Amyloid-β pathology and cerebral amyloid angiopathy are frequent in iatrogenic Creutzfeldt-Jakob disease after dural grafting
DOI: https://doi.org/10.4414/smw.2016.14287
Karl
Frontzek, Mirjam I.
Lutz, Adriano
Aguzzi, Gabor G.
Kovacs, Herbert
Budka
Summary
QUESTIONS UNDER STUDY: Alzheimer-type amyloid-β (Aβ) pathology was reported in brains of individuals developing iatrogenic Creutzfeldt-Jakob disease (iCJD) after treatment with human cadaveric growth hormone, and interpreted as evidence of human transmission of Aβ by the treatment. Here we investigated the prevalence of Aβ pathology in other instances of iCJD related to dura mater grafts.
METHODS: By use of immunohistochemistry for Aβ, we investigated seven brains of patients (age range 28–63) who succumbed to iCJD after dural grafting, which had been applied by means of neurosurgery between 11 and 25 years before death. For control, we examined a series of 21 brains of age-matched (40–63 years) patients with sporadic CJD (sCJD) and an additional series of 81 sCJD cases (55–85 years) with the same methods.
RESULTS: In five of seven iCJD brains, Aβ was deposited in meningeal vessels as congophilic amyloid angiopathy and brain parenchymal plaques. This was significantly (p <0.001) more frequent than in the age-matched sCJD controls and in the usual sCJD series.
CONCLUSIONS: We conclude that congophilic amyloid angiopathy and brain parenchymal Aβ plaques are frequent in iCJD after dural grafting. The presence of Aβ pathology in young individuals is highly unusual and suggests a causal relationship to the dural grafts. Further studies will be needed to elucidate whether such pathology resulted from the seeding of Aβ aggregates from the grafts to host tissues.
Introduction
Many neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinsons’s disease (PD), polyglutamine expansion diseases and prion diseases share fundamental pathogenetic similarities. In each of these diseases, proteins coalesce to form self-propagating oligomers that can recruit further monomers and elongate to form higher-order aggregates [1]. Such aggregates can then break, thereby multiplying their numbers [2]. This phenomenon is thought to represent the basis for the infectivity of prions [1, 3].
In prion diseases, the seeded propagation of the pathological, “scrapie” conformer of the prion protein PrPSc is dependent on the presence of its cellular counterpart PrPC [4]. During the pathogenesis of AD, Aβ displays behaviour similar to that described above, raising the legitimate question whether it may also behave as a prion and be infectious. Indeed, thorough experiments with transgenic mice overexpressing Aβ have repeatedly shown that Aβ aggregates (derived from mice or from humans) may be propagated in a prion-like manner [5–8]. Similar observations have been made for tau [9, 10], the other major protein involved in AD. Likewise, it was found that injection of α-synuclein aggregates in transgenic and even in wild-type mice can provoke a spreading pathology similar to that observed in human synucleinopathies [11–14]. However, epidemiological surveillance has thus far failed to detect any correlation between iatrogenic manipulations, which may conceivably introduce Aβ or α-synuclein seeds, and the development of AD or PD.
Recently, Jaunmuktane et al. [15] reported Alzheimer-type gray matter and vascular Aβ pathology in four of eight brains of individuals with iatrogenic Creutzfeldt-Jakob disease (iCJD) after treatment with human growth hormone (hGH) preparations from cadaveric pituitary glands. They interpreted their data as evidence of human transmission of Aβ by the treatment and suggested that healthy exposed individuals may be at risk of iatrogenic Alzheimer’s disease. These findings have raised the question whether other known iatrogenic routes of prion transmission may also be relevant to Aβ and other proteopathic seeds [15]. Another major transmission route known in iCJD has been the use of dural grafts during neurosurgery in the 1980s and early 1990s that resulted in more than 220 iCJD cases, a number similar to that caused by hGH treatment [3].
To answer this question, we have retrospectively analysed iCJD patients from two National Prion Reference Centres (namely Austria and Switzerland) and assessed brain specimens histologically for the presence or absence of AD-associated pathology.
