Figure 1
Practical approach to modern epilepsy care.
DOI: https://doi.org/10.4414/smw.2016.14310
Abbreviations
AED antiepileptic drug
ANT anterior nucleus of thalamus
DBS deep brain stimulation
EEG electroencephalogram
ESI electric source imaging
fMRI functional MRI
MRI magnet resonance imaging
PET positron emission tomography
PRE pharmacoresistent epilepsy
SISCOM subtraction Ictal SPECT Co-registered to MRI
SPECT single photon emission computed tomography
The prevalence of epilepsy is 0.5–1%, with an overall rate of complete seizure control in 40–50% of epileptic patients. “Epilepsy” has to be differentiated from acute symptomatic seizures (table 1) and diseases that mimic epileptic seizures (table 2). Acute symptomatic seizures, previously also called “provoked seizures”, do not meet the criteria of this definition, and therefore do not represent “epilepsy”. Table 1 gives an overview of the most frequent acute insults. In the majority of cases, they do not need antiepileptic treatment (exceptions include recurrent alcohol withdrawal or other recurrent acute toxicometabolic conditions that cannot be controlled).
| Table 1: Possible causes of acute symptomatic seizures. | |
| Insult | Clinical features |
| Illicit drugs | Amphetamine-like drugs, cocaine, crack, angel dust (phencyclidine) Less likely for heroin or cannabis |
| Infections | Within <7–14 d Viral encephalitis Bacterial meningitis Degenerative phase of neurocysticercosis |
| Medication | Chlorpromazine, clozapine, Maprotiline, clomipramine, Bupropion, meperidine, Flumazenil, cyclic antidepressants, Theophylline, isoniazid, alkylating antineoplastic agents, Ciclosporin; Overdose of medication |
| Metabolic | Renal or hepatic dysfunction, especially rapid changes Hyperammonaemia (35 mM) Na <115 mg/dl (<5 mM) Mg <0.8 mg/dl (<0.3 mM) Ca <5 mg/dl (<1.2 mM) Glucose <36 mg/dl (2.0 mM) or 450 mg/dl (25 mM) associated with keotacidosis |
| Traumatic | Within <7 d Associated with haemorrhage |
| Vascular | Subarachnoid bleeding Ischaemic stroke (<7 d) Intracerebral haemorrhage Cerebral vein thrombosis |
| Withdrawal, deficiency | Alcohol Benzodiazepine Barbiturate Rarely vitamin B12, Vitamin B6 (young children) |
| Other | Posterior reversible encephalopathy syndrome Cerebral anoxia Eclampsia Multiple sclerosis within 7 d of relapse |
| Adapted from [1, 2]. | |
| Table 2: Differential diagnosis of seizures. | |
| Diagnosis | Symptoms |
| Cardiac events with falls and loss of consciousness | Not necessarily with prodomes, due to cardiac arrhythmia, or carotid sinus hypersensitivity (see also below under syncope) |
| Migraine | – Progression of neurologic symptoms >5–15 min followed by headaches (but not always) – Personal and/or family history of migraine – Basilar migraine: confusion, bilateral blindness; headache may be minimal or absent |
| Movement disorders | – Rare, specific syndrome, like paroxysmal dyskinesia |
| Psychogenic | – Is not equal to absence of “organic” findings! → positive psychiatric history or findings mandatory – Semiology suggestive of psychogenic onset: eyes closed, resistant to forceful opening, eye fluttering, rhythmic horizontal head movement, pelvic thrusting – Paradoxical response to antiepileptic drug introduction (“even worse”) or no change at all |
| Syncope | – Brief loss of consciousness, with rapid recovery – Typical prodromes: nausea, “spots before the eyes”, sweating, light-headedness, cardiac palpitations – At the end muscle jerks at the end of event, usually less rhythmic, only few (“convulsive syncope”). – Precipitating circumstances often identifiable. Cave in the elderly: recovery may take 10–60 min in elderly, mimicking postictal state loss of consciousness left often, syncope may present as transient ischaemic attack (e.g. speech difficulties) |
| Transient global amnesia | – More often in patients >50 years – Prolonged duration (several hours) without alteration of consciousness; isolated memory deficit excluding long-term memory before onset of the symptoms |
| Transient ischaemic attack | – More often in patients >50 years – Rather negative symptoms (e.g., weakness, aphasia), but usually no loss of consciousness |
The new definition of epilepsy no longer requires the occurrence of two seizures (table 3), but rather one seizure plus an enduring high likelihood that a second seizure will occur, if the patient is not treated accordingly [3]. This is the case, for example, in a patient whose first seizure led to the discovery of a tumour on magnetic resonance imaging (MRI), or an 18-year-old with generalised polyspike-wave discharges under hyperventilation on a standard electroencephalogram (EEG), indicating the presence of juvenile myoclonic epilepsy. The new definition of epilepsy changes the interpretation of older epidemiology studies, which still used the definition of two spontaneous seizures within >24 h, but matches clinical experience in most cases [4].
Especially, the important question of when to introduce an antiepileptic treatment is still subject of ongoing discussion. In 2015, the American Association of Neurology issued new recommendations [5], giving level A evidence for an increased chance of a recurrent seizure for adult patients, greatest within the first 2 years after a first seizure (21–45%). However, evidence in favour of risk reduction when directly starting antiepileptic drug (AED) therapy, as compared with a delay of treatment pending a second seizure, is only level B, compared with level B evidence for a 7–31% incidence of adverse effects from AED therapy, but which are mild and reversible. There is only weak evidence that immediate AED therapy, as compared with awaiting a second seizure, may not improve quality of life (level C) [6]. It has been claimed that some of this evidence is based on a very few old studies [7]. In the case of prior brain insult such as stroke or trauma, as well as in the presence of epileptiform abnormalities on the EEG, evidence is level A for the increased risk for seizure recurrence. However, for the association of clinical factors such as brain-imaging abnormality or a nocturnal seizure, evidence is only level B. The guideline’s summary conclusion is therefore that “… recommendations whether to initiate immediate AED treatment after a first seizure should be based on individualized assessments that weigh the risk of recurrence against the AEs [adverse events] of AED therapy, consider educated patient preferences, and advise that immediate treatment will not improve the long-term prognosis for seizure remission but will reduce seizure risk over the subsequent 2 years.” [6] This emphasises that guidelines summarise current evidence, but cannot replace individual judgment in the case of every specific patient [7].
| Table 3: Conceptual difference between seizures and epilepsy. |
| Conceptual definition of seizure and epilepsy – 2014 ILAE Annual Report |
| An epileptic seizure is a transient occurrence of signs and/or symptoms due to abnormal excessive or synchronous neuronal activity in the brain. |
| Epilepsy is a disorder of the brain characterized by an enduring predisposition to generate epileptic seizures, and by the neurobiological, cognitive, psychological and social consequences of this condition. The definition of epilepsy requires the occurrence of at least one epileptic seizure. |
| From [4]. |
The aim of any epilepsy therapy should always be the suppression of all seizures. Drug studies showed that 40–50% of patients with focal epilepsy, and about 15% of patients with idopathic generalised epilepsy (in the new terminology now called “genetic generalised epilepsy”) are refractory to medical treatment. Pharmacoresistant epilepsy (PRE) is diagnosed if two or more AEDs do not lead to complete seizure control, despite regular drug intake of sufficiently high dosages as determined by regular measurement of serum levels (preferentially in the morning or before next drug intake). The chance to control seizures with a third drug is only 2% [8]. There are several reasons why drugs do not work (table 4). Therefore, PRE requires prompt evaluation to determine possible reasons, usually as an inpatient evaluation.
| Table 4: Reasons of nonresponse to drug therapy (pharmacoresistant epilepsy). | |
| Cause | Recommendation |
| Patient did not get the right drug | Recording of seizures and work-up helps to determine correct epilepsy syndrome |
| Patient does not get enough drug | Determine the blood level of the AED/s, in particular for older AEDs (if possible before drug intake). NB: for most newer drugs there is no linear relationship between blood level and efficacy |
| Drug is not taken | Determine the blood level of the AEDs, perhaps repeatedly. Ask the patient how many times she/he forgets the AED/s |
| It is not epilepsy | Recording of habitual events |
| Epilepsy is not pharmacosensitive | In-patient evaluation in specialized centre: is epilepsy surgery possible? |
Risk of seizure recurrence can be significantly reduced by 30–40% when treatment is immediately introduced once epilepsy is diagnosed [9]. The reduction is effective for the first 2 years. However, long-term outcome is not affected, and in the UK study around 75% were seizure-free at 2 years, no matter whether treatment was delayed until after the second or third seizure or not. Overall, if patients relapse, 90% do so within the first 2 years.
Since chances of responding to a second drug are slim (14% [10]), PRE might already be suspected after the lack of response of the first AED, and possibly diagnosed within the first 2–4 years. It is of note that quality of life is better in patients with single seizures or early remission than in those with late remission or who never experienced any seizure control [11], which is another argument in favour of immediate treatment and rigorous seizure control.
If seizures persist and PRE is diagnosed, further treatment options should be explored. These are epilepsy surgery and neuromodulation, i.e. intra- or extracranial stimulation. While surgery can cure epilepsy, neuromodulation – up to now – is a palliative treatment strategy giving freedom from seizures in <10% of the cases. In both cases, careful evaluation in specialised centres is necessary to obtain a clear picture of the underlying syndrome and information on the chance for benefit from these alternative therapeutic strategies.
