Epigenetic biomarkers in rheumatology – the future?
DOI: https://doi.org/10.4414/smw.2016.14312
Summary
Epigenetic changes are stable modifications of DNA or histones that profoundly alter gene expression. They can be changed by environmental influences and can then be passed on to daughter cells or via the germ line to offspring. A variety of changes in epigenetic marks and in the expression of noncoding RNA has been found in cancer as well as in chronic inflammatory diseases. Interestingly, in both diseases similar mechanisms and pathways are affected albeit often to a different extent. DNA methylation is often lost in repetitive sequences, while in promoter regions hypo- as well as hypermethylation is found. Changes in microRNA levels typically affect microRNAs that are changed by an inflammatory environment, but disease specific changes have also been found in the blood and various cell types of patients with rheumatoid arthritis, systemic lupus erythematosus and other rheumatic diseases. Therefore, changes in the expression of microRNA in particular, but also demethylated gene loci, have been proposed as potential biomarkers in chronic inflammatory diseases and in cancer. Potentially, these changes could be used for early diagnosis and also to predict treatment response. Unfortunately most studies in rheumatology up to now were not designed to validate these epigenetic changes as biomarkers. Since the cancer field is much more advanced in the usage of biomarkers for disease subclassifications and subsequent therapeutic decisions, it is worthwhile to take a closer look at the biomarkers, methods and procedures used in oncology and to see which of these could also be applied to predicting disease severity and therapeutic response in rheumatic diseases.
This article will highlight common epigenetic pathways activated in cancer and various rheumatic diseases and summarise epigenetic changes that have the potential to become biomarkers in rheumatic diseases.
Summary and outlook
In summary, epigenetic biomarkers have entered the cancer field already and are also starting to be analysed in rheumatology. Even though the field is still in its infancy, studies regarding DNA methylation and microRNA measurements in particular are promising. However, to further advance this field, studies have to be conducted that are specifically designed to validate the use of epigenetic changes and microRNA as prognostic or diagnostic biomarkers.
In patients with glioblastoma, promoter methylation of the DNA repair gene O6-methylguanine-DNA methyltransferase (MGMT) is already used in clinics to predict the response to alkylating drugs, which shows that the use of DNA methylation as epigenetic biomarker is feasible. Methylation-specific PCR, as well as pyrosequencing, can be applied to measure the methylation status of a specific locus, with the first being cheap and fast and the latter being more reliable, but also more expensive. In rheumatic diseases the status of DNA methylation could be assessed in biopsies from synovial tissues of patients, in peripheral blood cells or even in cell-free DNA isolated from the blood, which is currently being explored in cancer patients [59].
In view of the costs that are spent on drugs in rheumatology and the time some patients have to wait for their diagnosis and efficient treatment, it is absolutely justified to intensify efforts to find the right diagnosis and the right treatment. This may mean that more biopsies have to be taken and more than one biomarker has to be measured – measures that are readily accepted for cancer diagnosis.
Efforts like the Encyclopedia Of DNA Elements (ENCODE) project, which aims to identify all functional elements of the genome including histone modifications and DNA methylation, have already brought valuable insights into transcript expression and are requisite for the understanding of the alterations in the epigenome that are measured in rheumatic diseases. But to understand epigenetic changes in rheumatic diseases is probably not enough. As in the cancer genome atlas (TCGA), the rheumatology research community should aim to map molecular pathways that drive rheumatic diseases and connect changes in the genome, the epigenome, transcription, protein expression and clinical outcomes. Potential biomarkers or combination of biomarkers must then be selected and tested in studies with an appropriate study design. This strategy would not only bring us closer to finding diagnostic, predictive and prognostic biomarkers, but would also help to find new therapeutic targets.
What is epigenetics?
Throughout the years many different, more or less strict definitions of epigenetics have been formulated. In a consensus meeting in 2008 in Cold Spring Harbor it was agreed that an epigenetic trait is a “stably heritable phenotype resulting from changes in a chromosome without alterations in the DNA sequence” [1]. Wider definitions also include nonheritable mechanisms [2]. In general, modifications of DNA and histones, as well as noncoding RNA transcripts and regulation of chromatin accessibility by nucleosome positioning, are regarded as epigenetic mechanisms. Epigenetic marks determine which genes in our organism are expressed in a particular cell type or activation state and which are silenced. In contrast to the genetic code, which is more or less stable throughout the lifetime, epigenetic marks can be altered by environmental factors, such as toxins or nutrition, and thus allow the cell to adapt to long-lasting changes in its environment. On the downside, epigenetic changes induced by the environment might have detrimental effects and lead to disease development. In particular, in diseases with a missing heritability and low concordance rates between monozygotic twins, like many rheumatic diseases, a strong environmental influence on disease development is assumed. Therefore epigenetic changes induced over time, together with a genomic risk background, might lead to disease development and persistence.
