Current management of pulmonary arterial hypertension
DOI: https://doi.org/10.4414/smw.2016.14305
Patrick
Yerly, Maura
Prella, John-David
Aubert
Summary
Pulmonary arterial hypertension (PAH) is a vascular disease of unknown aetiology, characterised by an abnormal thickening of the arterial wall that is responsible for an increase in pulmonary vascular resistance. The haemodynamic consequence of PAH is an increased afterload for the right ventricle and, eventually, right heart failure. When untreated, PAH has a grim prognosis with a median survival of about 2 to 4 years from diagnosis. In the last 10 years new orally administered compounds have demonstrated clinical efficacy in controlled trials using various surrogate endpoints to survival. Although the disease remains without cure until now, the available phase III trials have allowed evidence-based recommendations for the medical management of these patients to be established. It appears, however, that none of the compounds from the three main therapeutic classes, endothelin receptor antagonists, agents acting on the nitric oxide–cyclic guanosine monophosphate pathway (including phosphodiesterase type 5 inhibitors and guanylate cyclase stimulator), and prostanoid receptor agonists are able alone to control disease progression in every patient. Therefore combination therapy with two or three drugs may be necessary in a significant number of patients in order to maintain patients in, or bring them to, a low risk profile. Several recent studies have now validated this approach for specific double or triple drug regimens. It remains, however, unclear whether an upfront combination is preferable to a sequential step-up approach based on clinical response. In addition, some specific combination therapies have failed to demonstrate superiority to single drug alone in randomised controlled trials. Besides PAH-specific treatment, the place of nonspecific pharmaceutical and nonpharmaceutical treatment has been also recently clarified.
Abbreviations
6MWD 6-minute walking distance
CCB calcium channel blockers
CTEPH chronic thromboembolic pulmonary hypertension
ERA endothelin receptor antagonist
ERS European Respiratory Society
ESC European Society of Cardiology
NO nitric oxide
NYHA New York Heart Association
OAC oral anticoagulation
PAH pulmonary arterial hypertension
PDE-5 phosphodiesterase type 5
RCT randomised controlled trial
Introduction
Since the last review on the therapy for pulmonary arterial hypertension (PAH) in this Journal [1], several new substances have been studied in randomised controlled trials (RCTs), and guidelines on the overall management of the disease have been extensively updated [2, 3]. In this review we will focus exclusively on the treatment of pulmonary arterial hypertension group 1 according to the Nice 2013 classification [4], and restrict the analysis to adult patients. Comprehensive guidelines addressing specifically the paediatric population have been published recently [5]. Results from RCTs performed in patients with PAH category 1 cannot be extrapolated to other types of pulmonary hypertension, which have been much less thoroughly studied.
Nonspecific therapy
Nonspecific supportive therapies in PAH, including anticoagulation, oxygen, diuretics and digoxin are addressed by expert opinion, owing to the lack of RCTs for most of these drugs and despite the recent advances in PAH-specific drugs.
Anticoagulation
PAH is characterised by a prothrombotic state and its pathophysiology includes in-situ thrombosis of small pulmonary arteries [6]. PAH patients often have nonspecific increased risk factors for venous thromboembolism including heart failure, long-term intravenous catheters and immobility. This, together with few old observational studies showing favourable effects of oral anticoagulation (OAC) explains why OAC became widely used in group 1 PAH [7].
However, the extent to which thrombosis plays a role in the progression of PAH is unknown and data assessing the survival effect of OAC are mostly limited to retrospective and single-centre studies. OAC needs to be balanced against the risk of bleeding, the major concern being gastrointestinal haemorrhage, which affects particularly patients with connective tissue disease PAH (CTD-PAH) with endoluminal telangiectasia and patients with porto-pulmonary hypertension.
In PAH the indication for OAC is still debated. In CTD-PAH, the two recent analyses of the European and American pulmonary hypertension registries, which were not designed to evaluate the effect of OAC on survival, concluded that OAC may not be beneficial [8, 9]. For idiopathic PAH these two studies show conflicting data with a survival benefit in the European COMPERA registry and no survival advantage in the USA REVEAL registry. In both analyses, only some of the patients were anticoagulated for the entire observation period, 55% in the COMPERA study and 25% in the REVEAL study. The difference in the target prothrombin time international normalised ratio (INR) range (2–3 in COMPERA, 1.5–2.5 in REVEAL) may also have played a role in the diverging results.
The latest European Society of Cardiology (ESC) / European Respiratory Society (ERS) guidelines consider OAC as a class IIb recommendation with a level C of evidence for PAH groups 1.1 to 1.3 [3]. The use of OAC should then be individualised. OAC may be considered in selected PAH patients with increased risk factors for venous thrombosis. Concomitant conditions with a need for OAC such as atrial fibrillation should be the main reason to treat these patients.
Digitalis and diuretics
Diuretics are beneficial in the case of volume overload. Loop diuretics are the first-line therapy. In patients with clinical right heart failure with oedema of the gastrointestinal tract mucosa, intravenous administration may be more efficient. Similarities in the effects of left and right ventricular failure on the renin-angiotensin system suggest that for patients in functional class 3 and 4, the addition of aldosterone antagonists may be beneficial [10].
The long-term effect of digitalis in PAH is unknown, except for patients with atrial arrhythmia in whom digoxin slows the ventricular rate. Its use is controversial because of its potential toxicity, which is increased in the event of renal failure, and drug interactions.
Oxygen therapy
The use of oxygen therapy has been extrapolated from data in patients with chronic obstructive pulmonary disease. In hypoxaemic PAH patients, oxygen administration reduces pulmonary venous resistance and is already recommended when PaO2 is consistently <60 mm Hg [3]. However this indication is controversial in congenital heart disease-associated PAH as a randomised controlled study in 23 adult patients showed no improvement in 6-minute walking distance, quality of life and survival with nocturnal oxygen administration [11]. However, the only RCT of oxygen therapy in PAH and inoperable chronic thromboembolic pulmonary hypertension (CTEPH) has shown that patients benefit from nocturnal oxygen therapy when they have a mean nocturnal oxygen saturation <90%, even with preserved daytime oxygenation [12].
Altitude exposure may worsen pre-existing hypoxaemia and current air travel recommendations propose in-flight oxygen administration for patients with New York Heart Association (NYHA) functional class III and IV [3]. However an uncontrolled study in 36 PAH patients suggested that functional class does not predict hypoxaemia during a hypoxic challenge testing and that its assessment does not identify who may have symptoms during aircraft travel [13]. Oxygen therapy should also be considered in patients living at altitude equal or superior to 1500 to 2000 m above sea level.
Nonpharmacological treatment: rehabilitation
Despite optimal medical treatment, most patients remain symptomatic with exercise intolerance and fatigue [14, 15]. During exercise, mean pulmonary arterial pressure increases dramatically while cardiac output does not rise as much as expected. PAH patients may present deconditioning and have impaired peripheral oxygen utilisation, which explains decreased peak oxygen consumption and early onset of anaerobic threshold.
A recent meta-analysis including 16 studies, among them 4 that were randomised, demonstrated that rehabilitation improves exercise capacity (6-minute walk distance and peak oxygen consumption [VO2]) and quality of life with only few minor adverse events [16]. Among the 434 studied patients, all but 19 were in NYHA functional class II and III, well-compensated under stable medication. More than 70% of the patients had PAH, the others had CTEPH. Exercise training protocols were all supervised; the majority were low workload (10–60 W) and aerobic with some form of resistance and respiratory training.
