Multidisciplinary discussion for diagnosis of interstitial lung disease in real life
DOI: https://doi.org/10.4414/smw.2016.14318
Sabina Anna
Guler, Sabina Anna
Berezowska, Andreas
Christe, Thomas
Geiser, Manuela
Funke-Chambour
Precise diagnosis of interstitial lung diseases, with multidisciplinary discussion (MDD) being a key element, is gaining importance in daily practice as novel antifibrotic therapies for idiopathic pulmonary fibrosis are emerging. We assessed the diagnostic impact of MDD in a real life setting by retrospective analysis. With a change or adjustment of diagnosis in 64% (overall diagnostic consensus in 88%), we support results of previously conducted controlled studies by reflection of our daily clinical routine.
Introduction and background
Interstitial lung diseases (ILDs) represent a variety of lung parenchymal disorders that are difficult to diagnose and distinguish in their specific subtypes. Since January 2016, two antifibrotic medications have become available for the treatment of patients with idiopathic pulmonary fibrosis (IPF) in Switzerland. This makes it even more crucial to distinguish IPF from other ILDs. MDD has been proven to increase consensus diagnosis among specialists involved in ILD patient care and to avoid further invasive investigations [1]. MDD has so far not been evaluated in daily clinical routine, outside a study protocol.
Methods and results
We retrospectively evaluated MDD as diagnostic approach in patients referred to our tertiary ILD clinic over 2 years. The patients’ cases were presented to the MDD either by their referring doctor or by internal pulmonologists, who had a consultation with the patient first and proposed a provisional ILD diagnosis. An extended patient history, physiological assessment (including pulmonary function tests, serology) and chest computed tomography (CT) scan were always available beforehand. If patients were suitable, bronchoscopy with bronchoalveolar lavage and transbronchial biopsy, and if necessary surgical lung biopsy, were performed before MDD, according to the initial estimation of the treating pulmonologist. Clinicians, radiologist and pathologist discussed the cases together in a common session, expressed their opinions on the specific ILD diagnosis, and had the possibility to change their initial diagnosis taking into account the findings and judgment of their colleagues. Diagnostic consensus was defined as agreement on diagnosis of all participating specialists.
We retrospectively evaluated the specific ILD diagnosis before and after MDD.
During 24 months a total of 90 cases were discussed. Patients were predominantly male (55/90, 61%) with an average age of 61.5 years (men 63.5 years; women 58.5 years). Fifty-six percent (50/90) of cases were referred by pulmonologists in private practice, 23% (21/90) by general practitioners and 21% (19/90) were internal referrals from inside our hospital. In addition to clinical evaluation and chest CT scan, pathological results were available in 74% (67/90) of cases. A total of 33/90 (36%) patients underwent transbronchial biopsy, of which 5 were diagnostic (15%); 39/90 (43%) patients had surgical biopsy, resulting in a histological diagnosis in 34/39 (87%) cases. Bronchoalveolar lavage was performed in 49/90 (54%) cases.
Overall, MDD achieved a precise diagnosis in 79/90 patients (88%), and the diagnosis was changed in 58/90 (64%) cases after MDD (figure). In 10 cases (11%) the suspected diagnosis was rejected and an alternative diagnosis was suggested. In total, 16 (18%) of the cases could not be assigned a confident diagnosis, with 5 already having a contributory biopsy (definitely unclassifiable) and the remaining 11 (12%) being provisionally unclassifiable, indicating that additional diagnostic steps could possibly lead to diagnosis. Interestingly, in patients with initially suspected IPF, IPF not always proved to be the final diagnosis: It was confirmed in 64%, but in 36% the diagnosis was rejected after MDD. In several of these cases (14%) nonspecific interstitial pneumonia was the final diagnosis instead.
Discussion and conclusion
We report on our real-life experience of MDD in our Swiss ILD centre. MDD provided a precise diagnosis in the majority of cases (88%). Diagnosis needed to be corrected in 64% by MDD (fig.).