Epigenetic modifications of DNA and histones are placed directly at the cytosine base of the DNA and at the protruding tails of the histones, which pack the DNA tightly into nucleosomes (fig. 1). Histone tails can be modified by numerous histone marks. Up to now histone acetylation, methylation, phosphorylation, sumoylation, ADP-ribosylation, ubiquitination, deiminiation/citrullination, proline isomerisation and O-GlcNAcylation have been described. Specific combinations of these different histone modifications are found at actively transcribed and silenced genes, respectively. The most studied modification of the DNA is cytosine methylation, which leads to gene silencing. Methylation of cytosine is often found in genomic regions that are rich in cytosine-phosphatidyl-guanine (CpG) dinucleotides, so-called CpG islands, in gene promoter regions.
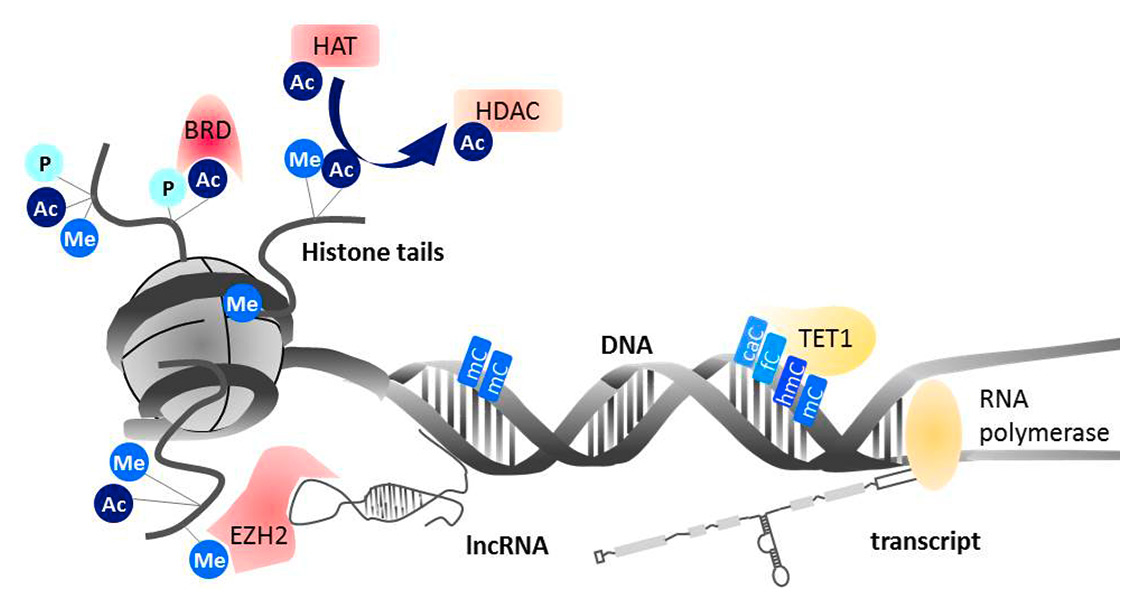
Figure 1
Acetyl (Ac), phospho (P) and methyl (Me) residues are placed on specific amino acids of the protruding histone tails by writer proteins. Histone acetyl transferases (HATs) and histone deacteylases (HDACs) are the readers and writers of acetylation, respectively. Bromodomains (BRDs) bind acetylated histone tails and promote the formation of multiprotein complexes. The methyltransferase EZH2 is bound to long non-coding RNA (lncRNA), which confers sequence specific location. On the DNA, ten-eleven translocation (TET) proteins convert methylated cytosines (mC) to carboxylcytosine (caC), formylcytosine (fC), and hydroxylmethylcytosine (hmC). Modified from [48] Kyburz D, et al. Epigenetic changes: the missing link. Best Pract Res Clin. 2014;28(4):577–87, with permission from Elsevier.