Since then a fifth randomised study including 86 stable patients with PAH (53%) and inoperable CTEPH (47%) in NYHA functional class II and III has showed a significant increase in peak VO2 and 6-minute walking distance along with haemodynamic improvement [17].
In the ERS/ESC guidelines, rehabilitation was downgraded from a class I to IIa recommendation with a B level of evidence because of absence of agreement regarding the methods of exercise, the intensity and duration of training and the characteristics of the supervision. In view of the latest publications, we believe that rehabilitation should be offered to group 1 and inoperable CTEPH stable patients in NYHA functional class II or III as add-on to optimal medical therapy. Highly supervised low-workload aerobic training should, however, be offered in centres with well-established experience in training pulmonary hypertension patients and in close collaboration with a pulmonary hypertension centre in order to avoid potentially deleterious side effects. Exercise should be discontinued in the case of light-headedness, chest pain, palpitations or syncope [18]. High-intensity exercise and Valsalva manoeuvres are not recommended. The usefulness of oxygen supply in patients desaturating during exercise and of increased physiotherapy in CTD-PAH has still to be demonstrated. As in other diseases, the challenge is to make severely affected patients change their life-style and continue regular exercise after the training programme.
Specific medical therapy for PAH
The current hypothesis addressing PAH pathogenesis is a model of acquired hits occurring on a favourable genetic background [19]. At some point, imbalance of the three major pathways of the endothelial function, namely the prostacyclin (PGI2), the endothelin (ET-1) and the nitric oxide (NO) pathways, has been consistently shown to be involved in the disease process, leading to a triad of constriction, remodelling and thrombosis of small pulmonary arteries [6, 20]. Presently, all drugs approved for the treatment of PAH in Switzerland target one of these three pathways, besides calcium channel blockers for reactive PAH (fig. 1). Only these drugs will be commented on in this review, including also selexipag, which will enter the review process for approval by regulatory authorities in Switzerland in 2016, and which has already been integrated in the 2015 guidelines [3].
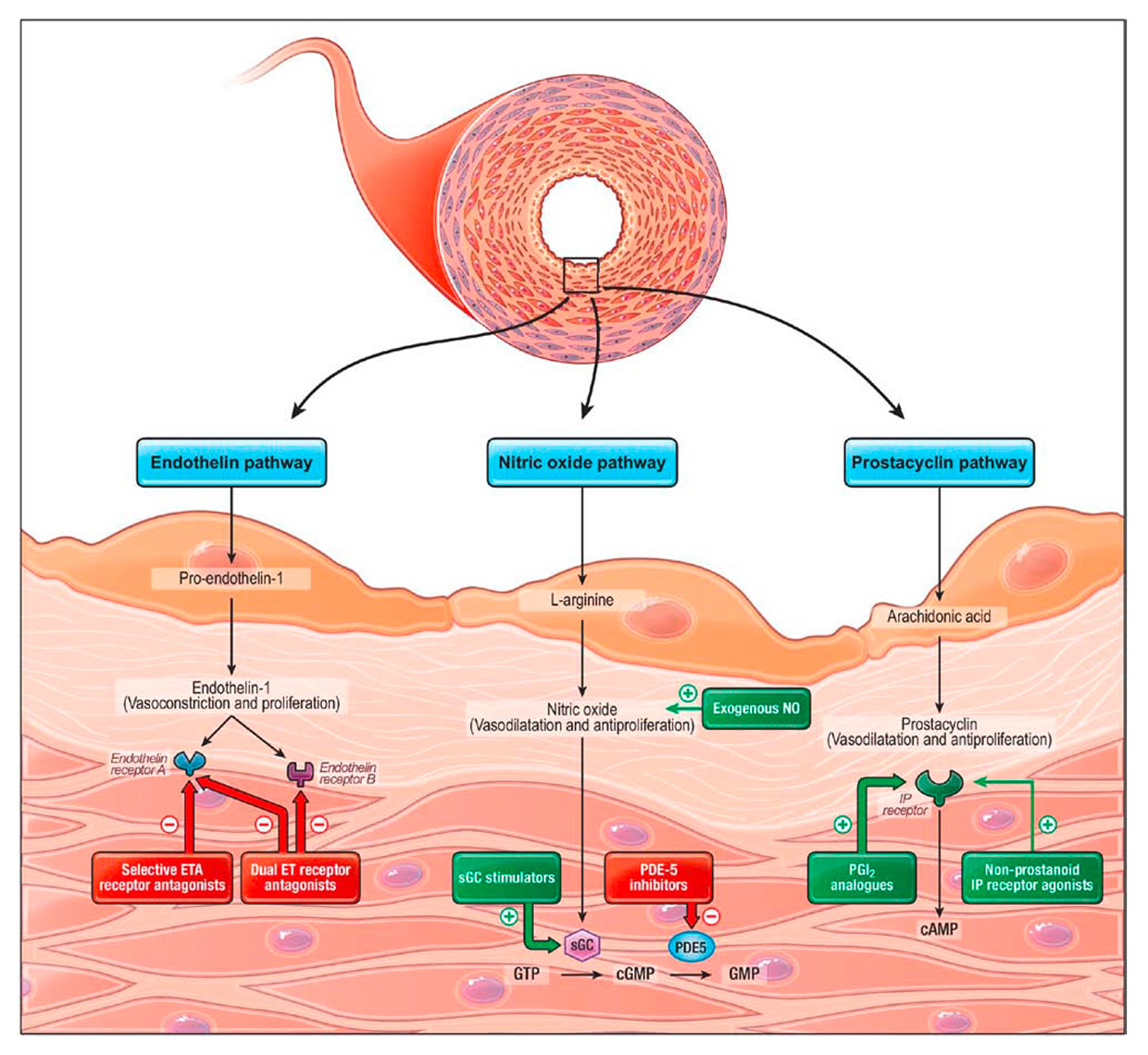
Figure 1
Established pathological pathways modulated by pharmacological substances currently used or in investigation for the treatment of pulmonary arterial hypertension. Antagonists or inhibitors are in red, activators or analogues are in green. (Reproduced with permission from: Humbert M, Lau EMT, Montani D, Jais X, Sitbon O, Simonneau G. Advances in therapeutic interventions for patients with pulmonary arterial hypertension. Circulation. 2014;130:2189–208.)
cAMP = cyclic adenosine monophosphate; cGMP = cyclic guanosine monophosphate; GTP = guanosine triphosphate; ET = endothelin; ETA = endothelin type A receptor; IP = prostaglandin I2; NO = nitric oxide; PDE-5 = phosphodiesterase type 5; sGC = soluble guanylate cyclase
ET-1 is the most potent vasoconstrictor synthesised in the human body and induces smooth muscle cell proliferation through the binding of endothelin receptors A and B (ETA/ETB) [21, 22]. In PAH, ET-1 biosynthesis is up-regulated, as demonstrated by both higher serum levels and increased ET-1 staining in the pulmonary arteries of lung specimens taken after transplantation [23]. On the side of vasodilators, NO bioavailability is reduced by numerous mechanisms in PAH, and soluble guanylate cyclase (sGC), which generates cyclic guanosine monophosphate (cGMP) in smooth muscles cells in response to NO, is partially inactivated because of high levels of oxidative stress [24]. Furthermore, phosphodiesterase type 5 (PDE-5) has been found to be upregulated in lung homogenates of patients with PAH, leading to quicker inactivation of cGMP [25]. Finally, PGI2 expression is also severely reduced in PAH [26], and to a lesser level its specific IP receptor in the smooth muscle cells of pulmonary arteries [27].
