Targeting the RAS pathway by mitogen-activated protein kinase inhibitors
DOI: https://doi.org/10.4414/smw.2015.14207
Michael
Kiessling, Gerhard
Rogler
Summary
Targeting of oncogenic driver mutations with small-molecule inhibitors resulted in powerful treatment options for cancer patients in recent years. The RAS (rat sarcoma) pathway is among the most frequently mutated pathways in human cancer. Whereas targeting mutant Kirsten RAS (KRAS) remains difficult, mutant B rapidly accelerated fibrosarcoma (BRAF) kinase is an established drug target in cancer. Now data show that neuroblastoma RAS (NRAS) and even Harvey RAS (HRAS) mutations could be predictive markers for treatment with mitogen-activated protein kinase (MEK) inhibitors. This review discusses recent preclinical and clinical studies of MEK inhibitors in BRAF and RAS mutant cancer.
RAS signalling pathway
RAS (rat sarcoma) kinases are evolutionarily conserved kinases that are important within the RAS–RAF–MEK–ERK pathway. Up to now, three members have been identified: neuroblastoma RAS (NRAS), Harvey RAS (HRAS), and Kirsten RAS (KRAS). The RAS family of small GTPases is stimulated either by upstream receptors or activating mutations. Upon activation, RAS changes its form from the inactive (GDP-bound) to the active (GTP-bound) form [1]. This process is regulated by guanine nucleotide exchange factors (GEFs) that favour the GDP to GTP exchange. The resulting conformational change leads to binding to rapidly accelerated fibrosarcoma (RAF) kinase, the first kinase downstream of RAS, and subsequent RAF dimerisation. Different forms of RAF kinases are known: ARAF, BRAF and CRAF (RAF-1). Activated RAF phosphorylates and activates the downstream kinase mitogen-activated protein kinase kinase (MEK), which in turn phosphorylates and activates the extracellular signal-regulated kinase (ERK) [1]. Phosphorylated ERK can translocate to the nucleus where it phosphorylates and activates various transcription factors. These include members of the ETS (E26 transformation-specific) family of transcription factors such as ELK1, which stimulates FOS expression. Further, ERK phosphorylates c-JUN leading to the formation of heterodimers of c-JUN and FOS. These heterodimers enable the expression of cell cycle proteins such as cyclines [2]. The RAS–RAF–MEK–ERK pathway has also been shown to activate other effector pathways like the phosphatidylinositol 3-kinase (PI3K) pathway [3]. Mutant RAS in particular was shown to activate the PI3K/mammalian target of rapamycin (mTOR) signalling [3]. Whether the PI3K pathway alone or in combination with the RAS pathway would provide a promising target for cancer therapy is currently under preclinical and early preclinical investigation [4].
The RAS–RAF–MEK–ERK pathway can be inactivated at level of the RAS kinases. Inactivation is secured by GTPase-activating proteins (GAPs) which lead to GTP hydrolysis and, thus, shift conformation to the inactive GDP-bound form [5]. This inactivation process is the target for mutations found in cancers. RAS mutations commonly occur at residues G12 and G13, which prevent binding between RAS and GAP and finally prevent hydrolysis of GTP [5]. Mutations at Q61 interfere with GTP hydrolysis directly by disturbing the orientation of a water molecule needed for hydrolysis. The consequence of these mutations is that the RAS kinase is kept in its active state, leading to continuous activation of the subsequent downstream effectors, which is often referred to as hyperactivation.
Common mutations in the RAS pathway
First, NRAS mutations were discovered in 1983 on chromosome 1 of neuroblastoma tumors; this was then followed by identification of KRAS and HRAS [6]. NRAS, HRAS and KRAS are mutated to a very different frequency depending on tissue background (table 1). KRAS mutations are predominantly found in pancreatic cancer (57%), in small and large intestinal cancers (34% and 23%, respectively) and lung cancer (17%) [7]. Interestingly, almost all mutations for pancreatic, colon, and lung cancer do occur at position G12 and very rarely at position G13 or Q61 (7).
NRAS mutations are often found in melanomas (11%) and tumours of the haematopoietic and lymphoid system (10%). Nearly all mutations of NRAS in melanoma are found at position Q61, whereas in haematopoietic and lymphoid tumours the mutations are distributed between G12, G13 and Q61 [7].
HRAS mutations are predominantly found in bladder cancer (9%), upper digestive tract (6%), and skin and cervix cancer (16% and 5%, respectively) (table 1) [7]. Whereas HRAS mutations in cervix cancer are predominantly found at G12, bladder, skin and upper digestive tract cancers show a more diverse pattern.