Methods
We performed immunohistochemistry for Aβ on brains of seven individuals with autopsy-confirmed iCJD (age range 28–63) after dural grafting, including two cases previously reported [16, 17]. Dural transplants (Lyodura, Braun-Melsungen, whenever specified) had been applied by neurosurgery between 11 and 25 years before death. Patient details are provided in table 1.
|
Table 1: Clinical details of patients suffering from iatrogenic CJD and indications for dural grafting. |
|
ID
|
Age
(years)
|
Gender
|
Aβ-CAA
|
Parenchymal
Aβ
|
Years from surgeryto symptoms
|
Years from surgeryto death
|
Indication for dural grafting
|
| 1 |
28 |
M |
+ |
+ |
21 |
22 |
Open craniocerebral injury |
| 2 |
33 |
F |
+ |
+ |
25 |
25 |
Craniocerebral injury |
| 3 |
47 |
M |
+ |
+ |
21 |
21 |
Brain trauma |
| 4 |
51 |
M |
– |
– |
12 |
12 |
Not available |
| 5 |
52 |
F |
+ |
+ |
22 |
22 |
Pituitary adenoma |
| 6 |
59 |
M |
– |
– |
11 |
11 |
Open craniocerebral injury |
| 7 |
63 |
F |
+ |
+ |
23 |
23 |
Meningeoma |
| Aβ-CAA = cerebral amyloid angiopathy with Aβ depositions; parenchymal Aβ = brain parenchymal Aβ depositions |
At least three different cortical and other brain regions were investigated. Monoclonal anti-Aβ antibodies 6F/3D from DAKO, Glostrup, Denmark, or 4G8 from Signet, Dedham, MA, USA, were used after paraffin section pretreatment with formic acid 80% for 1 hour or 100% for 5 minutes.
Each iCJD case was age-matched to three cases of sporadic CJD (sCJD) from the Swiss or Austrian National Reference Centres for Human Prion Diseases (age 40–63 years) (fig. 1B). As additional controls, Aβ pathology was studied in 81 sCJD cases in the expected age range (namely 55–85 years) [18] (fig. 1B) in the Austrian cohort.
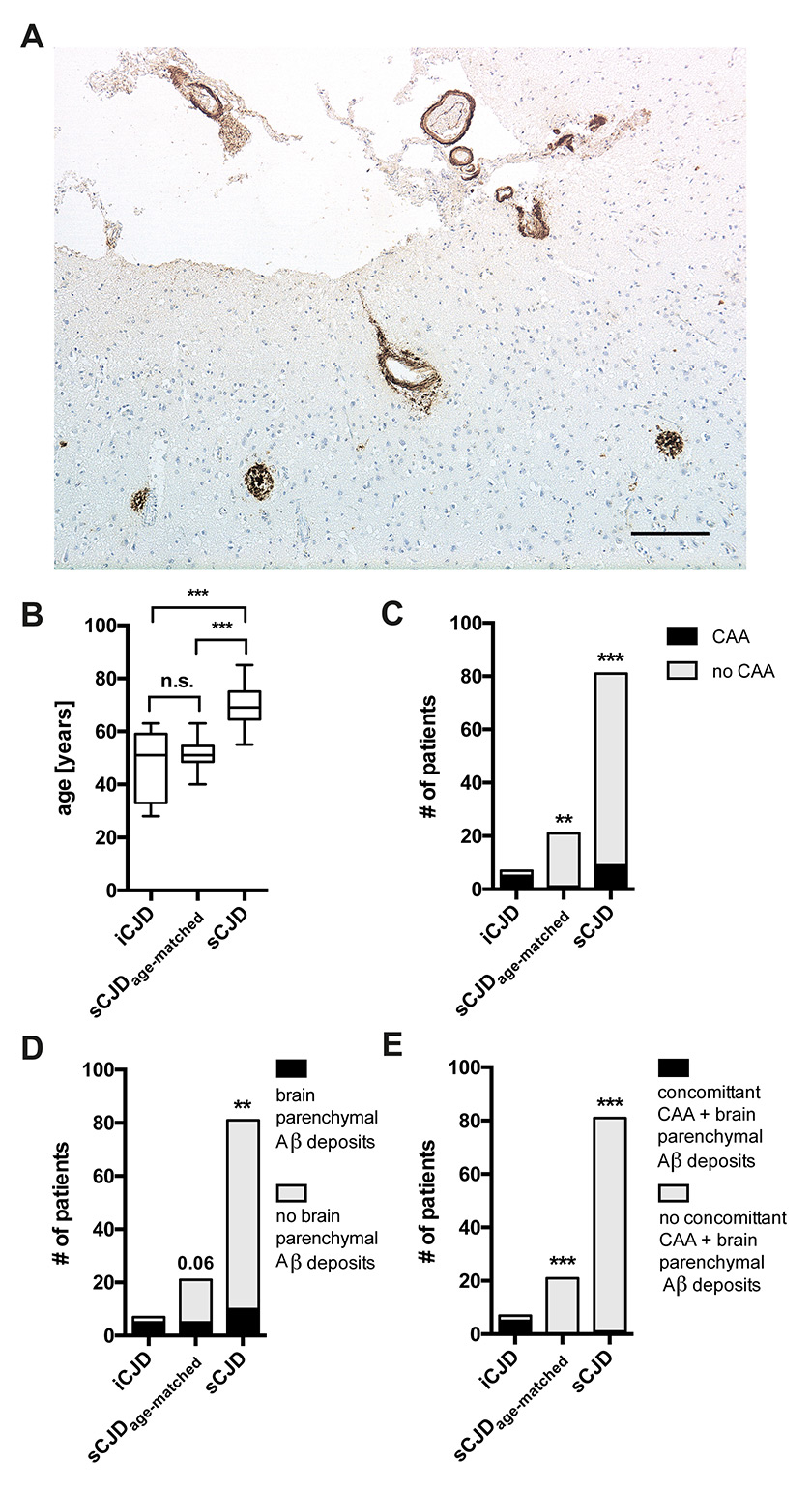
Figure 1
A: Immunohistochemistry for Aβ demonstrates prominent deposits (brown) in vessel walls and plaques in frontal cortex of iatrogenic CJD (iCJD) case (age 33 years). Scale bar = 200 μm.