Calcium channel blockers
The invasive haemodynamic assessment of PAH group 1.1 to 1.3 should always involve vasoreactivity testing [3] with inhaled NO as the standard of care or with alternative agents such as iloprost. In the case of substantial improvement under testing, calcium-channel blocker monotherapy is warranted. It must, however, be stated that about 50% of the initial responders may lose this characteristic and face worsening PAH after 1 year, with the need to initiate another PAH-specific therapy [28].
The prostacyclin pathway
Epoprostenol was historically the first treatment to have shown efficacy in nonreactive PAH and the only one to have brought a survival benefit in a RCT, with 100% survival at 12 weeks in the epoprostenol group as compared with 80% in the conventional treatment group (p = 0.003) [29]. This drug is a freeze-dried preparation of PGI2 restricted to continuous intravenous use through a central venous catheter and a dedicated pump. In spite of the dramatic improvement in haemodynamics, functional capacity and survival, the widespread use of epoprostenol is limited by the occurrence of a rather high rate of catheter-related infections and by the risk of a possibly fatal rebound pulmonary hypertensive crisis in the case of drug interruption due to catheter thrombosis or pump malfunction [30]. This latter risk is related to the particularly short half-life (≈6 min) of epoprostenol. Consequently, epoprostenol must be initiated only in expert centres, even with the improved convenience of a new thermostable formulation avoiding twice daily line manipulation [31].
In order to increase the safety of prostanoid therapy, prostacyclin analogues with longer half-lives such as tresprostinil and iloprost were developed and tested through alternative routes of administration. Subcutaneous tresprostinil is rapidly absorbed with complete bioavailability and it offers similar pulmonary vascular resistance reduction to intravenous epoprostenol or tresprostinil itself at maximum tolerated doses [32], as well as a dose-dependent increase in exercise capacity [33]. In practice, treprostinil, like the other prostanoids, is initiated at low dose and progressively up-titrated until typical side effects like jaw pain, flushing, hypotension or diarrhoea occur. The optimal infusion rate is considered as the maximum tolerated dose. Besides general prostanoid side effects, the continuous drug infusion under the skin also invariably produces local pain and cutaneous inflammation, and possible injection site infection, with high discontinuation rates over the mid- and long-term, which again limit wide implementation of prostanoids in clinical practice [34]. Intravenous infusion is also possible with tresprostinil, but there is so far no reliable multicentre randomised controlled trial to validate this option. Finally, iloprost is the third approved prostanoid for parenteral use in Switzerland. It can be used via inhalation with specific dedicated nebulisers, but plays a limited role in PAH therapy as a result of the six to nine daily administrations and possible mitigated long-term efficacy [35, 36].
The orally available nonprostanoid agonist at the IP receptor, selexipag, has been evaluated in the multicentre randomised placebo-controlled GRIPHON trial, whose results were published recently [37]. Selexipag is a prodrug that is rapidly transformed to an active metabolite with an 8-hour half-life, enabling administration twice daily. This active metabolite displays high specificity for the IP receptor, with positive vasodilator and antiproliferative effects on the smooth muscle cells of pulmonary arteries, but also with typical side effects related to IP receptor activation in the stomach and gut (nausea, diarrhoea), as well as in systemic vessels (hypotension, headache). Selexipag needs progressive up-titration and the optimal dose is considered to be the maximum tolerated dose. Under these conditions, the GRIPHON trial showed a significant 40% reduction of a combined morbidity-mortality endpoint in the active drug arm, with benefit both in naïve patients and in those already on background therapy [37]. The study was limited to patients in functional class II or III.
The endothelin pathway
Selective blocking of the endothelin receptor A (ETA) can be obtained with ambrisentan and dual blocking of the endothelin receptors A and B (ETA/B) with bosentan or macitentan, which are all oral drugs. Ambrisentan and bosentan were licensed on the basis of 12–16 weeks randomised placebo-controlled trials demonstrating improvements in exercise capacity as measured by the 6-minute walking distance (6MWD) (placebo-controlled increase by 31–76 m) [38–47]. Although the 6MWD can be considered as a predictor of clinical stability and mortality in PAH [48], its relevance to long-term clinical outcome is questionable. In a recent meta-analysis of 3112 patients from 22 clinical trials, 6MWD changes measured under treatment were not predictive of clinical outcome events like all-cause mortality, hospitalisation for PAH, lung transplantation and initiation of parenteral therapy [49]. Furthermore, improvement in 6MWD may not be apparent in less severe patients with normal or near-normal baseline walking distance because of a ceiling effect [50]. Nevertheless, long-term open label studies at 24 to 51 months confirmed the benefit of both ambrisentan and bosentan [39, 44], including for NYHA stage II (mildly symptomatic) patients [46].
Increased incidence of peripheral oedema, elevation of liver aminotransferases, mild anaemia and headaches are the major side effects of endothelin receptor antagonists (ERAs). With bosentan, the annual rate of aminotransferase elevation above 3 times the upper limit of normal is 10.1%, and discontinuation of therapy was required in 3.2% of patients in a 30-month post-marketing study on 4994 patients [51]. Aminotransferase elevation is dose-dependent, which warrants starting with low doses, and fully reversible after drug dose reduction or discontinuation. In spite of this hepatoxicity, first-line bosentan therapy can be safely administered in porto-pulmonary hypertension with Child-Pugh A and B cirrhosis, but less is known about Child-Pugh C cirrhosis [52]. Bosentan is cleared by CYP3A4 and CYP2C9 and is also an inducer of these two enzymes. Serum concentrations of concomitant drugs metabolised by CYP3A4 and CYP2C9 may thus be decreased, but most importantly, administration of strong inhibitors of these enzymes would enhance bosentan toxicity. With ambrisentan, the risk of liver injury is lower [38]; therefore it can safely replace bosentan in cases of liver toxicity [53]. Drug-drug interactions are also of less importance with ambrisentan, which is mainly cleared by glucuronide conjugation, but peripheral oedema is more common.
Macitentan is the last member of the ERA family developed for PAH and, through a modification of its chemical structure, is able to achieve enhanced tissue penetration, long-lasting pharmacologically active metabolites, an increased receptor affinity and more sustained receptor binding [54]. These properties allow a once-a-day regimen with lower doses and optimised safety profile, and with no effect on liver enzymes in phase II trials [55], but as ERAs may still induce hepatotoxicity after months or years, longer trials are needed to demonstrate truly the lack of delayed liver-related side effects [56].
With the phase III SERAPHIN trial, macitentan is the first PAH-specific therapy to have been studied in a long-term event-driven trial, which is a true paradigm shift as compared with the earlier short-term trials based on the surrogate 6MWD endpoint [57]. The SERAPHIN primary endpoint was designed to reflect the true progression of PAH and was defined as the time to a composite of mortality, atrial septostomy, lung transplantation, initiation of prostanoid therapy or PAH worsening. PAH worsening was further defined as the simultaneous occurrence of three criteria, namely persistent (>15%) 6MWD decrease, worsening PAH symptoms (increase in functional class or symptoms of right heart failure) and need for additional PAH therapy including intravenous diuretics. Actually, after a mean follow-up of about 2 years, macitentan reduced this morbidity–mortality endpoint by 45% (relative risk) and by 12.9% (absolute risk) (p <0.001) in the 10-mg group. The effect was positive in both naïve patients and patients on background therapy (mostly PDE-5 inhibitors). Of note, all-cause mortality occurring as the first clinical event was not significantly different with macitentan as compared to placebo (hazard ratio 0.64; 97.5% confidence interval 0.29–1.42) and it must also be emphasised that there were almost no patients in functional class IV in this study. Furthermore, although the robustness of the SERAPHIN trial may drive physicians to choose macitentan in naïve patients, there are currently no direct comparison studies to support the switch from an “old” ERA to the new compound, especially in stable patients. Regarding safety, macitentan was overall well tolerated, but anaemia occurred in 13.2% of treated patients, with haemoglobin level falling <8 g/l in 4.3% of subjects [57].
