J Gastroenterol. 2010;105:978–80.
36 Parker BA, Capizzi JA, Grimaldi AS, Clarkson PM, Cole SM, Keadle J, et al. Effect of statins on skeletal muscle function. Circulation. 2013;127:96–103.
37 Rosenson RS, Baker SK, Jacobson TA, Kopecky SL, Parker BA, The National Lipid Association's Muscle Safety Expert Panel. An assessment by the Statin Muscle Safety Task Force: 2014 update. J Clin Lipidol. 2014;8:S58–71.
38 Norata GD, Ballantyne CM, Catapano AL. New therapeutic principles in dyslipidaemia: focus on LDL and Lp(a) lowering drugs. Eur Heart J. 2013;34:1783–9.
39 Cannon CP, Blazing MA, Giugliano RP, McCagg A, White JA, Theroux P, et al.; IMPROVE-IT Investigators. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015;372:2387–97.
40 Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–67.
41 Taylor BA, Lorson L, White CM, Thompson PD. A randomized trial of Coenzyme Q10 in patients with confirmed statin myopathy. Atherosclerosis. 2015;238:329–35.
42 Fukumoto Y, Libby P, Rabkin E, Hill CC, Enomoto M, Hirouchi Y, et al. Statins alter smooth muscle cell accumulation and collagen content in established atheroma of watanabe heritable hyperlipidemic rabbits. Circulation. 2001;103:993–9.
43 Kini AS, Baber U, Kovacic JC, Limaye A, Ali ZA, Sweeny J, et al. Changes in plaque lipid content after short-term intensive versus standard statin therapy: the YELLOW trial. J Am Coll Cardiol. 2013;62:21–9.
44 Ridker PM, Cannon CP, Morrow D, Rifai N, Rose LM, McCabe CH, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med. 2005;352:20–8.
45 Arnaud C, Braunersreuther V, Mach F. Toward immunomodulatory and anti-inflammatory properties of statins. Trends Cardiovasc Med. 2005;15:202–6.
46 Tsujita K, Sugiyama S, Sumida H, Shimomura H, Yamashita T, Yamanaga K, et al. Impact of dual lipid-lowering strategy with ezetimibe and atorvastatin on coronary plaque regression in patients with percutaneous coronary intervention: the multicenter randomized controlled PRECISE-IVUS trial. J Am Coll Cardiol. 2015;66:495–507.
47 Bohula EA, Giugliano RP, Cannon CP, Zhou J, Murphy SA, White JA, et al. Achievement of dual low-density lipoprotein cholesterol and high-sensitivity C-reactive protein targets more frequent with the addition of ezetimibe to simvastatin and associated with better outcomes in IMPROVE-IT. Circulation. 2015;132:1224–33.
48 Abifadel M, Varret M, Rabes JP, Allard D, Ouguerram K, Devillers M, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–6.
49 Hovingh GK, Davidson MH, Kastelein JJ, O’Connor AM. Diagnosis and treatment of familial hypercholesterolaemia. Eur Heart J. 2013;34:962–71.
50 Gencer B, Lambert G, Mach F. PCSK9 inhibitors. Swiss Med Wkly. 2015;145:w14094.
51 Robinson JG, Farnier M, Krempf M, Bergeron J, Luc G, Averna M, et al.; ODYSSEY LONG TERM Investigators. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1489–99.
52 Sabatine MS, Giugliano RP, Wiviott SD, Raal FJ, Blom DJ, Robinson J, et al.; Open-Label Study of Long-Term Evaluation against LDL Cholesterol (OSLER) Investigators. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1500–9.
53 Lipinski MJ, Benedetto U, Escarcega RO, Biondi-Zoccai G, Lhermusier T, Baker NC, et al. The impact of proprotein convertase subtilisin-kexin type 9 serine protease inhibitors on lipid levels and outcomes in patients with primary hypercholesterolaemia: a network meta-analysis. Eur Heart J. 2016;37:536–45.
54 EMA. European Public Assessment Report (EPAR) for Rapatha (http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/003766/WC500191400.pdf).
55 EMA. European Public Assessment Report (EPAR) for Praluent (http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/003882/WC500194524.pdf).
56 https://www.swissmedic.ch/zulassungen/00153/00189/00200/03284 Accessed March 10, 2016.
57 Jacobson TA, Maki KC, Orringer CE, Jones PH, Kris-Etherton P, Sikand G, et al.; NLA Expert Panel. National Lipid Association recommendations for patient-centered management of dyslipidemia: Part 2. J Clin Lipidol 2015;9:S1–S122.
58 Navarese EP, Kolodziejczak M, Schulze V, Gurbel PA, Tantry U, Lin Y, et al. Effects of proprotein convertase subtilisin/kexin type 9 antibodies in adults with hypercholesterolemia: A systematic review and meta-analysis. Ann Intern Med. 2015;163:40–51.
59 Tice JA, Kazi DS, Pearson SD. Proprotein convertase subtilisin/kexin type 9 (pcsk9) inhibitors for treatment of high cholesterol levels: Effectiveness and value. JAMA Intern Med. 2016;176:107–8.
60 Choudhry NK, Patrick AR, Glynn RJ, Avorn J. The cost-effectiveness of C-reactive protein testing and rosuvastatin treatment for patients with normal cholesterol levels. J Am Coll Cardiol. 2011;57:784–91.