Of note, mutations of BRAF, KRAS, and NRAS are usually mutually exclusive whereas mutations of the PI3K pathway may overlap with mutations in BRAF, KRAS, and NRAS [8].
The reason for the varying distribution of NRAS, HRAS and KRAS mutations between different cancer types and the complex pattern of hotspot mutations is not well understood so far. Some in-vitro and preclinical data show that one RAS mutation might substitute for another, whereas other studies report the opposite. These studies will be further discussed in the following paragraph.
In a carcinogen-induced mouse tumour model, it was demonstrated that HRAS can fully substitute for KRAS Q61L, indicating that both have comparable capacity to promote tumour growth [9]. The authors also found that specificity of KRAS mutations for lung cancer or HRAS mutations for skin cancer is dependent on regulatory elements of the corresponding gene [9]. This shows that tissue-specific regulated expression and tissue-specific regulatory elements might be the reason if KRAS, NRAS or HRAS are undergoing mutations. However, in a colon cancer mouse model, only KRAS G12D, but not NRAS G12D, stimulated proliferation of colonic epithelial cells [10]. In addition, another study in a tumour mouse model observed that NRAS G12D could not equal the aggressive myeloproliferative disorder caused by the KRAS G12D mutation [11]. Although NRAS G12D mice ultimately died of haematological cancers, they did not develop as aggressive a course of leukaemic disease as KRAS G12D mice did [11]. A recent publication showed that only the NRAS Q61R mutation had the potency to generate melanomas and activate downstream signalling, whereas the NRAS G12D mutation did not [12]. This finding might explain the predominance of NRAS Q61 mutations over other oncogenic NRAS mutations in human melanoma. Taken together, the oncogenic potential of NRAS, HRAS and KRAS and the respective sites of the mutation might vary among the different isoforms and could be dependent on the tissue context.
This conclusion is supported by the following example of targeting mutant BRAF. BRAF mutations can be found in melanoma (43%), papillary thyroid cancer (41%), colon cancer (13%) and haematological malignancies such as Langerhans cell histiocytosis (57%) (table 1) [7, 13]. BRAF V600E is a remarkable target for BRAF inhibitors in melanoma patients, with response rates of about 50% [14]. Early clinical data of BRAF V600E in lung cancer also indicate successful treatment options, but less strong than those seen for BRAF mutant melanoma. However, preclinical data from colon cancer shows that BRAF inhibitors alone are not sufficient to block tumour growth and require combination therapy with upstream epidermal growth factor receptor (EGFR) antibodies in cell culture and xenograft models [15, 16]. These data demonstrate that BRAF V600E is a promising target; however, the efficacy of BRAF inhibitors very much depends on tissue type and the respective signalling. In conclusion, it should be taken into consideration that the various mutations and their potential as drug targets should be investigated separately. This should include the form of mutant RAS involved, the position of the mutation, tissue lineage and maybe even subgroups thereof.
|
Table 1: Frequency of mutations in HRAS, KRAS, NRAS, and BRAF in different cancers. Data accessed from the Catalogue of somatic mutations in cancer (COSMIC) [7]. |
|
Disease
|
KRAS
|
NRAS
|
HRAS
|
BRAF
|
| Biliary tract |
31% (1679) |
1% (287) |
0% (153) |
5.3% (982) |
| Bone |
1% (252) |
0% (207) |
2% (199) |
7.9% (716) |
| Breast |
4% (782) |
2% (504) |
1% (716) |
1.1% (3836) |
| Brain |
1% (1054) |
1% (1017) |
0% (964) |
8% (490) |
| Cervix |
7% (1009) |
2% (132) |
5% (527) |
9.9% (802) |
| Endometrium |
14% (2251) |
0% (4544) |
1% (291) |
0% (4544) |
| Haematopeotic and lymphoid tissue |
5% (5978) |
9% (15226) |
0% (3076) |
10% (8671) |
| Kidney |
1% (704) |
0% (435) |
0% (273) |
0% (7339) |
| Large Intestine |
34% (53826) |
0% (2211) |
0% (671) |
13% (9391) |
| Lung |
17% (4742) |
0.6% (12053) |
0.5% (4031) |
2.2% (14682) |
| Oesophagus |
4% (375) |
0% (161) |
1% (161) |
0.5% (1517) |
| Ovary |
12% (5653) |
5% (191) |
0% (152) |
7.1% (4476) |
| Pancreas |
57% (8691) |
0.6% (1838) |
0.2% (1459) |
2% (2308) |
| Pleura |
3% (169) |
3% (77) |
0% (1551) |
2% (197) |
| Prostate |
4% (2210) |
2% (588) |
2% (1457) |
1.4% (1981) |
| Salivary Gland |
2% (402) |
0.7% (276) |
9% (401) |
1% (322) |
| Skin |
2.3% (3729) |
11% (508) |
16% (10377) |
43% (9048) |
| Small Intestine |
23% (656) |
0% (232) |
1% (132) |
4% (413) |
| Stomach |
6% (281) |
2% (215) |
4% (384) |
1% (262) |
| Testis |
3.4% (146) |
3% (283) |
2.2% (356) |
2.4% (325) |
| Thymus |
2% (261) |
0% (46) |
0% (49) |
0% (53) |
| Thyroid |
2% (7717) |
7% (7154) |
7% (7154) |
41% (19449) |
| Upper aerodigestive tract |
2% (3845) |
1.4% (2.40) |
6% (153) |
0.9% (2671) |
| Urinary tract |
4% (1953) |
2% (873) |
9% (2914) |
2% (1124) |
Blocking the RAS pathway by rapidly accelerated fibrosarcoma (RAF) kinase inhibitors
The first attempts to block hyperactivated RAS signalling in cancer were to address directly mutant RAS. Unfortunately, these attempts remained unsuccessful, as described below.