The NO-cGMP Pathway
As incorpus cavernosum, the type 5 isoform is the most abundant phophodiesterase in pulmonary arteries, and two orally active PDE-5 inhibitors licensed for erectile dysfunction are also approved for PAH treatment. Both sildenafil and tadalafil induce pulmonary vasodilatation in PAH (maximal effect after 60 and 75–90 minutes, respectively [58]. Sildenafil has also been shown to inhibit the proliferation and to stimulate the apoptosis of pulmonary artery smooth muscle cells in vitro [59, 60]. As for the first ERA generation, sildenafil and tadalafil were licensed on the basis of 12–16-week randomised placebo-controlled trials [61–63] demonstrating efficacy by improving 6MWD. Because the SUPER-1 study showed no statistical difference in 6MWD between 20, 40 or 80 mg sildenafil, USA and European regulatory authorities approved the drug at 20 mg three times daily (t.i.d), but because pulmonary vascular resistance decreases linearly with increasing sildenafil dose, many physicians up-titrate their patients up to 80 mg t.i.d. if necessary [64]. Also, the open-label extension of the SUPER-1 study (SUPER-2) suggested long-term efficacy of sildenafil given at 80 mg t.i.d but uncertainty remains with the 20 mg dose [62]. As compared with sildenafil, tadalafil has a longer plasma half-life (17.5 hours as compared with 4 hours) and can be given once a day. It also increases 6MWD in a dose-dependent manner with maximal effect at 40 mg (table 1) [63]. In patients receiving tadalafil 20 or 40 mg, the improvements in 6MWD demonstrated in the 16-week PHIRST study appeared sustained for up to 52 additional weeks of treatment in PHIRST-2 [65]. Both drugs are generally well tolerated and most side effects are essentially related to vasodilatation, such as headache, flushing, epistaxis, dyspepsia, diarrhoea, myalgia and hypotension. In some cases such as PAH associated with human immunodeficiency virus infection, the PDE-5 ihibitor dose may have to be reduced, because of interaction with antiretroviral therapy. Ocular complications with blurred vision and altered colour perception due to an effect of sildenafil on retinal PDE type 6 were described, but doses up to 80 mg t.i.d have recently been shown to be safe from an ocular perspective [66].
Active on the NO-cGMP pathway, riociguat is the most recently approved drug for PAH and the only one approved in Switzerland for inoperable CTEPH. Conceptually, it represents a novel class of drugs that sensitizes sGC to low NO levels and further activates oxidised sGC independently of NO [67]. The ability to produce cGMP even in the absence of NO might confer on riociguat a potential advantage over PDE-5 ihibitors, whose effect may be hampered by the reduced NO availability in PAH. At a 1.0–2.5 mg dose given t.i.d. and titrated according to blood pressure, riociguat decreased pulmonary arterial pressure while cardiac index increased in a phase II study involving 33 PAH patients [68]. The phase III PATENT-1 trial kept to the traditional 12-week 6MWD primary endpoint, and time to clinical worsening was only one of the secondary endpoints [69]. This study included almost only class II and III patients and showed a placebo-controlled 6MWD increase by 36 m, a significant delay in clinical worsening (p = 0.005) as well as a favourable safety profile. The most common serious adverse event was syncope and hypotension which occurred in 10% of patients in the 2.5-mg maximum group. The 6MWD improvement persisted at 1 year (+51 m) as compared with baseline, and functional class improved in 33%, stabilised in 66% and worsened in 6% [70]. Of note, combining riociguat with sildenafil in order to look for a synergistic effect was recently shown to bring excessive systemic hypotension with no favourable effect on haemodynamics or exercise capacity, and is therefore not recommended [71].
|
Table 1: Trials investigating monotherapy in PAH. Only numbers (N) from naïve patients are reported while data from patients on background therapy have been omitted. Trials that did not describe separately naïve patients are not shown. |
|
Short Term
|
Study 251; 2001 [41]
N = 32; 12 wks
Placebo-contr.
6’WD (+76 m)
BREATHE-1; 2002 [42]
N = 213; 16 wks
Placebo-contr.
6’WD (+44 m)
EARLY; 2008 [74]
N = 185 (classII) 24 wks
Placebo-contr.
PVR (–22.6%) |
ARIES-1; 2008 [40]
N = 202; 12 wks
Placebo-contr.
6’WD
(+31 m/5 mg)
(+51 m/10 mg)
"ARIES-2; 2008 [40]
N = 192; 12 wks
Placebo-contr.
6’WD (+59 m/5 mg) |
|
SUPER-1 2005[61]
N = 278; 12 wks
Placebo-contr.
6’WD
(+45 m/20 mg)
(+46 m/40 mg)
(+50 m/80 mg)
SINGH 2006[72]
N = 20; 6 wks
Placebo-contr.
6’WD (+65 m) |
PHIRST 2009[63]
N = 144; 16 wks
Placebo-contr.
6’WD (+44 m) |
PATENT-1 2013[73]
N = 89; 12 wks
Rioc 2.5max
Vs placebo
6’WD (+38 m) |
BARST 1996[29]
N = 81 (class III–IV)
12 wks
Placebo-contr.
6’WD (+113 m)
Survival
(100% vs 80%) |
Simonneau 2002 [33]
N = 470; 12 wks
Placebo-contr.
6’WD
(+16 m/overall)
(+36 m/>13.8 ng/kg) |
AIR 2002[35]
N = 203 (class III–IV) 12 wks
Placebo-contr.
6’WD (+59 ml)
FC improved (+17.2%) |
|
Long Term
|
McLaughlin 2005 [44]
N = 169; 24 mths
Open label observat.
70% alive monoRx
7% alive combiRx
12% alive otherRx
EARLY long -t 2014 [46]
N = 144 (classII) 4 yrs
Open label observat
52% alive monoRx
31% alive combiRx
15% mortality |
Oudiz 2009 [39]
N = 383; 2 yrs
Open label obs.
56% alive monoRx
12% alive combiRx
15% mortality
AMBITION 2015 [75]
[75] N = 126; ~484 days
Ambri-contr. arm observational
37% clin failure |
SERAPHIN; 2013 [57]
N = 184; 36 mths
Maci 10 mg vs plac.
Clinical worsenirg
HR = 0.45
(95%CI 0.28–0.72) |
SUPER-2 2011[62]
N = 259; 3 years
Open label obs
58% alive monoRx
12% alive combiRx
23% mortality |
AMBITION 2015 [75]
N = 121; ~484 days
tada-contr. arm observational
31% clin failure |
|
McLaughlin 2002 [30]
N = 162; 36 mths
Open label observ.
62.5% survival |
Barst 2006 [34]
N = 860; 4.5 yrs
Open label observ.
23% stopped (AE)
14% stopped
(disease progr.)
15% alive on combiRx
26% alive on monoRx
16% mortality |
Opitz 2005 [36]
N = 76; 5 yrs
Open label observ.
87% events (death, lung Tx, switch to IV, combination therapy)
Survival 40% |
| |
Bosentan
|
Ambrisentan
|
Macitentan
|
Sildenafil
|
Tadalafil
|
Riociguat
|
Epoprostenol
|
Treprostinil
|
Iloprost
|
| Grey: observational data; Orange: short-term randomised controlled trial with soft endpoints (PVR or 6’WD or functional class); Green: short/long-term randomised controlled trial with hard endpoints.