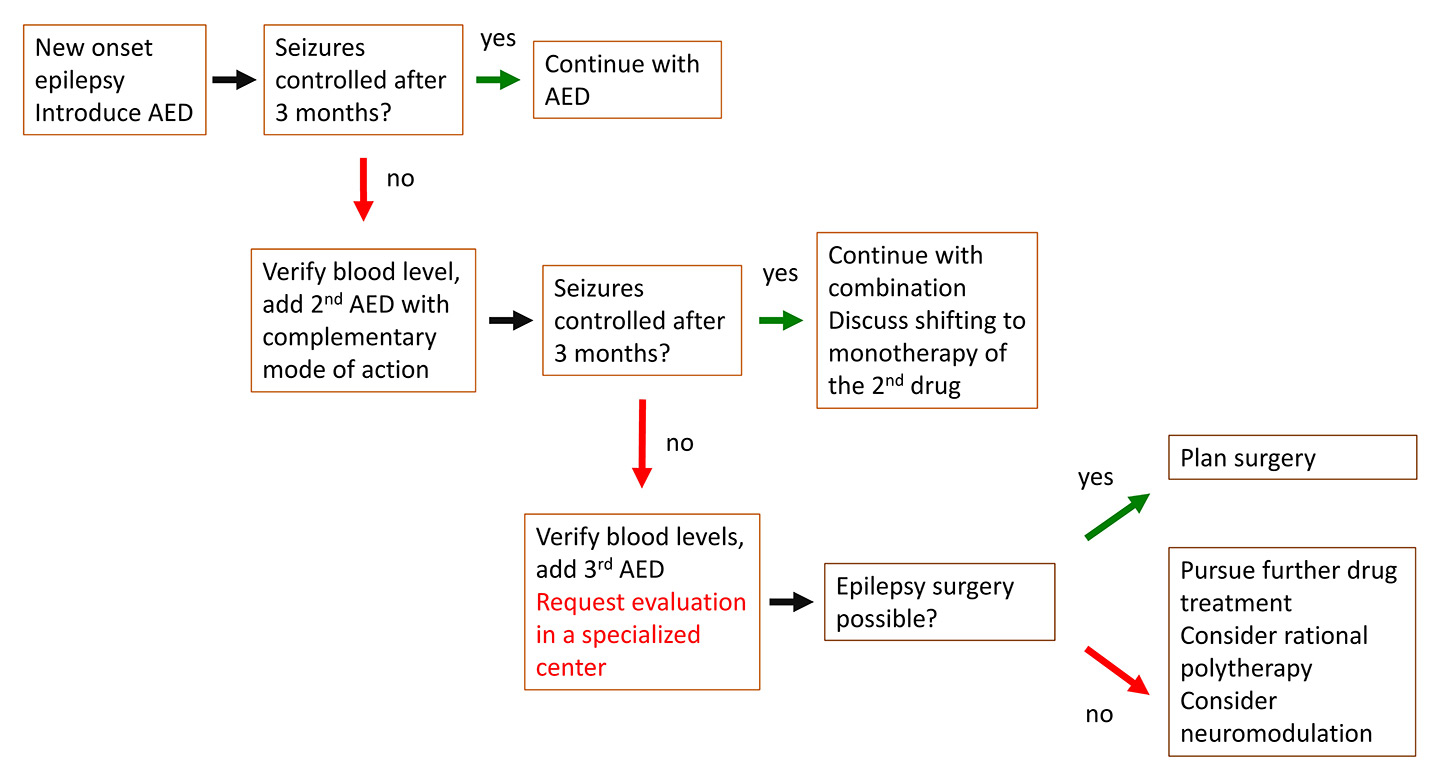
A practical approach to modern epilepsy care is summarised in figure 1.
In the present article, we summarise the most clinically relevant data on the newer AEDs (i.e., introduced after 1997) in alphabetical order, as well as epilepsy surgery and neuromodulation available in Switzerland: vagal nerve stimulation (VNS) and deep brain stimulation in the anterior thalamic nuclei (DBS-ANT).
Figure 1
Practical approach to modern epilepsy care.
Until 1993 only phenobarbital, primidone, phenytoin, carbamazepine and valproate were available. Of these, only carbamazepine and valproate are still used in a significant number of patients. Today, the administration of phenytoin is limited to difficult cases of status epilepticus, given its availability in an intravenous formulation. Phenytoin is considered neurotoxic and related to cerebellar atrophy and polyneuropathy with chronic treatment. Phenobarbital and primidone are sometimes used in patients with major compliance problems, because of their long half-lives. Both drugs frequently lead to vitamin D deficiency and osteoporosis, so they should be considered as a last resort.
In 2012, Swissmedic issued the recommendation to determine HLA-A*3101 before the introduction of carbamazepine. Studies in northern European and Japanese populations have found an association between Stevens-Johnson syndrome / toxic epidermal necrolysis with carbamazepine use and the presence of the HLA-A*3101 allele [12, 13]. In a southeast Asian population (of Han Chinese descent), these side effects seemed to be related to HLA-B*1502 [12]. Oxcarbazepine might also be associated with this risk; however, there is currently insufficient data to support a recommendation for testing the presence of both alleles in patients prior to treatment. There is also ongoing discussion of the cost-benefit of a regular HLA testing, given the high costs of the test, that this complication is extremely rare and that most patients in Switzerland are clinically monitored by their neurologists or family doctors.
In 1994 lamotrigine and in 1996 topiramate became available, both with a wide spectrum, for children and as monotherapy. Lamotrigine is very well tolerated in doses up to 500–700 mg (as monotherapy), and has a favourable effect on mood and cognitive functions. However, it requires very slow introduction extending over 2 months and more, which is not convenient in patients with a high seizure count. Also the high incidence of allergies was an issue during early use of the molecule, but can be largely avoided if this slow titration route is chosen. Topiramate is known for its anorexic effect (noted in around 20% of patients) due to decreased appetite. In some patients and children this can be severe, requiring drug withdrawal. Other side effects are hyperthermia, irritability and depression (which is the reason why it is sometimes used for mania). Rarely, glaucoma or kidney stones are also described. Topiramate probably has teratogenic effects, in particular in polytherapy [14, 15]. The most teratogenic AED is valproate, and children exposed in utero to valproate are at a high risk of major developmental disorders and congenital malformations (10% of cases). In 2015, the Medicines & Healthcare products Regulatory Agency of the UK suggested that valproate should not be used in female children and young women for this reason, unless other treatments are ineffective or not tolerated [16].
Before initiating antiepileptic drugs, the underlying syndrome needs to be determined, usually by a neurologist or neuropaediatrician. Two broad categories are distinguished: focal epilepsy and genetic (idiopathic) generalised epilepsy. This distinction is important since the latter can significantly worsen if, for example, with carbamazepine, oxcarbazepine or pregabalin are erroneously given. In fact, patients may describe symptoms suggestive of focal epilepsy. Work-up with EEG, sleep/long-term EEG and MRI usually helps to differentiate between focal and nonfocal epilepsy.
Fortunately, more antiepileptic drugs became available in Switzerland in the last 10–15 years. However, most of the evidence points to a lack of significantly augmented efficacy with newer AEDs: the chances of obtaining sufficient seizure control remain the same, no matter whether two new or two old AEDs are used. Nevertheless, newer drugs are often better tolerated and therefore should be considered early in the course of the disease in the context of patient-oriented tailored treatment, since this is crucial for the patient’s compliance.
In table 5, the main features of the drugs are summarised.
| Table 5: Summary of antiepileptic drugs | |||
| Lacosamide – Vimpat® | Levetiracetam – several brands | Oxcarbazepine – Trileptal®, Apydan® | |
| Approval in Switzerland | 2009 | 2000 | 1997 |
| Indication | Add-on in focal seizures ± generalisation(>18 y) | Add-on in focal seizures ± generalisation (>1 month) Monotherapy in focal seizures ± generalisation (>16 y) Monotherapy in IGE with myoclonic seizures (>12 y) Add-on in IGE with tonic-clonic seizures (>12 y) | Monotherapy or add-on therapy in focal onset seizures ± generalisation in adults and children (aged >1 month) |
| Price for daily dose | 9.6 CHF/d at 300 mg/d | 1.6 CHF/d at 1000 mg/d | 4.2 CHF at 1500 mg/d |
| Administration | PO, IV | PO, IV | PO |
| Mode of action | Na+-channel blocker, binding CRMP-2 | Unknown Suspected release of presynaptic Ca++; enhancement of GABAergic inhibition | Keto-analogue of CBZ, Blocks voltage gated Na+ channels, potentiates K+ conduction Inhibits Ca++channels Inhibits NMDA receptors |
| Frequent adverse effects | Dizziness, fatigue, ataxia, vertigo Rarely: increased PR interval on ECG | Behavioural changes, somnolence, | Hyponatraemia |
| Oral contraception | No interaction | No interaction | Can lower the contraceptive effect |
| Other interactions | No interaction | No interaction | OXC level is lowered by PHT, PB, VPA OXC can lower the level of PHT, PB |
| Remarks | Apydan®: extended version 9 h | ||
| Perampanel – Fycompa® | Pregabalin – several brands | Rufinamide – Inovelon® | |
| Approval in Switzerland | 2013 | 2005 | 2009 |
| Indication | Add-on in focal seizures ± generalisation age (>12 y) | Add-on in focal seizures ± generalisation (>18 y) | Add-on in Lennox-Gastaut syndrome (4 y) |
| Prize for (most frequent) daily dose | 8.4 CHF/d at 8 mg/d | 4.8 CHF/d at 600 mg/d | 18 CHF/d at 2400 mg/d |
| Administration | PO | PO | PO |
| Mode of action | Noncompetitive selective AMPA inhibitor of postsynaptic glutamate transmission | Structural derivative of GABA-inhibitor, acts on Ca++ channels | Uncertain – reduces Na+ channel activity |
| Frequent adverse effects | Dizziness, somnolence, irritability, headache, ataxia | Weight gain, somnolence, dizziness, irritability | Headache, dizziness, fatigue, nausea, somnolence, diplopia nasopharyngitis, tremor. |
| Oral contraception | Can lower the contraceptive effect | No interaction | Can lower the contraceptive effect |
| Other interactions | Decreases the level of CBZ, PHT, LTG, midazolam, clobazam etc. Increases the level of OXC. PER level is decreased by >50% in the presence of CBZ, PHT or OXC. | No interaction | Increases VPA and PHT, Level is increased by VPA Level is lowered by CBZ |
| Remarks | Indication also for neuropathic pain, fibromyalgia, general anxietys | ||
| AMPA = α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; CBZ = carbamazepine; CRMP-2 = collapsin response mediator protein 2; GABA = gamma-amino butyrate; IGE = idiopathic generalised epilepsy; IV = intravenous administration; LTG = lamotrigine; NMDA = N-methyl-D-aspartate; OXC = oxcarbazepine; PB = phenobarbital; PER = perampanel; PHT = phenytoin; PO = per os, oral administration; VPA = valproate | |||
Lacosamide is a sodium-channel modulator with a different mechanism of activity from the other known sodium-channel blockers like carbamazepine, lamotrigine or phenytoin. Unlike those drugs, lacosamide selectively enhances the slow inactivation of voltage-gated sodium channels without affecting their fast inactivation [17]. This normalises activation thresholds and therefore controls neuronal hyperexcitability and pathological neuronal activity [17].
Lacosamide was approved by the US Food and Drug Administration (FDA) in 2007 and has been available in Switzerland since 2009 as add-on therapy in adult patients with focal seizures with and without secondary generalisation. All galenic forms are available: tablets, syrup and an intravenous formulation. The most often used daily doses are 200–400 mg. Given its intravenous formulation, lacosamide can be used effectively against nonconvulsive and convulsive status epilepticus [18].
The elimination of lacosamide is essentially renal. It has a low potential for drug-drug interactions and is minimally bound to albumin. During the three pivotal clinical studies [19–22], lacosamide did not alter the mean plasma concentrations of concomitantly administered AEDs, including carbamazepine and the monohydroxy derivative of oxcarbazepine, lamotrigine, levetiracetam, phenytoin, topiramate or valproic acid.