B–E: Statistical comparisons of iCJD, age-matched sporadic CJD (sCJD) and non-age-matched sCJD for age (B), cerebral amyloid angiopathy (CAA) (C), brain parenchymal deposits (D), and concomitant CAA and brain deposits (E). B: Bars are median + 25%/75% percentile (box) and min/max (whiskers). ** p <0.01, *** p <0.001.
Statistical comparisons were made by one-way analysis of variance with Bonferroni correction (all groups vs all groups, fig. 1B) and Fisher’s exact test with Bonferroni correction (fig. 1C–E).
This study was approved by the Cantonal Ethics Commission of Zurich (KEK-ZH-Nr. 2015-0672) and the Ethics Commission of the Medical University Vienna (396/2011).
Results
Aβ was detectable in meningeal vessels as cerebral amyloid angiopathy (CAA) and as plaques in gray matter of five of seven iCJD brains (71%, age 28, 33, 47, 52, 63 years) (fig. 1A), but in only one as CAA (5%, age 55) and in five as parenchymal deposits (24%, age 49, 51, 62, 62, 63) of the 21 age-matched sCJD controls (fig. 1C & D). Of note, no conspicuous tau pathology was observed in all iCJD patients. Moreover, all five iCJD brains with Aβ pathology showed the combined presence of CAA and brain Aβ depositions, while age-matched sCJD cases showed either CAA or parenchymal Aβ (fig. 1E). The larger sCJD series showed CAA in 9/81 (11%), brain Aβ deposits in 9/81 other cases, and both in one more case (fig. 1C–E). Statistically, CAA was significantly more frequent in iCJD than in age-matched sCJD (p <0.01) and sCJD (p <0.001); brain parenchymal deposits were significantly more frequent in iCJD than in sCJD (p <0.01); and their combination was significantly more frequent in iCJD than in both age-matched sCJD and sCJD (p <0.001).
Discussion
We report here that CAA and brain parenchymal Aβ plaques are frequent in iCJD after dural grafting, even in young individuals. Similarly to what was previously reported in iCJD after hGH treatment [15], we failed to detect any marked tau pathology in our series after dural grafting. The presence of Aβ pathology in young individuals who present with neither a family history of early-onset dementia or prominent AD-related tau pathology is highly unusual and suggests a causal relationship to the dural grafts [19]. It is plausible that such pathology may have resulted from the seeding of Aβ aggregates from the grafts to host tissues, yet alternative explanations are also possible.
The Aβ pathology was observed many years after neurosurgery that applied a graft of dura mater. It is intriguing that all cases with particularly long intervals after dural grafting (more than 20 years) were the five who had Aβ pathology, whereas the two brains without Aβ had much shorter intervals of 11 and 12 years, respectively. This does not seem to be a function of age, as both cases without Aβ had ages in the 50s, whereas Aβ brains included three cases younger than 50. Such prolonged incubation over decades would be another striking similarity with prion diseases. As data on the site of the applied dural graft were not available for all cases, we were unable to investigate conclusively whether the severity of the induced Aβ pathology had a topographic relationship to the site of grafting. For the same reason, it was not possible to assert any local difference between meningeal vs parenchymal Aβ according to graft site.
The clinical signs and symptoms in all patients reported here were typical of CJD [3]; there was no report of previous mild or slowly progressive cognitive impairment that might have been the result of Aβ pathology prior to the onset of rapidly progressive iCJD. All brains had prominent and widespread deposition of PrPSc; in comparison, Aβ was less prominent. Thus, any striking local co-occurrence suggestive of potential cross-seeding was not discernible.