6’WD = 6-minute walking distance; AE = adverse event; Ambri = ambrisentan; clin = clinical; contr = controlled; combiRx = combination therapy; FC = functional class; Maci = macitentan; monoRx = monotherapy; mths = months; observ = observational; Plac = placebo; progr = progression; Rioc = riociguat; tada = tadalafil; Tx = transplantation; wks = weeks; yrs = year |
Treatment strategies for PAH
Monotherapy
Monotherapy refers to treating PAH with a single drug. This strategy relies on a solid background of numerous studies showing clinical benefit with any of the drugs described above when tested versus placebo in naïve patients. Table 1 shows the published evidence for monotherapy (trial data concerning patients on background therapy have been omitted on purpose) with all drugs approved in Switzerland, with numbers of patients, study duration and results for the primary endpoint. What may first appear on looking over the table is that the sub-analysis restricted to naïve patients recruited in the SERAPHIN trial currently offers the only evidence that oral monotherapy may delay disease progression in PAH [57]. However, while other randomised-controlled trials also validate some short-term benefit on soft endpoints, long-term follow-up studies consistently show a significant number of patients initially on monotherapy who are either dead (15–60%), or on combination therapy (7–31%). This most probably reflects worsening PAH or a clinical failure (31–37%) after 1.5 to 4 years. These considerations, added to results from meta-analyses concluding that monotherapy alone is unsatisfactory regarding hard clinical outcomes [76, 77], indicates that oral monotherapy is not enough for the long term, at least in a subset of patients with more severe disease. Regarding parenteral therapy, only epoprostenol showed increased survival in monotherapy class III and IV patients, and the effects of none of the other parenteral prostanoids were assessed on hard clinical endpoints.
Currently, initial monotherapy is a recommended option for the management of PAH patients at low or intermediate risk. Patients on monotherapy should then be carefully and regularly reassessed for disease progression, and combination therapy should be applied for all deteriorating subjects and for all subjects who persist with an unsatisfactory response to treatment (see below) [3].
Combination therapy
Randomised trials as well as clinical practice have shown that a single drug regimen is frequently not sufficient to control the patient’s symptoms and to decrease the risk of clinical deterioration. Pulmonary hypertension experts have empirically used combinations of drugs for the most severely ill, despite initial lack of published evidence [78]. Since then, several RCTs have attempted to address the issue, albeit with different designs, endpoints and durations, making comparisons hazardous. These studies are summarised in table 2. When considering the drugs presently available (including selexipag) and excluding combinations within the same class of medication, about 30 possible double drug regimens are available. It is important to note that not all combinations have been tested in RCTs and, among those evaluated, not all proved to be positive. In particular, double-drug regimens including the ERA bosentan were negative in four trials including the recently published COMPASS-2 trial [79]. Whether these failures were due to inadequate study design, insufficient statistical power or the consequence of a specific drug interaction, leading to decreased blood levels of either substance, is not known. However the two most recent substances riociguat and selexipag in association with bosentan demonstrated positive results [37, 69].
Based on these data, the ESC/ERS guidelines let the physician the choice to prescribe either a monotherapy or a double therapy to a patient newly diagnosed with PAH, in functional NYHA class II–III [3]. In contrast, a patient diagnosed at NYHA stage IV should be treated with an initial combination that includes an intravenous prostanoid. It is not known at the present time if selexipag might replace intravenous prostanoids in this setting and this point will have to be addressed by specific RCTs that include patients in NYHA class IV. Although NYHA functional class is a major determinant of prognosis in PAH, other independent risk factors have been validated, including the subtype of PAH, systemic blood pressure, the diffusing capacity of the lung for carbon monoxide and pulmonary vascular resistance [3] (table 3). In particular, scleroderma-associated PAH has been shown to have a worse prognosis [80].
These risk factors have been extensively studied in the USA-based REVEAL registry [81] and may provide additional help to the clinician for the choice of treatment in patients with NYHA class II or III. The benefit of such a risk stratified treatment has, however, not been validated prospectively and is therefore not yet included in the most recent guidelines.
Concerning initial (upfront) double combination therapy, the association ambrisentan + tadalafil, which has been specifically analysed in the AMBITION study [75], receives grade I, level B in the European guidelines, whereas other combinations with ERAs and PDE-5 inhibitors are graded II, level C [3]. Moreover, in scleroderma-associated PAH, this combination has shown significant efficacy in an open long-term trial [82].
Sequential versus simultaneous (upfront) combined therapy
The AMBITION study demonstrated that upfront combination of ambrisentan with tadalafil was superior to either drug alone [75]. Therefore the initiation of monotherapy with one of these two medications should be restricted to patients with a low risk profile. However, the AMBITION study has not demonstrated that upfront therapy was superior to a sequential design, where the second drug is introduced when the target of PAH treatment has not been met after an observational period of 3 to 6 months. One might fear that with this step-wise approach the clinical worsening of a patient receiving a single drug would be eventually impossible to recover. However the PATENT-2 study, the open-label extension of the RCT for riociguat, demonstrated that patients initially on placebo during the blinded phase recovered to a similar extent to the active group during the extended phase, without increased mortality [71]. Another inconvenience of upfront double therapy is the occurrence of adverse events without knowing which one of the two medications is the culprit. Finally, an upfront treatment does not allow identification of patients who are entirely nonresponsive to a class of substance.
Apart from patients in NYHA IV, it appears therefore wise to adopt a sequential regimen, while taking care to shorten the delay when the initial risk assessment is elevated (e.g., REVEAL score ≥10).
An upfront triple drug strategy has not been tested in RCTs, but an observational study has been recently reported by the French group in 19 severe PAH patients. These results demonstrated an excellent clinical response under this aggressive approach [83]. Such a design should be restricted, however, to highly selected class IV patients.
|
Table 2: Published clinical studies in which double therapy was compared with single therapy plus placebo. In the second to lowest row are the drugs tested against placebo, and in the left-hand column is shown the patients’ existing regimen. In the BREATHE-2 and the AMBITION studies, both drugs were introduced simultaneously. |
|
Ambrisentan
|
|
|
|
|
AMBITION+
TCW n = 253
positive |
Patent-1
6WD n = (194)1
positive |
|
Griphon
TCW n = (94)1
positive |
|
Bosentan
|
– |
|
|
NCT297+
6MWD n = 103
negative |
Phirst
6MWD n = 42
negative |
Patent-1
6WD n = (194)1
positive |
Triumph3
6MWD n = 82
positive |
Griphon
TCW n = (94)1
positive |
| Freedom-C3
6MWD n = 32
negative |
|
Macitentan
|
|
|
|
|
|
|
|
|
|
Sildenafil
|
|
Compass-2
TCW n = 167
negative |
Seraphin
TCW n = (150)1
positive |
|
|
|
Triumph
6MWD n = 35
positive |
Griphon
TCW n = (189)1
positive |
| Freedom-C
6MWD n = 26
negative |
|
Tadalafil
|
AMBITION+
TCW n = 253
positive |
|
Seraphin
TCW n = (150)1
positive |
|
|
|
|
Griphon
TCW n = (189)1
positive |
|
Riociguat
|
|
|
|
|
|
|
|
|
|
Prostanoid
|
|
Breathe-2+
PVR n = 33
negative |
Seraphin
TCW n = 152
|
Paces
6MWD n = 267
positive |
|
Patent-1
6MWD n = 282
|
|
|
| Drug |
Ambrisentan
|
Bosentan
|
Macitentan
|
Sildenafil
|
Tadalafil
|
Riociguat
|
Prostanoid
|
Selexipag
|
| Class |
Endothelin receptor antagonist
|
Phosphodiesterase V inhibitor
|
cGMP activ.
|
Prostacyclin receptor pathway
|
| Green = positive studies. Red = negative studies.