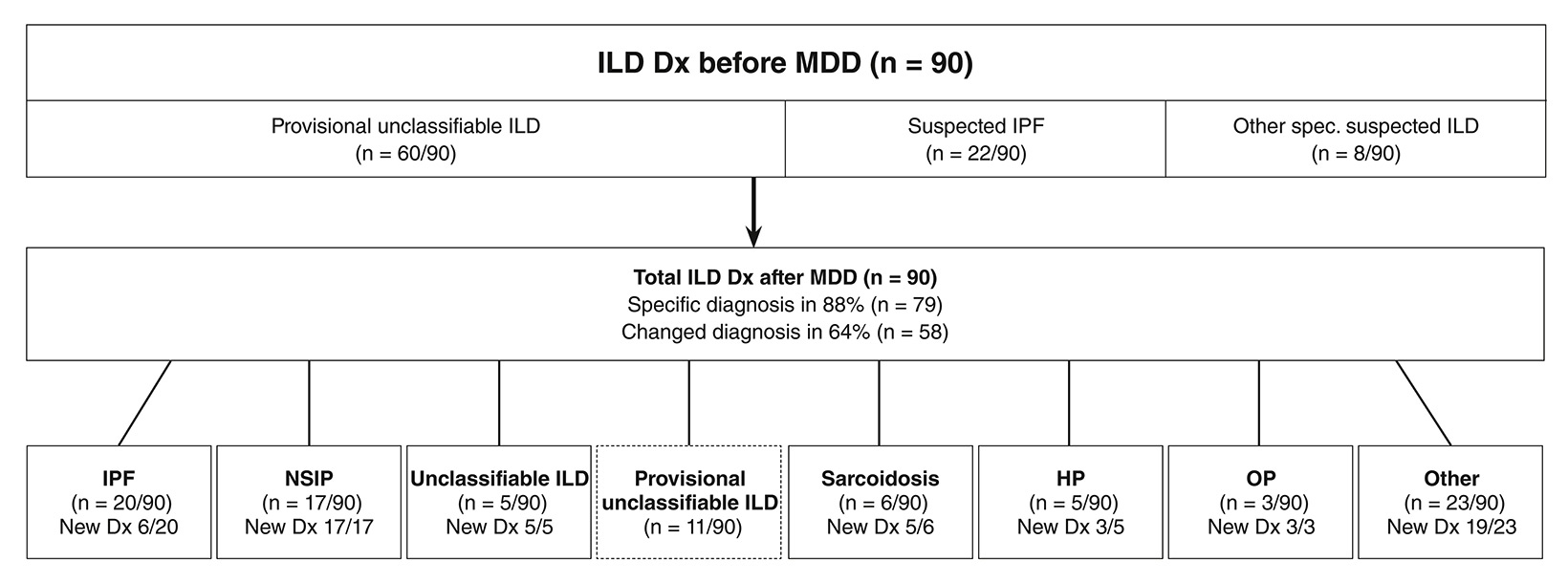
Figure
Diagnosis (Dx) before and after multidisciplinary discussion (MDD) for interstitial lung disease (ILD).
IPF = idiopathic pulmonary fibrosis; NSIP = nonspecific interstitial pneumonia; HP = hypersensitivity pneumonitis; OP = organising pneumonia.
Other diagnosis (1–2 cases each) included: combined pulmonary fibrosis and emphysema, Langerhans cell histiocytosis, vasculitis, respiratory bronchiolitis ILD, pulmonary lymphangioleiomyomatosis, Birt-Hogg-Dubé syndrome and diffuse idiopathic pulmonary neuroendocrine cell hyperplasia.
In oncology, multidisciplinary boards are widely applied and have demonstrated a significant impact on treatment decisions through collaboration between specialists [2]. IPF, a devastating disease, shows similarities with cancer: both diseases respond poorly to medical therapy, with IPF having a lower 5-year survival rate than many malignant neoplasms (e.g. colon, breast, kidney) [3]. Genetic changes and signalling pathways that are targets for new therapies (e.g. tyrosine kinase inhibitors) are overlapping [4]. Furthermore, as with cancer, decision making about diagnostic steps, final diagnosis and treatment benefits from the opinion of different specialists. This emphasises the role of MDD as an obvious approach. Nevertheless, MDD is not routinely used for all ILD patients.
The 2013 updated guidelines of the American Thoracic Society and the European Respiratory Society on classification of idiopathic interstitial pneumonias [5] strongly recommend the dynamic interaction and information exchange between clinicians, radiologists and pathologists in the diagnostic process of suspected ILD. MDD can be considered as the diagnostic gold standard in ILD management today. The combination of clinical with histological information is an asset for diagnosis [6]. MDD increased diagnostic confidence and agreement between specialists in a study setting where information was presented in portions in a strict stepwise protocol to the board members [1].