Most treatment-emergent adverse events were of mild or moderate intensity and lacosamide was generally well tolerated in adult patients with focal-onset seizures. The adverse events appeared dose-related and included dizziness, nausea, fatigue, ataxia, abnormal vision, vertigo, diplopia and nystagmus [19–23]. Their incidence and intensity decreased over time. Most importantly for clinical use, a dose-related increase in the PR interval of the electrocardiogram (ECG) during treatment with lacosamide was noticed, but a very recent study has demonstrated the safety of lacosamide below 400 mg/d in cardiac patients [24]
The exact mechanism of action of levetiracetam is unknown, but it appears that its affinity to the synaptic vesicle protein 2A seems to play a crucial role, in that it inhibits presynaptic calcium channels [25]. This synaptic vesicle protein is involved in vesicle exocytosis and neurotransmitter release [26].
Levetiracetam was approved by the FDA in 1999 and has been available in Switzerland since 2000 as add-on therapy (for patients ≥4 years of age) and since 2008 also as monotherapy in focal-onset seizures (>16 years) as well as an adjunctive therapy for primary generalised epilepsy onset (≥12 years). It is available as tablets, syrup and an intravenous solution, making it nowadays one of the most widely used first-line AEDs, including for status epilepticus [27, 28]. The effective dose of levetiracetam varies between 1000 mg and 3000 mg.
Levetiracetam is not bound to plasma proteins and is not metabolised in the liver [29]. Elimination is renal and therefore its dosage needs adaptation to the creatinine clearance. The half-life is around 7 h. Levetiracetam is not extensively metabolised, only 25% undergoes enzymatic hydrolysis of the acetamide group by a plasma hydroxylase. It has no known clinically significant pharmacological interactions [30]. A study of oral anticoagulants showed that the prothrombin time international normalised ratio (INR) values measured after repeated administration of placebo or levetiracetam were not statistically different [31].
Levetiracetam is well tolerated. Side-effects include somnolence and, especially, behavioural changes of varying intensity in around 15%. [32]. In particular, patients with learning disabilities may occasionally show an increase in aggressive behaviour [33, 34]. However, the psychiatric side effects resolve quickly after drug discontinuation.
Oxcarbazepine is a keto-analogue of carbamazepine, with a similar spectrum of anticonvulsive activity and efficacy: it blocks voltage-gated sodium channels, potentiates potassium conduction and inhibits calcium channels and N-methyl-D-aspartate (NMDA) receptors.
Oxcarbazepine has been available in Switzerland since 1997. It is used as monotherapy [35–37] or add-on therapy [40] for focal-onset seizures with or without secondary generalisation in adults [37–39] and children aged >1 month [40, 41]. It was shown to be equivalent to carbamazepine and phenytoin in its efficacy with a higher tolerability (class I and II evidence) [42, 43].
Oxcarbazepine is available as tablets or an oral suspension, with dosages up to 1800–2400 mg/d [38]. Previous treatment with carbamazepine can be switched to oxcarbazepine rapidly in a 2:3 fashion: 400 mg carbamazepine should be replaced by 600 mg oxcarbazepine [44]. In patients receiving additional AEDs other than carbamazepine, a gradual switch over 2–3 weeks is more appropriate [38].
Oxcarbazepine is metabolised mainly through reductive biotransformation and glucuronidation to the active 10-monohydroxy derivate, the pharmacologically active metabolite [45]. It is eliminated via the liver; it inhibits CYP2C19 and induces CYP3A4, therefore carbamazepine, phenytoin, phenobarbital and valproic acid can lower oxcarbazepine levels, whereas oxcarbazepine may lower phenobarbital and phenytoin levels [46].
Oxcarbazepine is generally well tolerated, except by patients with myoclonic and absence epilepsy, which can be worsened under oxcarbazepine treatment [47]. Adverse effects are similar to those known from carbamazepine, although less severe and less frequent [38, 48, 49]. Due to its effect on antidiuretic hormone in the distal convoluted tubule of the kidney, hyponatraemia is a well-known adverse effect. It is usually asymptomatic and returns to normal with fluid restriction, dose reduction or oxcarbazepine discontinuation [50].
Perampanel is a noncompetitive selective blocker of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) glutamate receptor, acting on post-synaptic glutamate transmission [51].
Perampanel was FDA-approved in 2012 and has been available in Switzerland since the end of 2013. It is used as add-on treatment of focal-onset seizures in patients ≥12 years of age [51–54]. In the European Union, but not in Switzerland, perampanel has recently been licensed for the adjunctive treatment of genetic tonic-clonic seizures following the promising results of multicentre, double-blind study [55]. A long-term safety study lasting 3 years showed good tolerability [54].
Perampanel is primarily metabolised in the liver via CYP3A4 and also CPY3A52. However, it only weakly affects the activities of some cytochrome P450 isoenzymes. CYP3A4 inhibitors/inducers affect the availability of perampanel: carbamazepine, oxcarbazepine, and phenytoin reduce perampanel serum levels by about 67%, 50% and 50%, respectively, all of which are considered clinically important [56]. Perampanel only weakly decreases the levels of carbamazepine, clobazam, lamotrigine, midazolam and valproate, whereas it can increase the level of oxcarbazepine [29, 57].
Perampanel is available as coated tablets. Effective dosages are mostly between 4–8 mg/d, but can be up to 12 mg/d. It has a long half-life of around 100 hours, which allows a single daily dose. Adverse events include dizziness, somnolence, irritability, headache and ataxia [58]. Note that with the FDA approval a special warning was issued concerning “serious psychiatric and behavioural adverse reactions including aggression, hostility, irritability, anger and homicidal ideation“, however, in a follow-up study neuropsychiatric adverse effects occurred in only a small percentage [59]. And in a very recent review of patients’ safety and efficacy during perampanel treatment [60], slow introduction and close clinical monitoring was recommended in order to prevent these effects and guarantee an overall excellent tolerability.
Pregabalin is a structural derivative of the inhibitory neurotransmitter gamma-amino-butyrate (GABA). Its effect is supposed to be based on its action on the alpha2 delta subunit of the P/Q-type voltage-sensitive calcium channels, which are present in presynaptic neurons and modulate depolarisation-induced calcium influx [61, 62].
Pregabalin has been available in Switzerland since 2005 as add-on therapy of focal seizures with or without generalisation in adults (>18 years), but is also indicated for the treatment of neuropathic pain, fibromyalgia and generalised anxiety. Dosages of 150–600 mg/d are used.
Pregabalin is not protein bound and is eliminated unchanged by renal excretion. Careful monitoring of creatinine clearance is therefore mandatory, even in moderate renal failure. The bioavailability of pregabalin is 90%. Given a half-life of approximately 6 hours, pregabalin can be given in two or three divided doses. Pregabalin had a low risk of drug-drug interactions when added to phenytoin, valproate or lamotrigine [63].
Most common adverse effects reported were somnolence, dizziness, ataxia, diplopia and weight gain. Irritability and euphoria are also described frequently. Most of them tend to resolve with slow continuation of treatment.
For women using oral contraception, no adaptation of treatment is necessary; oral contraceptives remain effective under pregabalin. No data are available to our knowledge about its use in patients taking anticoagulants, but theoretically there should be no significant interaction.
The exact mechanisms of action of rufinamide are not yet elucidated. Most evidence suggests that rufinamide, a triazole derivative, reduces the capacity of sodium channels to recover from inactivation and limits sodium-dependant action potential firing [64].
Rufinamide has been available in Switzerland since 2009 as add-on therapy for Lennox-Gastaut syndrome in patients aged 4 years or older [65]. The dosage is 400–3200 mg/d.
Elimination is predominantly via the renal pathway (85%), although it does not seem to be affected by renal impairment. Rufinamide is 34% plasma protein bound, the majority to albumin. There is no study in patients with severe hepatic dysfunction [64]. Being a triazole derivative, rufinamide is extensively metabolised by non-CYP450 systems (it is a weak inhibitor of CYP2E1 and a weak inducer of CYP3A4), with a half-life of 8–12 h. Rufinamide increases the serum levels of phenytoin and valproate. Since valproate reduces the clearance of rufinamide, lower doses are recommended (i.e. 600–1200 mg/d). In contrast, the effects of rufinamide might be less in patients who take carbamazepine [66]. Important interactions with oral contraceptives must be mentioned to the patient and caregiver, although the precise mechanism is unknown. Adding a second contraceptive device is therefore recommended to protect against pregnancy during rufinamide therapy.
Rufinamide was well tolerated at dosages up to 1600 mg/d. Adverse effects are dose-dependent and more frequent with higher doses. Pooled analysis of all data revealed the following adverse effects with a frequency >10%: headache, dizziness, fatigue, nausea, somnolence, diplopia, nasopharyngitis and tremor. The most frequent adverse events were somnolence (24.3% with rufinamide vs 12.5% with placebo) and vomiting (21.6% vs 6.3%) [65]. No worsening of cognitive function was found after 12 weeks of rufinamide treatment with daily doses of up to 1600 mg [67].
Zonisamide is a benzisoxazol derivative and sulfonamide, and has a wide spectrum of actions, among which are increased GABA release [68], inhibition of voltage sensitive sodium-channels [69] and T-type calcium-channels, as well as a reduction of potassium-mediated glutamatergic transmission [70, 71]. The inhibition of carbonic anhydrase does not contribute to the antiepileptic effect [72–74]. A previous US study (with doses of 300–500 mg) was halted because of an increased incidence of renal calculi (3.5%) [75]. Follow-up studies could not confirm this observation; however, avoiding zonisamide in patients with recurrent renal calculi is suggested.
The FDA approved zonisamide in 2000, although it has been well established in Japan since 1989, where it is used with success in a variety of seizure types, focal and non-focal tonic-clonic, tonic, clonic, atonic and myoclonic seizures as well as typical and atypical absences, with responder rates of around 55% for all seizure types together [76]. In Switzerland, zonisamide has been available since 2006 as add-on therapy of focal-onset seizures with or without secondary generalisation (age ≥6 years) and since 2014 also as monotherapy for partial seizures in adults (≥18 years). The dosage is usually between 300 and 500 mg/d (1 mg/kg in children), but patients may already respond at daily doses of 150–200 mg.
Zonisamide is eliminated via the renal pathway (97%) and only marginally via the liver (3%); however, it is extensively metabolised by the liver and 15–30% of zonisamide is excreted unchanged via the kidneys. Therefore, zonisamide is contraindicated in moderate to severe renal insufficiency (creatinine clearance of 50 ml/min or less) and also in patients with hepatic insufficiency (Child-Pugh B or C). Conversely, there is neither induction nor inhibition of CYP450 isoenzymes and no self-induction of metabolism [77], and there is no interaction with other AEDs. Zonisamide is therefore well suited to being added to a multidrug regimen, including older AEDs depending on the CYP450 system.