Methylation of cytosine in the DNA mediates the recruitment of transcriptional repressors and interferes with the binding of transcription factors. The bound repressor proteins can then form complexes with enzymes that place or remove modifications at the histone tails and thus stabilise DNA methylation-induced gene silencing. Thus, DNA methylation and histone modifications together reinforce dense packing and silencing of chromatin. In contrast to DNA methylation, however, some histone marks are also associated with actively transcribed chromatin, most prominently histone acetylation. The effect of histone modifications on gene expression is believed to be mediated by structural changes that alter accessibility of the transcription machinery to gene promoters as well as by recruitment of reader proteins that then form complexes with gene activating and repressive proteins. Even though the technology to study histone modifications has substantially improved in recent years, the complexity and dynamics of these epigenetic marks are far from being fully understood.
DNA methylation is crucial for the silencing of retrotransposable elements in the genome, inactivation of the second X chromosome in female cells and tissue specific gene expression. Accordingly, DNA methylation patterns have to be very stable and are faithfully restored after cell division. Nevertheless, changes in DNA methylation are commonly found in tumour cells and increasing numbers of studies have also found epigenetic changes in chronic inflammatory diseases [3–5]. In most cases, a state of global hypomethylation that mainly affects areas of repeated DNA elements such as long interspersed nuclear elements (LINE)-1 is combined with hypo- and hypermethylation in gene encoding areas [6–9]. Hypermethylation in tumour cells has been suggested to be either random due to aberrant activity of DNA methyltransferases (DNMTs) or selective due to guidance of DNMTs to DNA loci with specific cis features [10]. Studies of differentially methylated loci in promoters, 5’/3’ untranslated regions and gene bodies in synovial fibroblasts of rheumatoid arthritis (RA) patients suggest that differentially methylated genes are not random, but rather enriched for certain pathways, which supports a selective pathway introducing changes of DNA methylation in RA [11, 12]. In immune cells from patients with systemic lupus erythematosus (SLE), hypomethylated genes were enriched in interferon pathways, also pointing to selective loss of methylation [13]. Interestingly, CD4 T cells of patients with Sjögren’s syndrome also showed hypomethylation of genes of the interferon signature [14].
A mechanism for the global loss of methylation seen in repetitive genomic sequences in cancer as well as in chronic inflammation has been connected to the increased consumption of polyamines caused by increased rates of proliferation and cell growth [15–17]. Polyamine recycling and DNA methylation use S-adenosylmethionine as donor for the methyl group. Activated polyamine metabolism therefore can lead to a shortage of S-adenosylmethionine for the restoration of methyl marks after cell division, resulting in DNA hypomethylation of the daughter cells. Recent studies also showed that cytosine methylation can be converted to hydroxymethyl-cytosine, formyl-cytosine and carboxyl-cytosine by ten-eleven translocation (TET) enzymes [18]. Changes in TET expression in cancer and inflammation are therefore also assumed to promote changes in DNA methylation marks [19, 20].
In many tumours and chronic diseases, histone modifications, as well as the enzymes that write, read and erase these modifications, have been described as altered [21, 22]. Whether these changes are caused by altered genomic structures and changes in cell growth and metabolism in these conditions or are actually causing altered cell behaviour is still under debate.
New insights into how histone marks are placed at specific genomic loci has been gained through the study of long noncoding RNAs (lncRNA). LncRNAs form complexes with histone-modifying enzymes and guide them to a specific site in the genome in cis (at the site of their transcription) or in trans by binding DNA at a distant genomic site.
Also, small noncoding RNAs, such as microRNAs, play an important role in the regulation of gene expression. MicroRNA is only around 22 nucleotides long and can specifically bind to complimentary regions of messenger RNA (mRNA), thereby inhibiting its translation, i.e. protein production. It is suggested that the human genome encodes more than 25 000 microRNAs. One microRNA can bind to several target mRNAs and in turn one mRNA can be targeted by several microRNAs [23]. Many of these microRNAs are human and tissue specific, which makes them difficult to study in animal models, but also makes them particularly interesting in regard to disease development. In RA and other rheumatic diseases microRNAs have been shown to be altered in different cells types, as well as in serum and synovial fluid, and are considered to influence major disease pathogenic pathways in stromal and immune cells (for review see [24]).
Why epigenetic biomarkers?
Epigenetic modifications have not only been recognised as master regulators of many essential pathways in the cell, but have also been tested as potential biomarkers for disease states and treatment response. Epigenetic biomarkers have mainly been tested in the cancer field [25, 26]. However, in rheumatology we can learn from these studies regarding discovery approaches and measuring techniques, and we can envisage how epigenetic biomarkers could be used in the diagnosis and prognosis of rheumatic diseases.