6MWD = 6-minute walking distance; PVR = pulmonary vascular resistance; TCW = time to clinical worsening.
1 indicates that detailed numbers for each drug are not available. The figure in parentheses is for the whole class. 2 The number of patients is too low to prove efficacy. |
|
Table 3: Parameters associated with an increased morbidity and mortality risk in pulmonary arterial hypertension. Adapted from refs [3] and [81]. |
|
Nonevolving factorsPAH associated with connective tissue disorderPAH associated to portal hypertensionHeritable pulmonary hypertensionAge >60 years and male gender |
|
Evolving factorsNYHA class >IISystolic blood pressure <110 mm HgHeart rate >92/min6-minute walking distance <440 mDLCO <80% predictedBrain-type natriuretic peptide >50 ng/l, NT-ProBNP >300 ng/mlPeak VO2 <15 ml/kg*minPericardial effusionRight atrium area >18 cm2Right atrium pressure >8 mm Hg, cardiac index <2.5 l/min*kg, SvO2 <65% |
| DLCO = diffusing capacity of the lung for carbon monoxide; NT-Pro-BNP = N-terminal peptide of pro-brain natriuretic peptide; NYHA = New York Heart Association |
Monitoring the therapy
Follow-up of patients has been recently emphasised and structured into treatment goals [3] based on validated markers of morbidity and mortality in distinct cohorts [81, 84, 85] (table 3). It is assumed that the correction of these risk factors under therapy will have a positive impact on survival. We should, however, remain cautious as these risk factors are surrogate markers for mortality and, consequently, the correction of one of them does not guarantee automatically that survival will be better, as several unfortunate examples in the medical literature have previously shown (e.g., arrhythmia in left heart failure). However, taking into account not one but several surrogate markers together, including clinical signs, imaging studies and laboratory tests, makes the assumption more robust even if this strategy has not been validated in a prospective randomised trial. In clinical practice it is recommended that if several modifiable risk factors persist or appear under treatment, the medication be increased from a one- to two-drug regimen or from double to triple drug therapy [3]. When the goals are not met under a triple drug regimen, including intravenous prostanoids, lung transplantation should be considered and the patients referred to a lung transplant centre, if they qualify for such a treatment. Contrary to a common belief, follow-up of systolic pulmonary pressure by means of echocardiography is not valid for risk assessment. Indeed, a decreasing systolic pulmonary arterial pressure may indicate the onset of right ventricular failure.
Occasionally, the clinician may face the question of decreasing the number of PAH-specific drugs. Apart from side effects problems, this may arise in some drug-induced PAH [86], or HIV-associated PAH or portopulmonary PH after successful liver transplantation, but is exceptional in idiopathic PAH. Such a decision should be made only after a thorough assessment including repeated right heart catheterisation.
Conclusion
The number of substances available for the treatment of PAH has increased during the last 8 years, but remains within the three therapeutic targets identified in the early 2000s [87], and PAH remains a disease with poor prognosis. Combination therapy has been more and more validated through fairly large randomised trials with clinically relevant endpoints and with registry data. Today, because therapy is more and more complex and includes potentially harmful drugs, global management in expert centres with ambitious treatment goals and comprehensive functional, biological and haemodynamic follow-up is the standard of care.
Acknowledgments: we thank the executive board and scientific committee of the Swiss Society for Pulmonary Hypertension for critical and constructive reading of this manuscript.
References
1 Ulrich S, Fischler M, Speich R. Update on therapies for pulmonary hypertension. Swiss Med Wkly. 2007;137:73–82.
2 Galie N, Corris PA, Frost A, Girgis RE, Granton J, Jing ZC, et al. Updated treatment algorithm of pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62:D60–72.
3 Galie N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J. 2015;46:903–75.
4 Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D34–41.
5 Abman SH, Hansmann G, Archer SL, Ivy DD, Adatia I, Chung WK, et al. Pediatric Pulmonary Hypertension: Guidelines From the American Heart Association and American Thoracic Society. Circulation. 2015;132:2037–99.
6 Huber LC, Bye H, Brock M. The pathogenesis of pulmonary hypertension – an update. Swiss Med Wkly. 2015;145:w14202.
7 Johnson SR, Mehta S, Granton JT. Anticoagulation in pulmonary arterial hypertension: a qualitative systematic review. Eur Respir J. 2006;28:999–1004.
8 Olsson KM, Delcroix M, Ghofrani HA, Tiede H, Huscher D, Speich R, et al. Anticoagulation and survival in pulmonary arterial hypertension: results from the Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA). Circulation. 2014;129:57–65.
9 Preston IR, Roberts KE, Miller DP, Sen GP, Selej M, Benton WW, et al. Effect of Warfarin Treatment on Survival of Patients with Pulmonary Arterial Hypertension (PAH) in the Registry to Evaluate Early and Long-Term PAH Disease Management (REVEAL). Circulation. 2015.
10 Elinoff JM, Rame JE, Forfia PR, Hall MK, Sun J, Gharib AM, et al. A pilot study of the effect of spironolactone therapy on exercise capacity and endothelial dysfunction in pulmonary arterial hypertension: study protocol for a randomized controlled trial. Trials. 2013;14:91.
11 Sandoval J, Aguirre JS, Pulido T, Martinez-Guerra ML, Santos E, Alvarado P, et al. Nocturnal oxygen therapy in patients with the Eisenmenger syndrome. Am J Respir Crit Care Med. 2001;164:1682–7.
12 Ulrich S, Keusch S, Hildenbrand FF, Lo Cascio C, Huber LC, Tanner FC, et al. Effect of nocturnal oxygen and acetazolamide on exercise performance in patients with pre-capillary pulmonary hypertension and sleep-disturbed breathing: randomized, double-blind, cross-over trial. Eur Heart J. 2015;36:615–23.
13 Burns RM, Peacock AJ, Johnson MK, Church AC. Hypoxaemia in patients with pulmonary arterial hypertension during simulated air travel. Respir Med. 2013;107:298–304.
14 Mainguy V, Provencher S, Maltais F, Malenfant S, Saey D. Assessment of daily life physical activities in pulmonary arterial hypertension. PLoS One. 2011;6:e27993.
15 Pugh ME, Buchowski MS, Robbins IM, Newman JH, Hemnes AR. Physical activity limitation as measured by accelerometry in pulmonary arterial hypertension. Chest. 2012;142:1391–8.
16 Pandey A, Garg S, Khunger M, Garg S, Kumbhani DJ, Chin KM, Berry JD. Efficacy and Safety of Exercise Training in Chronic Pulmonary Hypertension: A Systematic Review and Meta-Analysis. Circ Heart Fail. 2015.
17 Ehlken N, Lichtblau M, Klose H, Weidenhammer J, Fischer C, Nechwatal R, et al. Exercise training improves peak oxygen consumption and haemodynamics in patients with severe pulmonary arterial hypertension and inoperable chronic thrombo-embolic pulmonary hypertension: a prospective, randomized, controlled trial. Eur Heart J. 2015; 37:35–44.
18 Spruit MA, Singh SJ, Garvey C, ZuWallack R, Nici L, Rochester C, et al. An official American Thoracic Society/European Respiratory Society statement: key concepts and advances in pulmonary rehabilitation. Am J Respir Crit Care Med. 2013;188:e13–64.
19 Archer SL, Weir EK, Wilkins MR. Basic science of pulmonary arterial hypertension for clinicians: new concepts and experimental therapies. Circulation. 2010;121:2045–66.