The only exception is topiramate, with which co-medication is currently not recommended: Some adverse effects resemble those of topiramate, zonisamide and topiramate both having an inhibitory effect on carbonic anhydrase and inducing metabolic acidosis and urinary alkalosis, which increases the risk of renal calculi [77]. Zonisamide and topiramate also commonly induce oligohydrosis with hyperthermia (especially in children), due to an inhibition of the carbonic anhydrase isoenzymes I and II and aquaporin-1 (AQP-1), localised in the human exocrine sweat gland. Like topiramate, zonisamide can lead to weight loss, which is a “positive” side effect in overweight patients: 22% of the patients lost >2.3 kg during one study [78]. However, in the majority of patients, no significant change in weight is observed. The other most frequently reported adverse effects of zonisamide were fatigue/somnolence, dizziness, nausea, irritability and ataxia [78].
It is noteworthy that a study on teratogenicity in humans showed two malformations, anencephaly and atrial septal defect, in 26 studied pregnancies [79]. In animals some teratogenic effects have been demonstrated, but the malformation rate is still unclear given that they were seen in polymedicated patients only.
Eslicarbazepine acetate was accepted by the FDA in June 2009, and in most European countries, but approval in Switzerland has not been obtained so far. Since patients with this drug may enter Switzerland and require care, we briefly discuss this drug.
Eslicarbazepine has mechanisms of action distinct from carbamazepine and oxcarbazepine, and demonstrated anticonvulsant properties with a wider (1.5- to 2.5-fold) protective index than carbamazepine [80]. Eslicarbazepine is the L-enantiomer of the active compound 10-monohydroxy-carbazepine (li-carbazepine). The R-enantiomer (which causes the adverse effects of carbamazepine and oxcarbazepine, especially hyponatraemia) accounts for less than 10%. Therefore, there was initially hope that hyponatraemia would occur less frequently than with carbamazepine and oxcarbazepine; however, it appeared to be noted to a similar extent [81–84]. However, unlike carbamazepine, it is less susceptible to enzyme induction or autoinduction. Other adverse effects are comparable to those of carbamazepine and oxcarbazepine. Note that eslicarbazepine interacts with oral contraceptives, so complementary contraception is recommended.
Eslicarbazepine metabolites are eliminated primarily by renal excretion. Moderate hepatic impairment has no clinically relevant effect on eslicarbazepine pharmacokinetics [82], and consequently no dose adjustment is required in patients with mild to moderate hepatic impairment. However, adjustment of the daily dose is recommended for patients with impaired renal function (creatinine clearance <50–60 ml/min) [85]. Eslicarbazepine has a long half-life of 20–24 h, so a single dose per day (mostly 800–1600 mg) may be sufficient.
If the first monotherapy does not lead to seizure control, combination of drugs is often the first option. However, there are only a few studies on effective “rational” polytherapies. A recent large retrospective study on >8000 patients suggested that combining drugs with different modes of action is more efficient as measured by treatment persistence, number of hospitalisations or emergency room visits, than combining those with similar modes of action [86]. While this has been taught already for some time, it had never been shown in a real-life setting.
Overall, polytherapy is not necessarily more toxic, as has been shown in an elegant study with valproate and carbamazepine [87]. However, in this study dosages were low and may explain the lack of difference between carbamazepine monotherapy and combination therapy. In any case, therapeutic drug monitoring is strongly recommended whenever adding or removing a drug, including non-AEDs. Break-through seizures should be avoided as much as possible: they may cost the patients their driver’s licenses or cause professional and physical disadvantages.
The only combination which was found to be truly synergistic was the combination of lamotrigine and valproate [88]. Addition of valproate to a lamotrigine regimen was much more effective than the addition of phenytoin to a carbamazepine regimen. Moreover, when patients were switched back to monotherapy, lamotrigine was less effective than in combination. This effect is most likely due to the fact that carbamazepine and phenytoin decrease the level of lamotrigine, whereas valproate increases it, i.e. more often at more efficient levels.
Effective polytherapy with more than two drugs leading to seizure-free patients included a combination of several broad-spectrum AEDs such as lamotrigine, levetiracetam or valproate, as shown in a larger retrospective study, which should be preferred to drugs with a narrower spectrum, like oxcarbazepine [89].
Approximately 40% of all epilepsy patients do not attain freedom from seizures despite regular drug intake in sufficiently high dosages [8]. In these cases, in-depth evaluation is mandatory, to determine the reasons of the non-response to drugs.
It should be considered if two AEDs do not result in seizure freedom, i.e. complete control of all seizures, which represents a significant handicap for the patient regarding his or her socio-professional activities and physical integrity. It is of note that exclusively nocturnal seizures also represent a danger, due to an increased risk of suffocation and sudden unexpected death [90]. Patients with chronic epilepsy have a 3–10-fold higher probability of mortality compared with their nonepileptic peers of similar age, depending on the precise underlying syndrome [8, 91, 92]. In an older study, those with right temporal epilepsy had a 30-fold increased risk of mortality [93], probably because the right insular cortex, crucial in the cerebral control of heart rhythm (bradycardia), is easily recruited during a seizure.
However, there is still reluctance towards epilepsy surgery among caring physicians [94, 95]. Two studies [94, 95] have showed that the most frequent causes include underestimation of the danger of recurrent seizures, underestimation of the success rate of epilepsy surgery, overestimation of the complication rate of the surgery, and unclear criteria when to refer. For example, in a study among American neurologists, 19% thought that their patients had to fail all approved AEDs to be considered medically refractory and 55% considered surgery only if the patient had more than one seizure per month [94]. However, three generalised tonic-clonic seizures per year represent also a significant danger for the patient.
The high risks of uncontrolled epilepsy and the low yield of addition of a fourth or fifth AED speak in favour of an evaluation for epilepsy surgery in order to determine the optimal resection site and extent, determine the prognosis for success and verify the absence of non-epileptic events mistaken for epileptic fits. No epilepsy surgery should be undertaken without preceding in-depth evaluation.
During the evaluation, two basic questions have to be answered: the location of the epileptic focus and identity of possibly adjacent vital cortex. The goal of epilepsy surgery is safe removal of the epileptogenic tissue without inflicting a (new) neurological deficit. Neurological, neuropsychological and psychiatric evaluations are basic elements of the preoperative work-up. The evaluation procedure includes the recording of several habitual seizures with long-term video-EEG monitoring in a specialised centre, with 24-h surveillance by specialised personnel, rapid access to intensive care and emergency CT. A high resolution MRI scan and, if possible, examination with other imaging tools (e.g. position-emission tomography [PET], single photon-emission-computer-tomography [SPECT], high-density EEG / electric source imaging [ESI]) are crucial in the identification of the seizure onset zone [96]. The more imaging examinations are concordant, the more likely there will be postoperative seizure freedom [97]. Figure 2 provides an example of multimodal imaging in a patient with bitemporal discharges.
Surgical results depend critically on the definition of the ictal and interictal EEG onset zone as well as on the presence of a lesion in 1.5-Tesla or, better, 3-Tesla high-resolution MRI, obtained with an epilepsy protocol, i.e. 1 mm slices for most sequences, as detailed elsewhere [98]. If the lesion is highly suspected or functionally localised with PET, SPECT or ESI, but not visualised in the high-resolution MRI, additional statistical analysis of the MRI may be useful and unravel the underlying epileptogenic lesion [99, 100] (fig. 3).
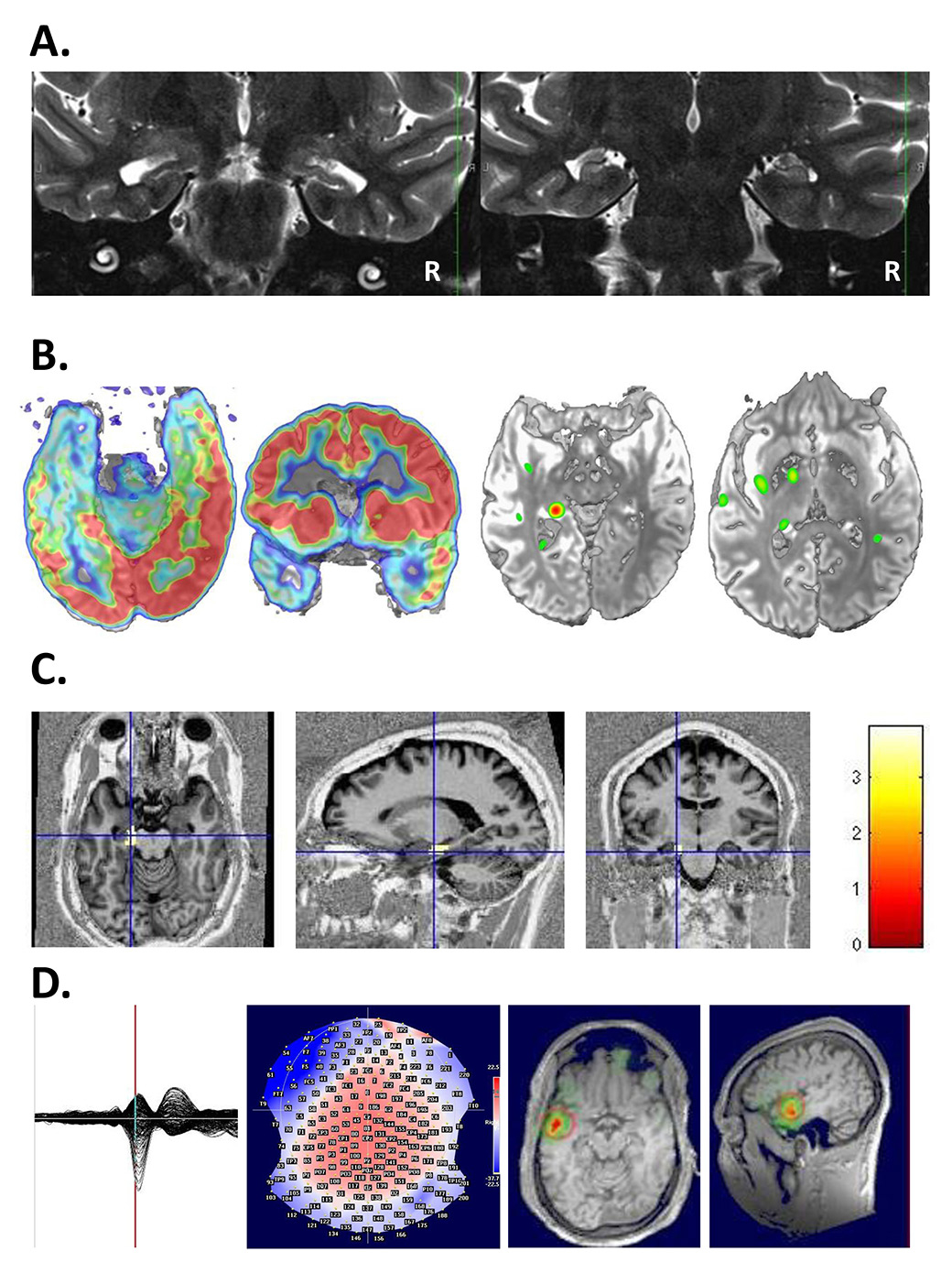
Figure 2
A 26-year-old patient undergoing multimodal imaging after a recording of bitemporal discharges with left predominance. A. MRI showed bilateral hippocampal atrophy with left predominance. B. left: PET showed a hypometabolism at the left mesioteporal lobe, right: Ictal-interictal substraction SPECT (SISCOM) showed an ictal hypermetabolism at the left mesiotemporal lobe. C: Combined EEG-fMRI showed a left hippocampal localization of spike-related BOLD changes. D. Electric source imaging (ESI) also showed a left localisation of spikes. With multimodal work-up left antero-mesial epilepsy was diagnosed.