In contrast to the cancer field, in rheumatology the usage of genetic biomarkers is very limited, since disease development is in the majority of cases not connected with a specific genomic mutation. Clinically relevant genetic markers such as HLA-B27 in spondyloarthritides or the shared epitope in RA can help to support a diagnosis in patients with symptoms. However, more than 95% of HLA-B27 positive individuals will never develop disease and around 30% of the healthy population carries the shared epitope.
One explanation for disease development in only a minority of the risk HLA subtypes might be changes in DNA methylation. Liu et al. identified several differentially methylated regions in the major histocompatibility complex (MHC) region [encoding for human leucocyte antigens (HLA)] in peripheral blood mononuclear cells (PBMCs) of RA patients, which were connected to RA risk single nucleotide polymorphisms (SNPs) [27]. The authors suggest that these changes in methylation mediate the genetic risk for developing RA, at least in regard to the MHC region. In order to see whether more genetic risk loci are affected by differential methylation and thus gain penetrance, other disease-relevant cell types, such as synovial fibroblasts, have also to be analysed in a similar way. Given the relatively weak connection between genetic background and rheumatic diseases, it has long been assumed that genetically predisposed individuals are affected by additional environmental factors that then lead to disease development. If we assume that epigenetic changes are induced by these causative environmental influences, alterations of epigenetic marks might be an early sign of disease development.
A major problem in biomarker discovery, in cancer as well as in rheumatic diseases, is the fact that disease-specific changes are accompanied by a strong nonnspecific inflammatory response [28]. Thus, many factors, such as simple C-reactive protein measurements can differentiate between healthy individuals and cancer patients or RA patients, but they are of little diagnostic, let alone prognostic value due to lack of specificity. Levels of interleukin-6 (IL-6), for instance, are increased in a number of pathological conditions, including RA. In RA patients, however, it could be shown that the promoter region of IL-6 is hypomethylated in PBMCs [29]. Even though elevated IL-6 levels are not specific, this loss of DNA methylation in the IL-6 promoter in PBMCs might be quite specific for RA patients. Unfortunately, epigenetic analysis of mixed cell populations such as PBMCs holds the risk that changes in cell composition are detected rather than changes at specific genomic sites. Furthermore, in this study RA PBMCs were only compared with healthy controls and not with PBMCs from patients with other chronic inflammatory conditions. Nevertheless, this example shows that even though a molecule might lack specificity to be used as biomarker, the reason for its increased expression might be quite specific. We know already that epigenetic changes cause altered expression of many factors in different cell types in rheumatic diseases and the appearance of these epigenetic marks is most probably more specific for rheumatic diseases than the expression of their target molecules. However, well-designed studies to prove this point are urgently needed (fig. 2).
Which epigenetic markers could be useful in rheumatology?
To date, almost all epigenetic biomarkers used in clinics come from cancer research, and DNA methylation marks are the most commonly used [30]. DNA methylation is stable and can even be measured in cell-free DNA in body fluids [31]. As mentioned above, several differentially methylated regions have been described in rheumatic diseases in various cell types. Already in the early 1990s a loss of DNA methylation was described in blood cells and synovial tissues of RA and SLE patients [32, 33]. Many reports of changes in DNA methylation, genome wide and at specific genomic sites, followed [7–9, 11, 12, 34, 35]. The earliest studies measured the total content of methylated cytosine in DNA of synovial fibroblasts and immune cells of RA and SLE patients, and found global DNA hypomethylation, which is similar to findings in cancer cells and most probably due to loss of methylation in repetitive sequences of the genome [7, 32, 33]. Later studies either looked at specific promoter sites, for example synovial fibroblasts of RA patients, or used array technology to analyse thousands of preselected methylation sites mainly in and around coding regions, but also in noncoding regions or regions encoding for microRNA [8, 9, 11, 12, 34, 36]. These studies found hypomethylated and hypermethylated regions, which points to a general disturbance of the epigenetic machinery in RA similar to findings in cancer cells [6]. In systemic sclerosis also, hypo- and hypermethylated regions were found in dermal fibroblasts and blood cells [37–39]. Interestingly, in blood cells from SLE patients, hypermethylation events are rarely found and hypomethylation seems to predominate [13]. This is also in line with studies showing that treatment of T cells with DNA demethylating agents can induce lupus-like symptoms in mice [40]. These results point towards a causative role for epigenetic changes in lupus. Studies in synovial fibroblasts from RA patients at a very early stage of disease also support the hypothesis that loss of DNA methylation, at least in synovial fibroblasts, has already occurred at a time point when destructive processes are not yet manifest and therefore could be causative [35, 41]. While these studies showed promising results, the question whether epigenetic changes are causes or consequences of chronic inflammation is not yet solved and the usage of DNA methylation as diagnostic biomarker in rheumatic diseases must be further followed up. Particularly needed are studies that analyse DNA methylation in peripheral blood cells from patients in the early stages of rheumatic diseases where late changes in DNA methylation have already been shown, and comparisons of DNA methylation not with healthy controls but with other rheumatic diseases. One such study recently showed that measurement of DNA methylation in the IFI44L promoter in whole blood could separate SLE patients from RA and systemic sclerosis patients with a sensitivity and specificity of around 90% [42].