20 Humbert M, Lau EM, Montani D, Jais X, Sitbon O, Simonneau G. Advances in therapeutic interventions for patients with pulmonary arterial hypertension. Circulation. 2014;130:2189–208.
21 Clarke JG, Benjamin N, Larkin SW, Webb DJ, Davies GJ, Maseri A. Endothelin is a potent long-lasting vasoconstrictor in men. Am J Physiol. 1989;257:H2033–5.
22 Davie N, Haleen SJ, Upton PD, Polak JM, Yacoub MH, Morrell NW, Wharton J. ET(A) and ET(B) receptors modulate the proliferation of human pulmonary artery smooth muscle cells. Am J Respir Crit Care Med. 2002;165:398–405.
23 Giaid A, Yanagisawa M, Langleben D, Michel RP, Levy R, Shennib H, et al. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med. 1993;328:1732–9.
24 Stasch JP, Schmidt PM, Nedvetsky PI, Nedvetskaya TY, H SA, Meurer S, et al. Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. J Clin Invest. 2006;116:2552–61.
25 Morrell NW, Adnot S, Archer SL, Dupuis J, Jones PL, MacLean MR, et al. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54:S20–31.
26 Christman BW, McPherson CD, Newman JH, King GA, Bernard GR, Groves BM, Loyd JE. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med. 1992;327:70–5.
27 Hoshikawa Y, Voelkel NF, Gesell TL, Moore MD, Morris KG, Alger LA, et al. Prostacyclin receptor-dependent modulation of pulmonary vascular remodeling. Am J Respir Crit Care Med. 2001;164:314–8.
28 Judge EP, Gaine SP. Management of pulmonary arterial hypertension. Curr Opin Crit Care. 2013;19:44–50.
29 Barst RJ, Rubin LJ, Long WA, McGoon MD, Rich S, Badesch DB, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med. 1996;334:296–301.
30 McLaughlin VV, Shillington A, Rich S. Survival in primary pulmonary hypertension: the impact of epoprostenol therapy. Circulation. 2002;106:1477–82.
31 Sitbon O, Delcroix M, Bergot E, Boonstra AB, Granton J, Langleben D, et al. EPITOME-2: An open-label study assessing the transition to a new formulation of intravenous epoprostenol in patients with pulmonary arterial hypertension. Am Heart J. 2014;167:210–7.
32 McLaughlin VV, Gaine SP, Barst RJ, Oudiz RJ, Bourge RC, Frost A, et al. Efficacy and safety of treprostinil: an epoprostenol analog for primary pulmonary hypertension. J Cardiovasc Pharmacol. 2003;41:293–9.
33 Simonneau G, Barst RJ, Galie N, Naeije R, Rich S, Bourge RC, et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med. 2002;165:800–4.
34 Barst RJ, Galie N, Naeije R, Simonneau G, Jeffs R, Arneson C, Rubin LJ. Long-term outcome in pulmonary arterial hypertension patients treated with subcutaneous treprostinil. Eur Respir J. 2006;28:1195–203.
35 Olschewski H, Simonneau G, Galie N, Higenbottam T, Naeije R, Rubin LJ, et al. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med. 2002;347:322–9.
36 Opitz CF, Wensel R, Winkler J, Halank M, Bruch L, Kleber FX, et al. Clinical efficacy and survival with first-line inhaled iloprost therapy in patients with idiopathic pulmonary arterial hypertension. Eur Heart J. 2005;26:1895–902.
37 Sitbon O, Channick R, Chin KM, Frey A, Gaine S, Galie N, et al. Selexipag for the Treatment of Pulmonary Arterial Hypertension. N Engl J Med. 2015;373:2522–33.
38 Galie N, Badesch D, Oudiz R, Simonneau G, McGoon MD, Keogh AM, et al. Ambrisentan therapy for pulmonary arterial hypertension. J Am Coll Cardiol. 2005;46:529–35.
39 Oudiz RJ, Galie N, Olschewski H, Torres F, Frost A, Ghofrani HA, et al. Long-term ambrisentan therapy for the treatment of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54:1971–81.
40 Galie N, Olschewski H, Oudiz RJ, Torres F, Frost A, Ghofrani HA, et al. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation. 2008;117:3010–9.
41 Channick RN, Simonneau G, Sitbon O, Robbins IM, Frost A, Tapson VF, et al. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled study. Lancet. 2001;358:1119–23.
42 Rubin LJ, Badesch DB, Barst RJ, Galie N, Black CM, Keogh A, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346:896–903.
43 Humbert M, Barst RJ, Robbins IM, Channick RN, Galie N, Boonstra A, et al. Combination of bosentan with epoprostenol in pulmonary arterial hypertension: BREATHE-2. Eur Respir J. 2004;24:353–9.
44 Girgis RE, Mathai SC, Krishnan JA, Wigley FM, Hassoun PM. Long-term outcome of bosentan treatment in idiopathic pulmonary arterial hypertension and pulmonary arterial hypertension associated with the scleroderma spectrum of diseases. J Heart Lung Transplant. 2005;24:1626–31.
45 Galie N, Beghetti M, Gatzoulis MA, Granton J, Berger RM, Lauer A, et al. Bosentan therapy in patients with Eisenmenger syndrome: a multicenter, double-blind, randomized, placebo-controlled study. Circulation. 2006;114:48–54.
46 Simonneau G, Galie N, Jansa P, Meyer GM, Al-Hiti H, Kusic-Pajic A, et al. Long-term results from the EARLY study of bosentan in WHO functional class II pulmonary arterial hypertension patients. Int J Cardiol. 2014;172:332–9.
47 Liu C, Chen J, Gao Y, Deng B, Liu K. Endothelin receptor antagonists for pulmonary arterial hypertension. Cochrane Database Syst Rev 2013, 2:CD004434.
48 Deboeck G, Scoditti C, Huez S, Vachiery JL, Lamotte M, Sharples L, et al. Exercise testing to predict outcome in idiopathic versus associated pulmonary arterial hypertension. Eur Respir J. 2012;40:1410–9.
49 Savarese G, Paolillo S, Costanzo P, D’Amore C, Cecere M, Losco T, et al. Do changes of 6-minute walk distance predict clinical events in patients with pulmonary arterial hypertension? A meta-analysis of 22 randomized trials. J Am Coll Cardiol. 2012;60:1192–201.
50 Frost AE, Langleben D, Oudiz R, Hill N, Horn E, McLaughlin V, et al. The 6-min walk test (6MW) as an efficacy endpoint in pulmonary arterial hypertension clinical trials: demonstration of a ceiling effect. Vascul Pharmacol. 2005;43:36–9.
51 Humbert M, Segal ES, Kiely DG, Carlsen J, Schwierin B, Hoeper MM. Results of European post-marketing surveillance of bosentan in pulmonary hypertension. Eur Respir J. 2007;30:338–44.
52 Savale L, Magnier R, Le Pavec J, Jais X, Montani D, O’Callaghan DS, et al. Efficacy, safety and pharmacokinetics of bosentan in portopulmonary hypertension. Eur Respir J. 2013;41:96–103.
53 McGoon MD, Frost AE, Oudiz RJ, Badesch DB, Galie N, Olschewski H, et al. Ambrisentan therapy in patients with pulmonary arterial hypertension who discontinued bosentan or sitaxsentan due to liver function test abnormalities. Chest. 2009;135:122–9.
54 Iglarz M, Binkert C, Morrison K, Fischli W, Gatfield J, Treiber A, et al. Pharmacology of macitentan, an orally active tissue-targeting dual endothelin receptor antagonist. J Pharmacol Exp Ther. 2008;327:736–45.