EEG = electroencephalogram; fMRI = functional MRI; MRI = magnetic resonance imaging; PET = positron emission tomography; SPECT = single photon emission computed tomography. (The left side of the image is left.)
In some cases, the seizure onset region cannot be determined with sufficiently high certainty with noninvasive tools, so intracranial EEG with implanted electrodes is proposed. The need to turn towards intracranial EEG depends crucially on the yield of the complementary imaging tools: the clearer the result in the noninvasive image modalities, the lower the need to implant patients with intracranial electrodes. In our centre Geneva-Vaud, this affects only around 15% of all patients; and for the majority of patients, noninvasive imaging tools are sufficient to determine eligibility for surgery. Intracranial exploration has certain important limits, because of the fact that implanted contacts “see” neuronal activity within a radius of only 5–10 mm. Therefore, if the intracerebral electrode is not placed directly in or very close to the epileptic focus, even intracranial EEG recordings can be false negative. For this reason, a careful noninvasive work-up (phase I) is mandatory to prepare optimally for invasive evaluation and increase its yield.
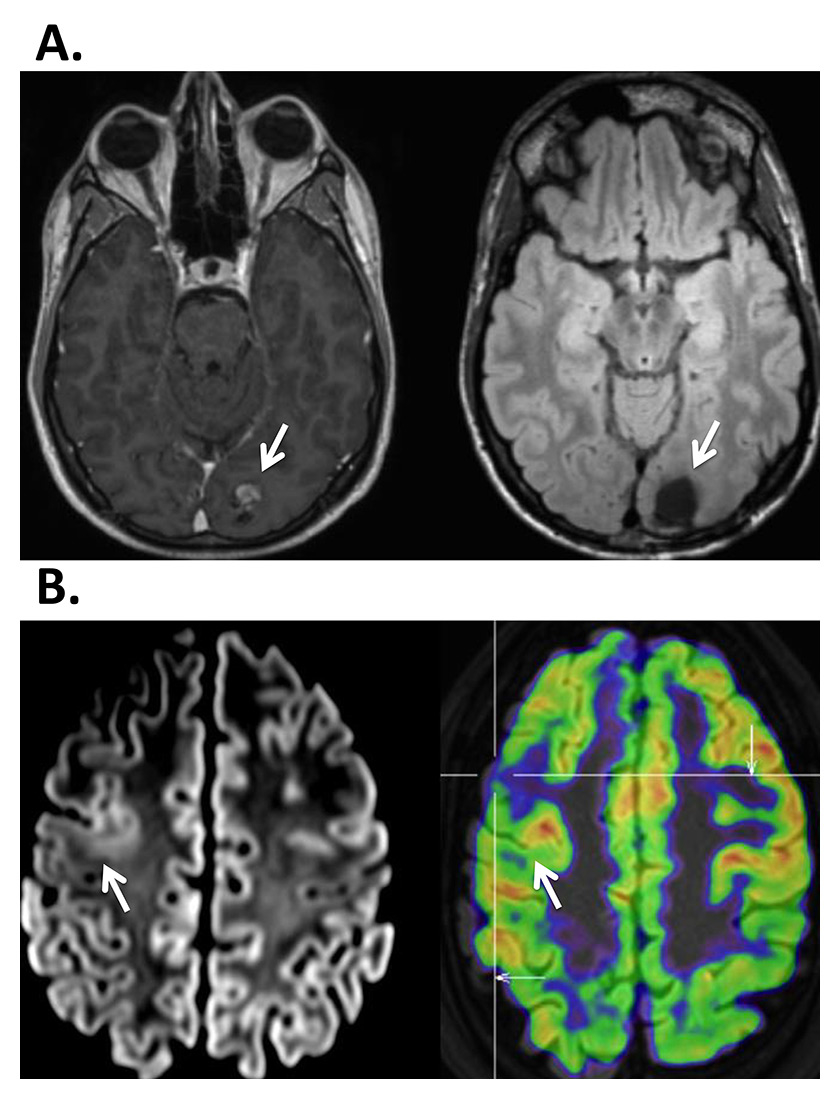
Figure 3
A. 37-year-old patient presenting with seizures with a visual aura in the right visual field. On MRI a left occipital cavernoma was found (à), which was surgically removed (right). Work-up did not show any other focus or area of dysfunction. B. 34-year-old patient presenting with generalized seizures, EEG showed a very active focus in the right frontal lobe, but MRI was considered normal. Right: PET identified a focal hypometabolism in the right frontal lateral area (à). Left: Review of the MRI showed blurring between gray and white matter indicating the presence of a dysplasia.
EEG = electroencephalogram; MRI = magnetic resonance imaging; PET = positron emission tomography
The risks of electrode implantation depend on the number of electrodes and sites to be explored; they should be discussed once the implantation strategy is clear. Due to rare occasions of (delayed) infections or bleeding, it is recommended to have round-the-clock in-house access to an intensive care unit and a neurosurgeon on call.
In general, temporal lobe epilepsy carries a better prognosis than extratemporal lobe epilepsy, as a result of less well defined anatomical limits. There is no need to avoid adjacent motor, sensory or language cortices, leading to less limited resection in temporal lobe epilepsy than in extratemporal. Nevertheless, the vast majority of temporal and extratemporal surgical candidates benefit clearly from the surgical procedure. Current surgical approaches are displayed in figure 4. In our centre, >200 patients have been operated on with the aim of complete seizure control. Overall, 78% were seizure-free at ≥1 year post-operatively and another 8% were almost seizure-free, which amounts to 84% who benefited from the procedure. Ten percent presented moderate seizure decrease or no change (3%). There was no patient with an aggravated seizure disorder after surgery [96].
In a recent review on 40 surgical series between 1995 and 2007, of a total of 3557 patients, 70% of patients with lesional epilepsy were postoperatively seizure-free [102], confirming the benefit of expert MR imaging and interpretation. Lesions included benign tumours, such as gangliogliomas, and developmental abnormalities, such as dysplasia or gliotic lesions (hippocampal sclerosis, post-traumatic, vascular insult). The outcome of nonlesional and extratemporal cases was markedly poorer, with 40–50% seizure-free patients. However, in most of the reviewed studies there was no access to PET, ictal SPECT or electric source imaging based on scalp EEG with >100 electrodes (ESI). In our experience, the chances of freedom from seizures are markedly higher in nonlesional epilepsy if a comprehensive work-up is done. For example, with full use of ESI, the number of seizure-free patients with no lesion in the MRI was 80% [103]. Even lesional epilepsy benefits from complementary imaging techniques other than MRI, which can lead to 80–85% being seizure-free, since not only the site but also the extent of the epileptogenic zone needs to be determined [96]. However, precise prognosis for the outcome of surgery can be given only after in-depth evaluation. Finally, not all patients are candidates for surgical resection; in our experience, 20–30% need treatments other than surgery.
Once patients have undergone the surgical procedure, they continue to be followed up by their referring neurologist. In about 50% of the cases drug therapy needs to be maintained. However, in most cases, only one drug is sufficient. In the other 50%, drugs can be withdrawn with success. Complete withdrawal should be tried, and it appears that the time-point of withdrawal is irrelevant: it can be tried, for example, after 6 months.
Overall, per year 30–40 patients are operated on in Switzerland. However, a conservative estimate (incidence of epilepsy of 50/100'000, 10% surgical candidates) suggests that there should be 400 patients operated per year in Switzerland. This mismatch is observed in most European and North American countries; the reasons are discussed above. In fact, the average epilepsy duration (in adults) on admission for evaluation is in most studies, as at our centre, around 20 years. Such a long duration might compromise surgical success. Shorter epilepsy duration is significantly related to better surgical outcome [104, 105].
Epilepsy surgery is an effective treatment option and the only one able to cure epilepsy. Surgery needs to be considered early in the work-up and not as “last resort” in order to obtain optimal results.
Neuromodulation is currently gaining importance for many diseases; the most established indication is the treatment of Parkinson’s disease. Regarding epilepsy, two neuromodulatory approaches are currently approved: vagal nerve stimulation and deep brain stimulation (DBS) of the anterior thalamic nuclei (ANT). Other techniques are still being researched and may represent possible future neuromodulatory modalities (transcranial magnetic stimulation, transcranial direct current stimulation, etc.). However, all approaches have in common that <5–10% of implanted patients become seizure-free [106]. Thus, neuromodulation should be considered as a palliative treatment and offered only after the surgical option is ruled out.