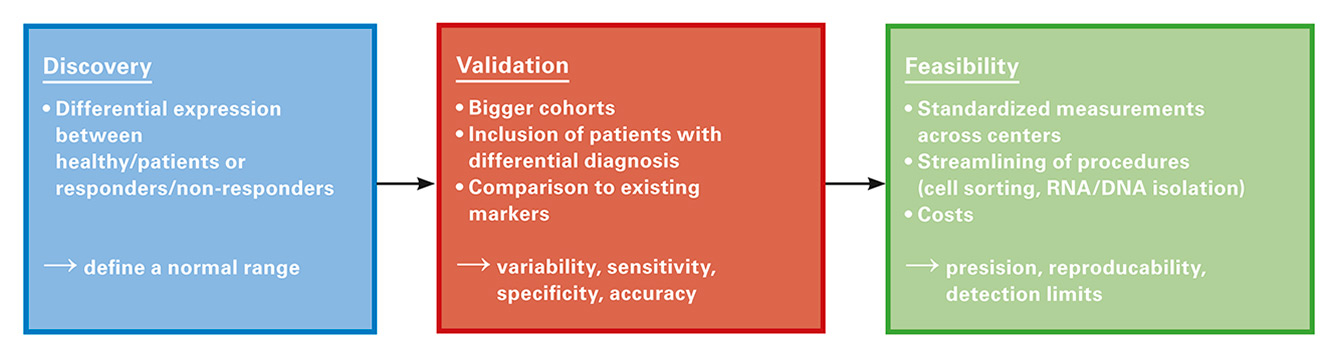
Figure 2
The development of a clinically useful biomarker requires several separate studies and analysis. Unfortunately in rheumatology most of the biomarker studies are in the discovery phase, but have neither been validated nor tested for their feasibility.
Recent studies also explored changes in DNA methylation as biomarker for treatment response [20, 43, 44]. None of the currently used therapies for RA are effective in all patients. In practice this means several months of expensive treatment with potential severe side effects and with no benefit for a substantial percentage of RA patients. There is an urgent need for biomarkers to stratify these patients before initiation of treatment, or at least to detect response faster than the currently used clinical measures. The fact that methotrexate treatment was found to influence DNA methylation gives hope that assessing affected DNA loci before and after treatment might be useful as predictor and/or early measure for response to treatment [20, 44].
Also certain histone modifications and histone-modifying enzymes were found to be altered in rheumatic diseases [45]. However, at the moment histone modifications at specific sites are still technically much more difficult to measure than DNA methylation. It is also not clear how stable these histone modifications are, throughout the disease as well as in the biological sample that is used for testing. Ideally a biomarker should be as robust as possible and reliably measurable with different methods and in different settings. Therefore histone modifications do not appear as ideal candidates for biomarkers at the moment.
MicroRNAs on the other hand fulfil most of the requirements of a biomarker. Their expression has been shown to be tissue- and disease-specific, they are very stable, and can easily be measured in tissues as well as body fluids by polymerase chain reaction (PCR) or array technology [24, 46]. Not surprisingly, in rheumatic diseases many of the microRNAs that are known to be induced or inhibited by an inflammatory environment, so called ‘inflamma-miRs’ such as miR-155 and miR-146a have been shown to be altered [47–51]. MicroRNA profiles measured in the serum/plasma of patients show significant changes of various microRNAs in RA, as well as in SLE, juvenile idiopathic arthritis and in systemic sclerosis [52–55]. Whether their expression levels are sufficiently sensitive and specific for them to be used as biomarkers remains to be proven, since the studies conducted up to now were not designed to validate microRNAs as diagnostic biomarkers.