The findings reported here extend a previous study of iCJD after hGH treatment [15] and suggest that both human dural tissue grafts and pituitary extracts are able to elicit Aβ pathology decades later. This would be in agreement with ample evidence of prion-like propagation of aggregated proteins in animal models of neurodegeneration [8]. However, as discussed previously [16], it is currently impossible to eliminate the possibility that head trauma or the underlying conditions which had led to dural grafting, or neurosurgery, may have contributed to the induction of Aβ pathology.
A previous study [20] demonstrated the presence of Aβ in human pituitary tissue, the source of prion-contaminated hGH preparations. However, no clinically manifest cases of AD or PD were identified among recipients of pituitary-derived hGH in review of the large US National Hormone and Pituitary Program cohort database [20]. Hence, further studies are needed to elucidate whether potential transmission and propagation of Aβ – and of other neurodegeneration-related proteins – from external sources is indeed able to induce a clinically manifest human disease.
Whilst the iatrogenic transmission of aggregated Aβ is one of several possible explanations for the findings reported here, the growing circumstantial evidence for such transmission should prompt a critical re-evaluation of the decontamination procedures for surgical instruments and drugs of biological origin, with the goal to ensure the complete absence of potentially transmissible contaminants.
References
1 Ashe KH, Aguzzi A. Prions, prionoids and pathogenic proteins in Alzheimer disease. Prion. 2013;7(1):55–9.
2 Knowles TPJ, Waudby CA, Devlin GL, Cohen SIA, Aguzzi A, Vendruscolo M, et al. An analytical Solution to the kinetics of breakable filament assembly. Science. 2009;326(5959):1533–7.
3 Budka H, Will RG. The end of the BSE saga: do we still need surveillance for human prion diseases? Swiss Med Wkly. 2015;145:w14212.
4 Büeler H, Aguzzi A, Sailer A, Greiner R-A, Autenried P, Aguet M, et al. Mice devoid of PrP are resistant to scrapie. Cell. 1993;73:1–20.
5 Eisele YS, Obermüller U, Heilbronner Gt, Baumann F, Kaeser SA, Wolburg H, et al. Peripherally applied Aβ-containing inoculates induce cerebral β-amyloidosis. Science. 2010;330(6006):980–2.
6 Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. 2013;501(7465):45–51.
7 Prusiner SB. Biology and genetics of prions causing neurodegeneration. Annu Rev Genet. 2013;47:601–23.
8 Jucker M, Walker LC. Neurodegeneration: Amyloid-β pathology induced in humans. Nature. 2015;525(7568):193–4.
9 Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench Jr, et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A. 2013;110(23):9535–40.
10 Lewis J, Dickson DW. Propagation of tau pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol. 2016;131(1):27–48.
11 Lee S-J, Masliah E. Neurodegeneration: Aggregates feel the strain. Nature. 2015;522(7556):296–7.
12 Bae E-J, Yang N-Y, Song M, Lee CS, Lee JS, Jung BC, et al. Glucocerebrosidase depletion enhances cell-to-cell transmission of α-synuclein. Nat Commun. 2014;5.
13 Amschl D, Neddens J, Havas D, Flunkert S, Rabl R, Römer H, et al. Time course and progression of wild type α-Synuclein accumulation in a transgenic mouse model. BMC Neurosci. 2013;14(1):1–14.
14 Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ, et al. Pathological α-synuclein transmission initiates parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338(6109):949–53.
15 Jaunmuktane Z, Mead S, Ellis M, Wadsworth JDF, Nicoll AJ, Kenny J, et al. Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature. 2015;525(7568):247–50.
16 Preusser M, Ströbel T, Gelpi E, Eiler M, Broessner G, Schmutzhard E, et al. Alzheimer-type neuropathology in a 28 year old patient with iatrogenic Creutzfeldt-Jakob disease after dural grafting. J Neurol Neurosurg Psychiatry. 2006;77(3):413–6.
17 Frontzek K, Moos R, Schaeper E, Jann L, Herfs G, et al. Iatrogenic and sporadic Creutzfeldt-Jakob disease in two sisters without mutation in the prion protein gene. Prion. 2015;9(6):444–8.
18 Ladogana A, Puopolo M, Croes EA, Budka H, Jarius C, Collins S, et al. Mortality from Creutzfeldt-Jakob disease and related disorders in Europe, Australia, and Canada. Neurology. 2005;64(9):1586–91.
19 Schellenberg GD, Montine TJ. The genetics and neuropathology of Alzheimer’s disease. Acta Neuropathol. 2012;124(3):305–23.
20 Irwin DJ, Abrams JY, Schonberger LB, Leschek EW, Mills JL, Lee VM, et al. Evaluation of potential infectivity of Alzheimer and Parkinson disease proteins in recipients of cadaver-derived human growth hormone. JAMA Neurol. 2013;70(4):462–8.