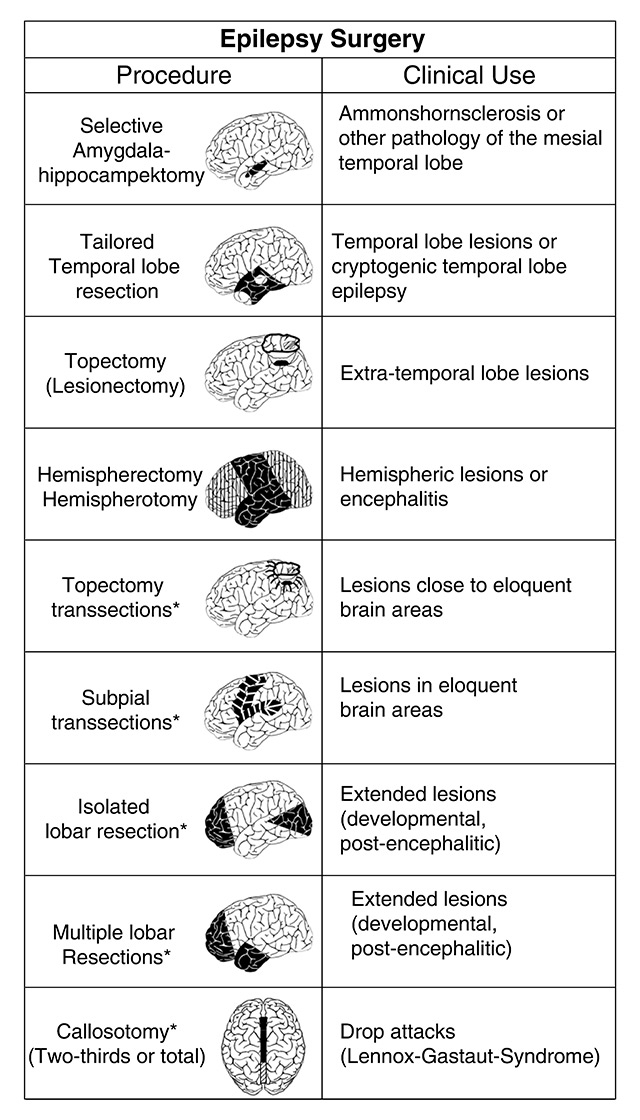
Figure 4
Current surgical approaches (reprinted from: Elger CE, Schmidt D. Modern management of epilepsy: a practical approach. Epilepsy Behav. 2008;12:501–39, with permission from Elsevier).
The most established among the neuromodulatory techniques is vagal nerve stimulation (VNS). It was approved by the FDA in 1997 for medically refractory epilepsy, and in 2005 also for treatment-resistant depression. The exact mechanisms on how VNS modulates seizures and mood are still not understood, and there are no indicators as to which patients are most likely to benefit.
A spiral-shaped electrode is implanted with its end around the vagus nerve in the left jugular cervical region (the right vagus innervates the sinoatrial node) and a pacer device is placed subcutaneously in the left clavicular or lateral thoracic region, similar to a cardiac pacemaker. Stimulation parameters are relatively well established but intensity needs to be progressively adjusted during the following weeks. The most frequently used protocol is the regular stimulation of 30 sec every 5 min. Patients have the possibility to apply additional stimulations with a hand-held magnet, if they feel a typical aura or an imminent seizure.
VNS is safe and relatively well tolerated; the most frequent side-effects are a cough during stimulation or brief modification of the voice during the short period of stimulation.
A recent review on 74 studies of a total of 3321 patients, including three of class I and two of class II quality, showed that seizure frequency was reduced on average by 45%. Only 4.6% of the patients became seizure free and another 7.6% had a major seizure decrease in the order of >90%. Most patients fall in the category “no change” (49.4%). The remainder experienced some decrease between 50–90% [107].
Open-label studies suggest that these benefits are increased with time and stimulation parameter adjustment [107–110], although not all studies report significant effects. It appears that generalised seizures respond better to VNS than partial seizures [107]. A new device, taking into account abrupt onset tachycardia as an indicator of a seizure, may be associated with greater effectiveness. We will see if this translates into higher responder rates.
Serious adverse events are very rare (infections, aspiration, cardiac arrhythmias); however, patients with VNS can undergo MRI only in very limited conditions, as outlined by the manufacturer. Also the use of surgical electrocautery in the thoraco-cervical region is only possible with special precautionary measures.
Deep brain stimulation (DBS) consists of the stereotactic implantation of an electrode into the deep structures of the brain, with a wire to an externally placed pacer similar to VNS. Since early pioneer studies [111], a number of studies have targeted many brain sites such as the cerebellum [112–114], the nonspecific activation system in the centromedial thalamus [115–118], the hippocampi [119–123], eloquent motor cortex [124–125], or caudate nucleus [127], the subthalamic nucleus [128, 129]. DBS comprises two principal types, the stimulation of a particular site in an automatic fashion and so-called “responsive neurostimulation” of the individual seizure foci (RNS) [126, 130]. Regarding the latter, the electrode which applies the stimulation is also capable of recording and analyzing the EEG and stimulating in a more targeted fashion. RNS, after a randomised double-blinded trial, was approved in the USA in 2013, but is not yet available in Europe. DBS of the anterior thalamus (DBS-ANT) was approved after the SANTE trial in 2010 [131].
The anterior thalamus represents a relay structure of the Papez circuit, which connects the structures of the limbic system such as hippocampus, parahippocampal gyrus and entorhinal cortex with the ipsilateral mammillary body, cingulate cortex and cingulum bundle, and is therefore an entry gate into the epileptogenic limbic circuit. Several favourable pilot studies resulted in the multicentre randomised controlled trial of bilateral stimulation of the ANT for epilepsy (SANTE) in 110 adult patients with focal seizures [131]. Seizure frequency dropped in the first month by 20% for all participants and thereafter significantly by 40% for the active group compared to only 14% in the control group. As in VNS, the effect of stimulation increased over time with a maximum typically seen 1–2 years after implantation, and up to 50-70% seizure reduction after several years [132]. Ten percent of the patients (6/59) were seizure-free for more than 2 years and 16% were without seizures for at least 6 months. Interestingly, better effects were obtained in patients with temporal lobe seizures, most likely due to the fact the ANT is part of the Papez circuit.
The main complications originate from the implantation procedure itself. There were also triggered seizures, electrode misplacement and mistargeting causing undesired effects such as stimulation-induced paraesthesias, and memory and mood decrease, but this was usually reversible [131, 132].
With so many new antiepileptic drugs we now have a much larger armamentarium to control seizures. Classical and new antiepileptic drugs differ in their profile of side effects. All are equally effective but some of the new AEDs result in weight loss, pain control or decreased anxiety, which may be welcome properties. Newly developed drugs are generally more expensive than established drugs, and this is also true for AEDs. However, fewer side-effects, fewer pharmacological interactions, long half-life / single daily dose regimen and an easier switch between oral and intravenous formulations, if available, may argue in favour using new AEDs.
The various drugs differ in their mechanisms of action, which should promote research towards a better understanding of the underlying cellular and molecular mechanisms. We still need to learn more about “good” polytherapy, i.e., which drug combinations are most efficient. If optimal combinations are known, lengthy trial-and-error treatments with low tolerability are avoided.
However, in patients for whom the first two AEDs do not control seizures, chances that the third or fourth drug will provide complete seizure control are weak. Already at this point, in-depth evaluation should be requested which may or may not lead to epilepsy surgery. It is not rare that recurrent fits are of nonepileptic origin and/or related to poor compliance, which may be difficult to identify in an outpatient.
The most important aspect of epilepsy care is that we should never be content with less than complete seizure freedom. Admittedly this may not be always possible, in particular in patients with neurological deficits or learning disabilities. Certainly, there should always be a cost-benefit analysis, but there is no doubt that a seizure free patient provides the best cost-benefit ratio both for direct medical costs as well as indirect costs for the society (unemployment etc.).
1 Beleza P. Acute symptomatic seizures: a clinically oriented review. Neurologist. 2012;18:109–19.
2 Delanty N, Vaughan CJ, French JA. Medical causes of seizures. Lancet. 1998;352:383–90.
3 Fisher RS, van Emde Boas W, Blume W, et al. Epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia. 2005;46:470–2.
4 Fisher RS, Acevedo C, Arzimanoglou A, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. 2014;55:475–82.
5 https://www.aan.com/Guidelines/home/GetGuidelineContent/688.
6 Krumholz A, Wiebe S, Gronseth GS, et al. Evidence-based guideline: Management of an unprovoked first seizure in adults: Report of the Guideline Development Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2015;84:1705–13.
7 Cole AJ, Cascino GD. First seizure management: I can see clearly now? Neurol Clin Pract. 2015;5:278–80.
8 Brodie MJ, Barry SJ, Bamagous GA, et al. Patterns of treatment response in newly diagnosed epilepsy. Neurology. 2012;78:1548–54.
9 Marson A, Jacoby A, Johnson A, et al. Immediate versus deferred antiepileptic drug treatment for early epilepsy and single seizures: a randomised controlled trial. Lancet. 2005;365:2007–13.
10 Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med. 2000;342:314–9.
11 Jacoby A, Lane S, Marson A, et al. Relationship of clinical and quality of life trajectories following the onset of seizures: findings from the UK MESS Study. Epilepsia. 2011;52:965–74.
12 McCormack M, Alfirevic A, Bourgeois S, et al. HLA-A*3101 and carbamazepine-induced hypersensitivity reactions in Europeans. N Engl J Med. 2011;364:1134–43.
13 Ozeki T, Mushiroda T, Yowang A, et al. Genome-wide association study identifies HLA-A*3101 allele as a genetic risk factor for carbamazepine-induced cutaneous adverse drug reactions in Japanese population. Hum Mol Genet. 2011;20:1034–41.
14 Vajda FJ, O'Brien TJ, Lander CM, et al. The teratogenicity of the newer antiepileptic drugs - an update. Acta Neurol Scand. 2014;130:234–8.
15 Campbell E, Kennedy F, Russell A, et al. Malformation risks of antiepileptic drug monotherapies in pregnancy: updated results from the UK and Ireland Epilepsy and Pregnancy Registers. J Neurol Neurosurg Psychiatry. 2014;85:1029–34.
16 https://www.gov.uk/drug-safety-update/medicines-related-to-valproate-risk-of-abnormal-pregnancy-outcomes.
17 Errington AC, Coyne L, Stohr T, et al. Seeking a mechanism of action for the novel anticonvulsant lacosamide. Neuropharmacology. 2006;50:1016–29.
18 Sutter R, Marsch S, Ruegg S. Safety and efficacy of intravenous lacosamide for adjunctive treatment of refractory status epilepticus: a comparative cohort study. CNS Drugs. 2013;27:–9.
19 Ben-Menachem E, Biton V, Jatuzis D, et al. Efficacy and safety of oral lacosamide as adjunctive therapy in adults with partial-onset seizures. Epilepsia. 2007;48:1308–17.
20 Halasz P, Kalviainen R, Mazurkiewicz-Beldzinska M, et al. Adjunctive lacosamide for partial-onset seizures: Efficacy and safety results from a randomized controlled trial. Epilepsia. 2009;50:443–53.
21 Chung S, Sperling MR, Biton V, et al. Lacosamide as adjunctive therapy for partial-onset seizures: a randomized controlled trial. Epilepsia. 2010;51:958–67.
22 Doty P, Hebert D, Mathy FX, et al. Development of lacosamide for the treatment of partial-onset seizures. Ann N Y Acad Sci. 2013;1291:56–68.
23 Cross SA, Curran MP. Lacosamide: in partial-onset seizures. Drugs. 2009;69:449–59.
24 Rudd GD, Haverkamp W, Mason JW, et al. Lacosamide cardiac safety: clinical trials in patients with partial-onset seizures. Acta Neurol Scand. 2015;132:355–63.