Several recent studies identified microRNAs that predicted therapeutic response in RA patients [56–58]. Krintel et al. showed that patients with low levels of miR-22 and high levels of miR-886-3p, measured in whole blood before initiation of therapy, can benefit more from treatment with adalimumab [57]. In the study of Duroux-Richard et al., high levels of miR-125b in whole blood at baseline predicted good response to rituximab treatment [58]. In both studies high/low expression in responders was defined as significantly different from that of nonresponders. However, for the use of microRNA as biomarker in an individual patient a clear separation of expression of the microRNA to a previously defined normal range of expression must be given. Such analyses have not been done in any study so far. Also, bigger cohorts are needed to validate these published results.
References
1 Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev. 2009;23(7):781–3.
2 Bird A. Perceptions of epigenetics. Nature. 2007;447(7143):396–8.
3 Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358(11):1148–59.
4 Koch MW, Metz LM, Kovalchuk O. Epigenetic changes in patients with multiple sclerosis. Nat Rev Neurol. 2013;9(1):35–43.
5 Klein K, Gay S. Epigenetics in rheumatoid arthritis. Curr Opin Rheumatol. 2015;27(1):76–82.
6 Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics. 2009;1(2):239–59.
7 Karouzakis E, Gay RE, Michel BA, Gay S, Neidhart M. DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2009;60(12):3613–22.
8 Karouzakis E, Rengel Y, Jungel A, Kolling C, Gay RE, Michel BA, et al. DNA methylation regulates the expression of CXCL12 in rheumatoid arthritis synovial fibroblasts. Genes Immun. 2011;12(8):643–52.
9 Takami N, Osawa K, Miura Y, Komai K, Taniguchi M, Shiraishi M, et al. Hypermethylated promoter region of DR3, the death receptor 3 gene, in rheumatoid arthritis synovial cells. Arthritis Rheum. 2006;54(3):779-87.
10 McCabe MT, Brandes JC, Vertino PM. Cancer DNA methylation: molecular mechanisms and clinical implications. Clin Cancer Res. 2009;15(12):3927–37.
11 Whitaker JW, Shoemaker R, Boyle DL, Hillman J, Anderson D, Wang W, et al. An imprinted rheumatoid arthritis methylome signature reflects pathogenic phenotype. Genome Med. 2013;5(4):40.
12 Nakano K, Whitaker JW, Boyle DL, Wang W, Firestein GS. DNA methylome signature in rheumatoid arthritis. Ann Rheum Dis. 2013;72(1):110–7.
13 Absher DM, Li X, Waite LL, Gibson A, Roberts K, Edberg J, et al. Genome-wide DNA methylation analysis of systemic lupus erythematosus reveals persistent hypomethylation of interferon genes and compositional changes to CD4+ T-cell populations. PLoS genetics. 2013;9(8):e1003678.
14 Altorok N, Coit P, Hughes T, Koelsch KA, Stone DU, Rasmussen A, et al. Genome-wide DNA methylation patterns in naive CD4+ T cells from patients with primary Sjogren’s syndrome. Arthritis Rheumatol. 2014;66(3):731–9.
15 Furumitsu Y, Yukioka K, Kojima A, Yukioka M, Shichikawa K, Ochi T, et al. Levels of urinary polyamines in patients with rheumatoid arthritis. J Rheumatol. 1993;20(10):1661–5.
16 Karouzakis E, Gay RE, Gay S, Neidhart M. Increased recycling of polyamines is associated with global DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2012;64(6):1809–17.
17 Gerner EW, Meyskens FL, Jr. Polyamines and cancer: old molecules, new understanding. Nat Rev Cancer. 2004;4(10):781–92.
18 Wu H, Zhang Y. Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell. 2014;156(1-2):45–68.
19 Cimmino L, Abdel-Wahab O, Levine RL, Aifantis I. TET family proteins and their role in stem cell differentiation and transformation. Cell Stem Cell. 2011;9(3):193–204.
20 de Andres MC, Perez-Pampin E, Calaza M, Santaclara FJ, Ortea I, Gomez-Reino JJ, et al. Assessment of global DNA methylation in peripheral blood cell subpopulations of early rheumatoid arthritis before and after methotrexate. Arthritis Res Ther. 2015;17:233.