55 Sidharta PN, van Giersbergen PL, Halabi A, Dingemanse J. Macitentan: entry-into-humans study with a new endothelin receptor antagonist. Eur J Clin Pharmacol. 2011;67:977–84.
56 Montani D, Chaumais MC, Guignabert C, Gunther S, Girerd B, Jais X, et al. Targeted therapies in pulmonary arterial hypertension. Pharmacol Ther. 2014;141:172–91.
57 Pulido T, Adzerikho I, Channick RN, Delcroix M, Galie N, Ghofrani HA, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369:809–18.
58 Ghofrani HA, Voswinckel R, Reichenberger F, Olschewski H, Haredza P, Karadas B, et al. Differences in hemodynamic and oxygenation responses to three different phosphodiesterase-5 inhibitors in patients with pulmonary arterial hypertension: a randomized prospective study. J Am Coll Cardiol. 2004;44:1488–96.
59 Wharton J, Strange JW, Moller GM, Growcott EJ, Ren X, Franklyn AP, et al. Antiproliferative effects of phosphodiesterase type 5 inhibition in human pulmonary artery cells. Am J Respir Crit Care Med. 2005;172:105–13.
60 Tantini B, Manes A, Fiumana E, Pignatti C, Guarnieri C, Zannoli R, et al. Antiproliferative effect of sildenafil on human pulmonary artery smooth muscle cells. Basic Res Cardiol. 2005;100:131–8.
61 Galie N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353:2148–57.
62 Rubin LJ, Badesch DB, Fleming TR, Galie N, Simonneau G, Ghofrani HA, et al. Long-term treatment with sildenafil citrate in pulmonary arterial hypertension: the SUPER-2 study. Chest. 2011;140:1274–83.
63 Galie N, Brundage BH, Ghofrani HA, Oudiz RJ, Simonneau G, Safdar Z, et al. Tadalafil therapy for pulmonary arterial hypertension. Circulation. 2009;119:2894–903.
64 Hoeper MM, Welte T. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2006;354:1091–3; author reply 1091–3.
65 Oudiz RJ, Brundage BH, Galie N, Ghofrani HA, Simonneau G, Botros FT, et al. Tadalafil for the treatment of pulmonary arterial hypertension: a double-blind 52-week uncontrolled extension study. J Am Coll Cardiol. 2012;60:768–74.
66 Wirostko BM, Tressler C, Hwang LJ, Burgess G, Laties AM. Ocular safety of sildenafil citrate when administered chronically for pulmonary arterial hypertension: results from phase III, randomised, double masked, placebo controlled trial and open label extension. BMJ. 2012;344:e554.
67 Grimminger F, Weimann G, Frey R, Voswinckel R, Thamm M, Bolkow D, et al. First acute haemodynamic study of soluble guanylate cyclase stimulator riociguat in pulmonary hypertension. Eur Respir J. 2009;33:785–92.
68 Ghofrani HA, Hoeper MM, Halank M, Meyer FJ, Staehler G, Behr J, et al. Riociguat for chronic thromboembolic pulmonary hypertension and pulmonary arterial hypertension: a phase II study. Eur Respir J. 2010;36:792–9.
69 Ghofrani HA, Galie N, Grimminger F, Grunig E, Humbert M, Jing ZC, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013;369:330–40.
70 Rubin LJ, Galie N, Grimminger F, Grunig E, Humbert M, Jing ZC, et al. Riociguat for the treatment of pulmonary arterial hypertension: a long-term extension study (PATENT-2). Eur Respir J. 2015;45:1303–13.
71 Galie N, Muller K, Scalise AV, Grunig E. PATENT PLUS: a blinded, randomised and extension study of riociguat plus sildenafil in pulmonary arterial hypertension. Eur Respir J. 2015;45:1314–22.
72 Singh TP, Rohit M, Grover A, Malhotra S, Vijayvergiya R. A randomized, placebo-controlled, double-blind, crossover study to evaluate the efficacy of oral sildenafil therapy in severe pulmonary artery hypertension. Am Heart J. 2006;151:851 e851–855.
73 Ghofrani HA, Simonneau G, Rubin LJ. Riociguat for pulmonary hypertension. N Engl J Med. 2013;369:2268.
74 Galie N, Rubin L, Hoeper M, Jansa P, Al-Hiti H, Meyer G, et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trial. Lancet. 2008:371:2093–100.
75 Galie N, Barbera JA, Frost AE, Ghofrani HA, Hoeper MM, McLaughlin VV, et al. Initial Use of Ambrisentan plus Tadalafil in Pulmonary Arterial Hypertension. N Engl J Med. 2015;373:834–44.
76 Zhang HD, Zhang R, Jiang X, Sun K, Wu DC, Jing ZC. Effects of oral treatments on clinical outcomes in pulmonary arterial hypertension: A systematic review and meta-analysis. Am Heart J. 2015;170:96–103, 103 e101–114.
77 Coeytaux RR, Schmit KM, Kraft BD, Kosinski AS, Mingo AM, Vann LM, et al. Comparative effectiveness and safety of drug therapy for pulmonary arterial hypertension: a systematic review and meta-analysis. Chest. 2014;145:1055–63.
78 Mueller-Mottet S, Stricker H, Domenighetti G, Azzola A, Geiser T, Schwerzmann M, et al. Long-term data from the Swiss pulmonary hypertension registry. Respiration. 2015;89:127–40.
79 McLaughlin V, Channick RN, Ghofrani HA, Lemarie JC, Naeije R, Packer M, et al. Bosentan added to sildenafil therapy in patients with pulmonary arterial hypertension. Eur Respir J. 2015;46:405–13.
80 Rhee RL, Gabler NB, Sangani S, Praestgaard A, Merkel PA, Kawut SM. Comparison of Treatment Response in Idiopathic and Connective Tissue Disease-associated Pulmonary Arterial Hypertension. Am J Respir Crit Care Med. 2015;192:1111–7.
81 Benza RL, Gomberg-Maitland M, Miller DP, Frost A, Frantz RP, Foreman AJ, et al. The REVEAL Registry risk score calculator in patients newly diagnosed with pulmonary arterial hypertension. Chest. 2012;141:354–62.
82 Hassoun PM, Zamanian RT, Damico R, Lechtzin N, Khair R, Kolb TM, et al. Ambrisentan and Tadalafil Up-front Combination Therapy in Scleroderma-associated Pulmonary Arterial Hypertension. Am J Respir Crit Care Med. 2015;192:1102–10.
83 Sitbon O, Jais X, Savale L, Cottin V, Bergot E, Macari EA, et al. Upfront triple combination therapy in pulmonary arterial hypertension: a pilot study. Eur Respir J. 2014;43:1691–7.
84 Benza RL, Miller DP, Foreman AJ, Frost AE, Badesch DB, Benton WW, McGoon MD. Prognostic implications of serial risk score assessments in patients with pulmonary arterial hypertension: a Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL) analysis. J Heart Lung Transplant. 2015;34:356–61.
85 Sitbon O, Benza RL, Badesch DB, Barst RJ, Elliott CG, Gressin V, et al. Validation of two predictive models for survival in pulmonary arterial hypertension. Eur Respir J. 2015;46:152–64.
86 Gibbons E, Promislow S, Davies RA, Chandy G, Stewart DJ, Vladamir CD, et al. Reversible pulmonary arterial hypertension associated with interferon-beta treatment for multiple sclerosis. Can Respir J. 2015;22:263–5.
87 Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med. 2004;351:1425–36.