25 Vogl C, Mochida S, Wolff C, et al. The synaptic vesicle glycoprotein 2A ligand levetiracetam inhibits presynaptic Ca2+ channels through an intracellular pathway. Mol Pharmacol. 2012;82:199–208.
26 Lynch BA, Lambeng N, Nocka K, et al. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc Natl Acad Sci U S A. 2004;101:9861–6.
27 Ruegg S, Naegelin Y, Hardmeier M, et al. Intravenous levetiracetam: treatment experience with the first 50 critically ill patients. Epilepsy Behav. 2008;12:477–80.
28 Jaques L, Rossetti AO. Newer antiepileptic drugs in the treatment of status epilepticus: impact on prognosis. Epilepsy Behav. 2012;24:70–3.
29 Patsalos PN. The pharmacokinetic characteristics of levetiracetam. Methods Find Exp Clin Pharmacol. 2003;25:123–9.
30 Contin M, Albani F, Riva R, et al. Levetiracetam therapeutic monitoring in patients with epilepsy: effect of concomitant antiepileptic drugs. Ther Drug Monit. 2004;26:375–9.
31 Ragueneau-Majlessi I, Levy RH, Meyerhoff C. Lack of effect of repeated administration of levetiracetam on the pharmacodynamic and pharmacokinetic profiles of warfarin. Epilepsy Res. 2001;47:55–63.
32 Weintraub D, Buchsbaum R, Resor SR, Jr., et al. Psychiatric and behavioral side effects of the newer antiepileptic drugs in adults with epilepsy. Epilepsy Behav. 2007;10:105–10.
33 Dinkelacker V, Dietl T, Widman G, et al. Aggressive behavior of epilepsy patients in the course of levetiracetam add-on therapy: report of 33 mild to severe cases. Epilepsy Behav. 2003;4:537–47.
34 Szucs A, Clemens Z, Jakus R, et al. The risk of paradoxical levetiracetam effect is increased in mentally retarded patients. Epilepsia. 2008;49:1174–9.
35 Schachter SC, Vazquez B, Fisher RS, et al. Oxcarbazepine: double-blind, randomized, placebo-control, monotherapy trial for partial seizures. Neurology. 1999;52:732–7.
36 Sachdeo R, Beydoun A, Schachter S, et al. Oxcarbazepine (Trileptal) as monotherapy in patients with partial seizures. Neurology. 2001;57:864–71.
37 Beydoun A, Sachdeo RC, Rosenfeld WE, et al. Oxcarbazepine monotherapy for partial-onset seizures: a multicenter, double-blind, clinical trial. Neurology. 2000;54:2245–51.
38 Schmidt D, Sachdeo R. Oxcarbazepine for Treatment of Partial Epilepsy: A Review and Recommendations for Clinical Use. Epilepsy Behav. 2000;1:396–405.
39 Beydoun A, Sachdeo RC, Kutluay E, et al. Sustained efficacy and long-term safety of oxcarbazepine: one-year open-label extension of a study in refractory partial epilepsy. Epilepsia. 2003;44:1160–5.
40 Glauser TA, Nigro M, Sachdeo R, et al. Adjunctive therapy with oxcarbazepine in children with partial seizures. The Oxcarbazepine Pediatric Study Group. Neurology. 2000;54:2237–44.
41 Coppola G. Treatment of partial seizures in childhood : an overview. CNS Drugs. 2004;18:133–56.
42 Christe W, Kramer G, Vigonius U, et al. A double-blind controlled clinical trial: oxcarbazepine versus sodium valproate in adults with newly diagnosed epilepsy. Epilepsy Res. 1997;26:451–60.
43 Bill PA, Vigonius U, Pohlmann H, et al. A double-blind controlled clinical trial of oxcarbazepine versus phenytoin in adults with previously untreated epilepsy. Epilepsy Res. 1997;27:195–204.
44 Albani F, Grassi B, Ferrara R, et al. Immediate (overnight) switching from carbamazepine to oxcarbazepine monotherapy is equivalent to a progressive switch. Seizure. 2004;13:254–63.
45 Gonzalez-Esquivel DF, Ortega-Gavilan M, Alcantara-Lopez G, et al. Plasma level monitoring of oxcarbazepine in epileptic patients. Arch Med Res. 2000;31:202–5.
46 Hwang H, Kim KJ. New antiepileptic drugs in pediatric epilepsy. Brain Dev. 2008;30:549–55.
47 Gelisse P, Genton P, Kuate C, et al. Worsening of seizures by oxcarbazepine in juvenile idiopathic generalized epilepsies. Epilepsia. 2004;45:1282–6.
48 Ferrendelli JA. Concerns with antiepileptic drug initiation: safety, tolerability, and efficacy. Epilepsia. 2001;42(Suppl 4):28–30.
49 Perucca E. Clinical pharmacology and therapeutic use of the new antiepileptic drugs. Fundam Clin Pharmacol. 2001;15:405–17.
50 Glauser TA. Oxcarbazepine in the treatment of epilepsy. Pharmacotherapy. 2001;21:904–19.
51 Krauss GL, Perucca E, Ben-Menachem E, et al. Perampanel, a selective, noncompetitive alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor antagonist, as adjunctive therapy for refractory partial-onset seizures: interim results from phase III, extension study 307. Epilepsia. 2013;54:126–34.
52 French JA, Krauss GL, Steinhoff BJ, et al. Evaluation of adjunctive perampanel in patients with refractory partial-onset seizures: results of randomized global phase III study 305. Epilepsia. 2013;54:117–25.
53 French JA, Krauss GL, Biton V, et al. Adjunctive perampanel for refractory partial-onset seizures: randomized phase III study 304. Neurology. 2012;79:589–96.
54 Krauss GL, Serratosa JM, Villanueva V, et al. Randomized phase III study 306: adjunctive perampanel for refractory partial-onset seizures. Neurology. 2012;78:1408–15.
55 French JA, Krauss GL, Wechsler RT, et al. Perampanel for tonic-clonic seizures in idiopathic generalized epilepsy A randomized trial. Neurology. 2015;85:950–7.
56 Gidal BE, Laurenza A, Hussein Z, et al. Perampanel efficacy and tolerability with enzyme-inducing AEDs in patients with epilepsy. Neurology. 2015;84:1972–80.
57 Novy J, Rothuizen LE, Buclin T, et al. Perampanel: a significant liver enzyme inducer in some patients? Eur Neurol. 2014;72:213–6.
58 Krauss GL, Bar M, Biton V, et al. Tolerability and safety of perampanel: two randomized dose-escalation studies. Acta Neurol Scand. 2012;125:8–15.
59 Krauss GL, Perucca E, Ben-Menachem E, et al. Long-term safety of perampanel and seizure outcomes in refractory partial-onset seizures and secondarily generalized seizures: results from phase III extension study 307. Epilepsia. 2014;55:1058–68.
60 Schulze-Bonhage A, Hintz M. Perampanel in the management of partial-onset seizures: a review of safety, efficacy, and patient acceptability. Patient Prefer Adherence. 2015;9:1143–51.
61 Taylor CP, Angelotti T, Fauman E. Pharmacology and mechanism of action of pregabalin: the calcium channel alpha2-delta (alpha2-delta) subunit as a target for antiepileptic drug discovery. Epilepsy Res. 2007;73:137–50.
62 Dooley DJ, Taylor CP, Donevan S, et al. Ca2+ channel alpha2delta ligands: novel modulators of neurotransmission. Trends Pharmacol Sci. 2007;28:75–82.
63 Brodie MJ, Wilson EA, Wesche DL, et al. Pregabalin drug interaction studies: lack of effect on the pharmacokinetics of carbamazepine, phenytoin, lamotrigine, and valproate in patients with partial epilepsy. Epilepsia. 2005;46:1407–13.
64 Deeks ED, Scott LJ. Rufinamide. CNS Drugs. 2006;20:751–60; discussion 61.
65 Glauser T, Kluger G, Sachdeo R, et al. Rufinamide for generalized seizures associated with Lennox-Gastaut syndrome. Neurology. 2008;70:1950–8.
66 Brodie MJ, Rosenfeld WE, Vazquez B, et al. Rufinamide for the adjunctive treatment of partial seizures in adults and adolescents: a randomized placebo-controlled trial. Epilepsia. 2009;50:1899–909.
67 Aldenkamp AP, Alpherts WCJ. The Effect of the New Antiepileptic Drug Rufinamide on Cognitive Functions. Epilepsia. 2006;47:1153–9.
68 Kawai M, Hiramatsu M, Endo A, et al. Effect of zonisamide on release of aspartic acid and gamma-aminobutyric acid from hippocampal slices of E1 mice. Neurosciences. 1994;20:115–9.
69 Rock DM, Macdonald RL, Taylor CP. Blockade of sustained repetitive action potentials in cultured spinal cord neurons by zonisamide (AD 810, CI 912), a novel anticonvulsant. Epilepsy Res. 1989;3:138–43.
70 Okada M, Kawata Y, Mizuno K, et al. Interaction between Ca2+, K+, carbamazepine and zonisamide on hippocampal extracellular glutamate monitored with a microdialysis electrode. Br J Pharmacol. 1998;124:1277–85.
71 Suzuki S, Kawakami K, Nishimura S, et al. Zonisamide blocks T-type calcium channel in cultured neurons of rat cerebral cortex. Epilepsy Res. 1992;12:21–7.
72 Thone J, Leniger T, Splettstosser F, et al. Antiepileptic activity of zonisamide on hippocampal CA3 neurons does not depend on carbonic anhydrase inhibition. Epilepsy Res. 2008;79:105–11.
73 Masuda Y, Karasawa T. Inhibitory effect of zonisamide on human carbonic anhydrase in vitro. Arzneimittelforschung. 1993;43:416–8.
74 Masuda Y, Noguchi H, Karasawa T. Evidence against a significant implication of carbonic anhydrase inhibitory activity of zonisamide in its anticonvulsive effects. Arzneimittelforschung. 1994;44:267–9.
75 Leppik IE, Willmore LJ, Homan RW, et al. Efficacy and safety of zonisamide: results of a multicenter study. Epilepsy Research. 1993;14:165–73.
76 Ohtahara S. Zonisamide in the management of epilepsy – Japanese experience. Epilepsy Res. 2006;68(Suppl 2):S25–33.
77 Sills GJ, Brodie MJ. Pharmacokinetics and Drug Interactions with Zonisamide. Epilepsia. 2007;48:435–41.
78 Faught E, Ayala R, Montouris GG, et al. Randomized controlled trial of zonisamide for the treatment of refractory partial-onset seizures. Neurology. 2001;57:1774–9.