21 Quintero-Ronderos P, Montoya-Ortiz G. Epigenetics and autoimmune diseases. Autoimmune Dis. 2012;2012:593720.
22 Sandoval J, Esteller M. Cancer epigenomics: beyond genomics. Curr Opin Genet Dev. 2012;22(1):50–5.
23 Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM, et al. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126(6):1203–17.
24 Vicente R, Noel D, Pers YM, Apparailly F, Jorgensen C. Deregulation and therapeutic potential of microRNAs in arthritic diseases. Nat Rev Rheumatol. 2015.
25 Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003.
26 Weller M, Felsberg J, Hartmann C, Berger H, Steinbach JP, Schramm J, et al. Molecular predictors of progression-free and overall survival in patients with newly diagnosed glioblastoma: a prospective translational study of the German Glioma Network. J Clin Oncol. 2009;27(34):5743–50.
27 Liu Y, Aryee MJ, Padyukov L, Fallin MD, Hesselberg E, Runarsson A, et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol. 2013;31(2):142–7.
28 Chechlinska M, Kowalewska M, Nowak R. Systemic inflammation as a confounding factor in cancer biomarker discovery and validation. Nat Rev Cancer. 2010;10(1):2–3.
29 Nile CJ, Read RC, Akil M, Duff GW, Wilson AG. Methylation status of a single CpG site in the IL6 promoter is related to IL6 messenger RNA levels and rheumatoid arthritis. Arthritis Rheum. 2008;58(9):2686–93.
30 Mikeska T, Craig JM. DNA methylation biomarkers: cancer and beyond. Genes (Basel). 2014;5(3):821–64.
31 Schwarzenbach H, Hoon DS, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer. 2011;11(6):426–37.
32 Richardson B, Scheinbart L, Strahler J, Gross L, Hanash S, Johnson M. Evidence for impaired T cell DNA methylation in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum. 1990;33(11):1665–73.
33 Corvetta A, Della Bitta R, Luchetti MM, Pomponio G. 5-Methylcytosine content of DNA in blood, synovial mononuclear cells and synovial tissue from patients affected by autoimmune rheumatic diseases. J Chromatogr. 1991;566(2):481–91.
34 Karouzakis E, Trenkmann M, Gay RE, Michel BA, Gay S, Neidhart M. Epigenome Analysis Reveals TBX5 as a Novel Transcription Factor Involved in the Activation of Rheumatoid Arthritis Synovial Fibroblasts. J Immunol. 2014;193(10):4945–51.
35 Ai R, Whitaker JW, Boyle DL, Tak PP, Gerlag DM, Wang W, et al. DNA Methylome Signature in Synoviocytes From Patients With Early Rheumatoid Arthritis Compared to Synoviocytes From Patients With Longstanding Rheumatoid Arthritis. Arthritis Rheumatol. 2015;67(7):1978–80.
36 de la Rica L, Urquiza JM, Gomez-Cabrero D, Islam AB, Lopez-Bigas N, Tegner J, et al. Identification of novel markers in rheumatoid arthritis through integrated analysis of DNA methylation and microRNA expression. J Autoimmun. 2013;41:6–16.
37 Altorok N, Tsou PS, Coit P, Khanna D, Sawalha AH. Genome-wide DNA methylation analysis in dermal fibroblasts from patients with diffuse and limited systemic sclerosis reveals common and subset-specific DNA methylation aberrancies. Ann Rheum Dis. 2015;74(8):1612–20.
38 Dees C, Schlottmann I, Funke R, Distler A, Palumbo-Zerr K, Zerr P, et al. The Wnt antagonists DKK1 and SFRP1 are downregulated by promoter hypermethylation in systemic sclerosis. Ann Rheum Dis. 2014;73(6):1232–9.
39 Lian X, Xiao R, Hu X, Kanekura T, Jiang H, Li Y, et al. DNA demethylation of CD40l in CD4+ T cells from women with systemic sclerosis: a possible explanation for female susceptibility. Arthritis Rheum. 2012;64(7):2338–45.
40 Quddus J, Johnson KJ, Gavalchin J, Amento EP, Chrisp CE, Yung RL, et al. Treating activated CD4+ T cells with either of two distinct DNA methyltransferase inhibitors, 5-azacytidine or procainamide, is sufficient to cause a lupus-like disease in syngeneic mice. J Clin Invest. 1993;92(1):38–53.