Here we report our real-life experience in diagnosing ILDs at our tertiary centre and confirm the efficacy of MDD in a daily setting. We are aware that this retrospective observation carries bias and confounders, but we believe it to add to the information from the previous controlled study [1] and case based reviews [7] with a reflection of clinical routine and daily practice in a real-life scenario [8].
Current guidelines on diagnosis of IPF [9] suggest that MDD should include surgical lung biopsy that has been obtained beforehand; however, guidelines on diagnosis of idiopathic interstitial pneumonias [5] state that MDD should be used to discuss if lung biopsy is even appropriate. Depending on the question asked, the treating clinician decides about the time-point of MDD. After MDD the majority of cases could be labelled with one of the diverse specific ILD subtypes including the entity of unclassifiable ILD (fig.). Eleven “unresolved” cases (12%) were labelled as provisionally unclassifiable without a lung biopsy (e.g. not eligible for surgery) and theoretically another specific ILD might have been determined. Our clinical experience corresponds to a recent review [10] that defined the term “provisional”unclassifiable ILD and reported that 5.1–15.1% of all ILD cases remain unclassifiable.
Correct ILD diagnosis is of utmost importance since it entails different treatment options: immunosuppressive agents can be indicated for patients with non-IPF ILDs but can increase morbidity and mortality in IPF patients [11] for whom novel cost-intensive antifibrotic treatments are now available [4, 12]. We assume this paradigm shift has reignited diagnostic interest in IPF all over the medical community, reflected in the 24% of our cases referred with the specific suspicion of IPF.
In conclusion, by describing the diagnostic impact of our MDD integrated in clinical ILD routine we observed an adjustment in diagnosis in 64% and an overall diagnostic consensus in 88% of the discussed cases. It is crucial to identify patients with reversible ILDs [13] and distinguish them from IPF, to determine the moment of intervention and beginning of therapy, and to arrange accurate long-term monitoring. By demonstrating the pivotal role of MDD in the diagnosis of suspected ILD in real life we want to highlight the importance of early multidisciplinary assessment, which improves diagnostic precision.
References
1 Flaherty KR, King TE, Jr., Raghu G, Lynch JP, 3rd, Colby TV, Travis WD, et al. Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med. 2004;170(8):904–10.
2 Greer HO, Frederick PJ, Falls NM, Tapley EB, Samples KL, Kimball KJ, et al. Impact of a weekly multidisciplinary tumor board conference on the management of women with gynecologic malignancies. Int J Gynecol Cancer. 2010;20(8):1321–5.
3 Vancheri C, Failla M, Crimi N, Raghu G. Idiopathic pulmonary fibrosis: a disease with similarities and links to cancer biology. Eur Respir J. 2010;35(3):496–504.
4 Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071–82.
5 Travis WD, Costabel U, Hansell DM, King TE, Jr., Lynch DA, Nicholson AG, et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188(6):733–48.
6 Theegarten D, Muller HM, Bonella F, Wohlschlaeger J, Costabel U. Diagnostic approach to interstitial pneumonias in a single centre: report on 88 cases. Diagn Pathol. 2012;7:160.
7 Tomassetti S, Piciucchi S, Tantalocco P, Dubini A, Poletti V. The multidisciplinary approach in the diagnosis of idiopathic pulmonary fibrosis: a patient case-based review. Eur Respir Rev. 2015;24(135):69–77.
8 Roche N, Reddel H, Martin R, Brusselle G, Papi A, Thomas M, et al. Quality standards for real-world research. Focus on observational database studies of comparative effectiveness. Ann Am Thorac Soc. 2014;11(Suppl 2):S99–104.
9 Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824.
10 Skolnik K, Ryerson CJ. Unclassifiable interstitial lung disease: A review. Respirology. 2016;21(1):51–6.
11 Idiopathic Pulmonary Fibrosis Clinical Research N, Raghu G, Anstrom KJ, King TE, Jr., Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366(21):1968–77.
12 King TE, Jr., Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2083–92.
13 Elicker BM, Jones KD, Henry TS, Collard HR. Multidisciplinary Approach to Hypersensitivity Pneumonitis. J Thorac Imaging. 2016;31(2):92–103.