79 Kondo T, Kaneko S, Amano Y, et al. Preliminary Report on Teratogenic Effects of Zonisamide in the Offspring of Treated Women with Epilepsy. Epilepsia. 1996;37:1242–4.
80 Soares-da-Silva P, Pires N, Bonifacio MJ, et al. Eslicarbazepine acetate for the treatment of focal epilepsy: an update on its proposed mechanisms of action. Pharmacol Res Perspect. 2015;3:e00124.
81 Elger C, Bialer M, Cramer JA, et al. Eslicarbazepine acetate: a double-blind, add-on, placebo-controlled exploratory trial in adult patients with partial-onset seizures. Epilepsia. 2007;48:497–504.
82 Almeida L, Potgieter JH, Maia J, et al. Pharmacokinetics of eslicarbazepine acetate in patients with moderate hepatic impairment. Eur J Clin Pharmacol. 2007;64:267–73.
83 Sperling MR, Abou-Khalil B, Harvey J, et al. Eslicarbazepine acetate as adjunctive therapy in patients with uncontrolled partial-onset seizures: Results of a phase III, double-blind, randomized, placebo-controlled trial. Epilepsia. 2015;56:244–53.
84 Sperling MR, Harvey J, Grinnell T, et al. Efficacy and safety of conversion to monotherapy with eslicarbazepine acetate in adults with uncontrolled partial-onset seizures: a randomized historical-control phase III study based in North America. Epilepsia. 2015;56:546–55.
85 Maia J, Almeida L, Falcão A, et al. Effect of renal impairment on the pharmacokinetics of eslicarbazepine acetate. Int J Clin Pharmacol Ther. 2008;46:119–30.
86 Margolis JM, Chu BC, Wang ZJ, et al. Effectiveness of antiepileptic drug combination therapy for partial-onset seizures based on mechanisms of action. JAMA Neurol. 2014;71:985–93.
87 Deckers CL, Hekster YA, Keyser A, et al. Monotherapy versus polytherapy for epilepsy: a multicenter double-blind randomized study. Epilepsia. 2001;42:1387–94.
88 Brodie MJ, Yuen AW. Lamotrigine substitution study: evidence for synergism with sodium valproate? 105 Study Group. Epilepsy Res. 1997;26:423–32.
89 Stephen LJ, Forsyth M, Kelly K, et al. Antiepileptic drug combinations – have newer agents altered clinical outcomes? Epilepsy Res. 2012;98:194–8.
90 Lamberts RJ, Thijs RD, Laffan A, et al. Sudden unexpected death in epilepsy: people with nocturnal seizures may be at highest risk. Epilepsia. 2012;53:253–7.
91 Tomson T, Walczak T, Sillanpaa M, et al. Sudden unexpected death in epilepsy: a review of incidence and risk factors. Epilepsia 2005;46(Suppl 11):54–61.
92 Sperling MR, Feldman H, Kinman J, et al. Seizure control and mortality in epilepsy. Ann Neurol. 1999;46:45–50.
93 Hennessy MJ, Langan Y, Elwes RD, et al. A study of mortality after temporal lobe epilepsy surgery. Neurology. 1999;53:1276–83.
94 Hakimi AS, Spanaki MV, Schuh LA, et al. A survey of neurologists’ views on epilepsy surgery and medically refractory epilepsy. Epilepsy Behav. 2008;13:96–101.
95 Uijl SG, Leijten FS, Moons KG, et al. Epilepsy surgery can help many more adult patients with intractable seizures. Epilepsy Res. 2012;101:210–6.
96 Lascano AM, Perneger T, Vulliemoz S, et al. Yield of MRI, high-density electric source imaging (HD-ESI), SPECT and PET in epilepsy surgery candidates. Clin Neurophysiol. 2015.
97 Kurian M, Spinelli L, Delavelle J, et al. Multimodality imaging for focus localization in pediatric pharmacoresistant epilepsy. Epileptic Disord. 2007;9:20–31.
98 Woermann FG, Vollmar C. Clinical MRI in children and adults with focal epilepsy: a critical review. Epilepsy Behav. 2009;15:40–9.
99 Martin P, Bender B, Focke NK. Post-processing of structural MRI for individualized diagnostics. Quant Imaging Med Surg. 2015;5:188–203.
100 Wilke M, Kassubek J, Ziyeh S, et al. Automated detection of gray matter malformations using optimized voxel-based morphometry: a systematic approach. Neuroimage. 2003;20:330–43.
101 Elger CE, Schmidt D. Modern management of epilepsy: a practical approach. Epilepsy Behav. 2008;12:501–39.
102 Tellez-Zenteno JF, Hernandez Ronquillo L, Moien-Afshari F, et al. Surgical outcomes in lesional and non-lesional epilepsy: a systematic review and meta-analysis. Epilepsy Res. 2010;89:310–8.
103 Brodbeck V, Spinelli L, Lascano AM, et al. Electrical source imaging for presurgical focus localization in epilepsy patients with normal MRI. Epilepsia. 2010;51:583–91.
104 Engel J, Jr., McDermott MP, Wiebe S, et al. Early surgical therapy for drug-resistant temporal lobe epilepsy: a randomized trial. JAMA. 2012;307:922–30.
105 Simasathien T, Vadera S, Najm I, et al. Improved outcomes with earlier surgery for intractable frontal lobe epilepsy. Ann Neurol. 2013;73:646–54.
106 Fisher RS, Velasco AL. Electrical brain stimulation for epilepsy. Nat Rev Neurol. 2014;10:261–70.
107 Englot DJ, Chang EF, Auguste KI. Vagus nerve stimulation for epilepsy: a meta-analysis of efficacy and predictors of response. J Neurosurg. 2011;115:1248–55.
108 Orosz I, McCormick D, Zamponi N, et al. Vagus nerve stimulation for drug-resistant epilepsy: a European long-term study up to 24 months in 347 children. Epilepsia. 2014;55:1576–84.
109 Uthman BM, Reichl AM, Dean JC, et al. Effectiveness of vagus nerve stimulation in epilepsy patients: a 12-year observation. Neurology. 2004;63:1124–6.
110 Wheless JW, Baumgartner J. Vagus nerve stimulation therapy. Drugs Today (Barc) 2004;40:501–15.
111 Heath RG. Electrical Self-Stimulation of the Brain in Man. Am J Psychiatry. 1963;120:571–7.
112 Cooper IS, Amin I, Gilman S, et al. The Effect of Chronic Stimulation of Cerebellar Cortex on Epilepsy in Man. The Cerebellum, Epilepsy, and Behavior: Springer Science + Business Media; 1974. p. 119–71.
113 Van Buren JM, Wood JH, Oakley J, et al. Preliminary evaluation of cerebellar stimulation by double-blind stimulation and biological criteria in the treatment of epilepsy. J Neurosurg. 1978;48:407–16.
114 Velasco F, Carrillo-Ruiz JD, Brito F, et al. Double-blind, randomized controlled pilot study of bilateral cerebellar stimulation for treatment of intractable motor seizures. Epilepsia. 2005;46:1071–81.
115 Velasco F, Velasco M, Ogarrio C, et al. Electrical stimulation of the centromedian thalamic nucleus in the treatment of convulsive seizures: a preliminary report. Epilepsia. 1987;28:421–30.
116 Velasco F, Velasco M, Velasco AL, et al. Electrical stimulation of the centromedian thalamic nucleus in control of seizures: long-term studies. Epilepsia. 1995;36:63–71.
117 Valentin A, Nguyen HQ, Skupenova AM, et al. Centromedian thalamic nuclei deep brain stimulation in refractory status epilepticus. Brain Stimul. 2012;5:594–8.
118 Valentin A, Garcia Navarrete E, Chelvarajah R, et al. Deep brain stimulation of the centromedian thalamic nucleus for the treatment of generalized and frontal epilepsies. Epilepsia. 2013;54:1823–33.
119 Sramka M, Fritz G, Galanda M, et al. Some Observations in Treatment Stimulation of Epilepsy. Stereotactic Treatment of Epilepsy: Springer Science + Business Media; 1976. p. 257–62.
120 Velasco M, Velasco F, Velasco AL, et al. Subacute electrical stimulation of the hippocampus blocks intractable temporal lobe seizures and paroxysmal EEG activities. Epilepsia. 2000;41:158–69.
121 Boex C, Seeck M, Vulliemoz S, et al. Chronic deep brain stimulation in mesial temporal lobe epilepsy. Seizure. 2011;20:485–90.
122 Cukiert A, Cukiert CM, Burattini JA, et al. Seizure outcome after hippocampal deep brain stimulation in a prospective cohort of patients with refractory temporal lobe epilepsy. Seizure. 2014;23:6–9.
123 Vonck K, Boon P, Achten E, et al. Long-term amygdalohippocampal stimulation for refractory temporal lobe epilepsy. Ann Neurol. 2002;52:556–65.
124 Elisevich K, Jenrow K, Schuh L, et al. Long-term electrical stimulation-induced inhibition of partial epilepsy. Case report. J Neurosurg. 2006;105:894–7.
125 Velasco AL, Velasco F, Velasco M, et al. Neuromodulation of epileptic foci in patients with non-lesional refractory motor epilepsy. Int J Neural Syst. 2009;19:139–47.
126 Sun FT, Morrell MJ, Wharen RE, Jr. Responsive cortical stimulation for the treatment of epilepsy. Neurotherapeutics. 2008;5:68–74.
127 Chkhenkeli SA, Sramka M, Lortkipanidze GS, et al. Electrophysiological effects and clinical results of direct brain stimulation for intractable epilepsy. Clin Neurol Neurosurg. 2004;106:318–29.
128 Capecci M, Ricciuti RA, Ortenzi A, et al. Chronic bilateral subthalamic stimulation after anterior callosotomy in drug-resistant epilepsy: long-term clinical and functional outcome of two cases. Epilepsy Res. 2012;98:135–9.
129 Handforth A, DeSalles AA, Krahl SE. Deep brain stimulation of the subthalamic nucleus as adjunct treatment for refractory epilepsy. Epilepsia. 2006;47:1239–41.
130 Heck CN, King-Stephens D, Massey AD, et al. Two-year seizure reduction in adults with medically intractable partial onset epilepsy treated with responsive neurostimulation: final results of the RNS System Pivotal trial. Epilepsia. 2014;55:432–41.
131 Fisher R, Salanova V, Witt T, et al. Electrical stimulation of the anterior nucleus of thalamus for treatment of refractory epilepsy. Epilepsia. 2010;51:899–908.
132 Salanova V, Witt T, Worth R, et al. Long-term efficacy and safety of thalamic stimulation for drug-resistant partial epilepsy. Neurology. 2015;84:1017–25.
Disclosure statement: M.S. received consultation fees for EISAI Co. Ltd. and for UCB S.A.