41 Karouzakis E, Filer A, Eyre S, Raza K, Kolling C, Gay RE, et al. Genetic and Epigenetic Mapping of Very Early RA Synovial Fibroblasts [abstract]. Arthritis Rheumatol. 2015;67(supplement 10).
42 Zhao M, Zhou Y, Zhu B, Wan M, Jiang T, Tan Q, et al. IFI44L promoter methylation as a blood biomarker for systemic lupus erythematosus. Ann Rheum Dis. 2016.
43 Plant D, Webster A, Nair N, Oliver J, Smith S, Eyre S, et al. Differential methylation as a biomarker of response to etanercept in patients with rheumatoid arthritis. Arthritis Rheumatol. 2016. [epub ahead of print].
44 Cribbs AP, Kennedy A, Penn H, Amjadi P, Green P, Read JE, et al. Methotrexate Restores Regulatory T Cell Function Through Demethylation of the FoxP3 Upstream Enhancer in Patients With Rheumatoid Arthritis. Arthritis Rheumatol. 2015;67(5):1182–92.
45 Gay S, Wilson AG. The emerging role of epigenetics in rheumatic diseases. Rheumatology (Oxford). 2014;53(3):406–14.
46 Liang Y, Ridzon D, Wong L, Chen C. Characterization of microRNA expression profiles in normal human tissues. BMC Genomics. 2007;8:166.
47 Stanczyk J, Pedrioli DM, Brentano F, Sanchez-Pernaute O, Kolling C, Gay RE, et al. Altered expression of MicroRNA in synovial fibroblasts and synovial tissue in rheumatoid arthritis. Arthritis Rheum. 2008;58(4):1001–9.
48 Kyburz D, Karouzakis E, Ospelt C. Epigenetic changes: the missing link. Best Pract Res Clin Rheumatol. 2014;28(4):577–87.
49 Olivieri F, Rippo MR, Procopio AD, Fazioli F. Circulating inflamma-miRs in aging and age-related diseases. Front Genet. 2013;4:121.
50 Tang Y, Luo X, Cui H, Ni X, Yuan M, Guo Y, et al. MicroRNA-146A contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis RheumArthritis. 2009;60(4):1065–75.
51 Duroux-Richard I, Jorgensen C, Apparailly F. miRNAs and rheumatoid arthritis - promising novel biomarkers. Swiss Med Wkly. 2011;141:w13175.
52 Carlsen AL, Schetter AJ, Nielsen CT, Lood C, Knudsen S, Voss A, et al. Circulating microRNA expression profiles associated with systemic lupus erythematosus. Arthritis RheumArthritis. 2013;65(5):1324–34.
53 Steen SO, Iversen LV, Carlsen AL, Burton M, Nielsen CT, Jacobsen S, et al. The circulating cell-free microRNA profile in systemic sclerosis is distinct from both healthy controls and systemic lupus erythematosus. J Rheumatol. 2015;42(2):214–21.
54 Murata K, Furu M, Yoshitomi H, Ishikawa M, Shibuya H, Hashimoto M, et al. Comprehensive microRNA analysis identifies miR-24 and miR-125a-5p as plasma biomarkers for rheumatoid arthritis. PLoS One. 2013;8(7):e69118.
55 Kamiya Y, Kawada J, Kawano Y, Torii Y, Kawabe S, Iwata N, et al. Serum microRNAs as Potential Biomarkers of Juvenile Idiopathic Arthritis. Clin Rheumatol. 2015;34(10):1705–12.
56 Castro-Villegas C, Perez-Sanchez C, Escudero A, Filipescu I, Verdu M, Ruiz-Limon P, et al. Circulating miRNAs as potential biomarkers of therapy effectiveness in rheumatoid arthritis patients treated with anti-TNFalpha. Arthritis Res Ther. 2015;17:49.
57 Krintel SB, Dehlendorff C, Hetland ML, Horslev-Petersen K, Andersen KK, Junker P, et al. Prediction of treatment response to adalimumab: a double-blind placebo-controlled study of circulating microRNA in patients with early rheumatoid arthritis. Pharmacogenomics J. 2015.
58 Duroux-Richard I, Pers YM, Fabre S, Ammari M, Baeten D, Cartron G, et al. Circulating miRNA-125b is a potential biomarker predicting response to rituximab in rheumatoid arthritis. Mediators Inflamm. 2014;2014:342524.
59 Warton K, Samimi G. Methylation of cell-free circulating DNA in the diagnosis of cancer. Front Mol Biosci. 2015;2:13.