Since RAS has a very high affinity to GTP, in the picomolar range, inhibition of nucleotide binding is more challenging than with the ATP-binding pockets of other kinases. Another way of targeting RAS activation is to prevent RAS binding to the cell membrane, which is crucial for RAS activation. RAS is modified post-translationally by farnesylation, which allows binding to the cell membrane. Direct targeting of mutant RAS by farnesylation inhibitors was encouraging in preclinical studies, but did not show any benefit in clinical studies with unselected patient populations [17, 18]. Despite these failures, RAS as a drug target was not abandoned. In 2013, the National Institutes of Health (NIH) launched the RAS initiative, a multidisciplinary effort, including the biotechnology and pharmaceutical industries, to explore novel strategies to treat RAS-driven tumours.
So far, the most effective way to target RAS signalling in cancer is with RAF inhibitors. However, this possibility exists only if the BRAF downstream of RAS is mutated and activated. BRAF is most commonly mutated in melanoma, where the V600E mutations seem to be the predominant mutation [7]. Specific inhibitors for BRAF V600E like vemurafenib (Zelboraf®; Genentech/Plexxikon) or dabrafenib (Tafinlar®; GSK) proved to be clinically successful with response rates of 50%, significant progression-free survival and overall survival in patients with metastatic melanoma [14, 19]. These were impressive results after decades without therapies having an effect on overall survival. However, as anticipated, the responses were terminated by occurrence of resistant mutations such as an activating mutation in NRAS, MEK1 or AKT and appearance of a BRAF V600E splice variant, which favour RAF dimerisation [20]. Currently, phase III clinical trials have demonstrated that combination of the BRAF and MEK inhibitors could further improve clinical responses, which will be discussed below [21, 22].
Blocking RAS signalling with mitogen-activated protein kinase (MEK) inhibitors
Instead of inhibiting RAS, downstream MEK represents another opportunity as drug target. Interestingly, in a large phase III clinical study it was found that MEK inhibition alone is effective in BRAF V600E-positive melanoma patients [23]. The MEK inhibitor trametinib showed a better outcome for response rate, progression-free survival and overall survival than standard chemotherapy [23]. Although a head-to-head trial of a MEK inhibitor versus a BRAF inhibitor is lacking, the clinical efficacy of trametinib compared with vemurafenib (or dabrafenib) seems equal in BRAF mutant melanoma patients. This is in line with very early preclinical studies showing that BRAF mutant cell lines are highly sensitive to either RAF or MEK inhibition [24]. Therefore, sufficient preclinical and clinical data are available to target MEK in RAF mutated cancers. MEK is an interesting and promising target for small molecule inhibitors. MEK has a specific pocket structure, which is close to the MgATP-binding site. This pocket structure is conserved among MEK proteins and represents an ideal target for allosteric inhibitors [25]. After binding of a MEK inhibitor, conformational changes keep the unphosphorylated MEK in a nonfunctional state [26]. Since binding of the MEK inhibitor does not interfere with ATP binding to the active kinase site, MEK inhibitors are highly specific to MEK kinases at very low nanomolar concentrations and have fewer off targets than other inhibitors [25]. Several compounds that show very potent inhibitory activity of MEK1/2 are currently in clinical development or even approved (fig. 1). Whereas nonspecific binding of MEK inhibitors might not contribute to toxities, animal studies in MEK1/2 knock-out in mice led to perinatal death, showing that MEK activity is required in adult animals [27]. This indicates that MEK inhibitors may have specific toxicities, which will be discussed later.
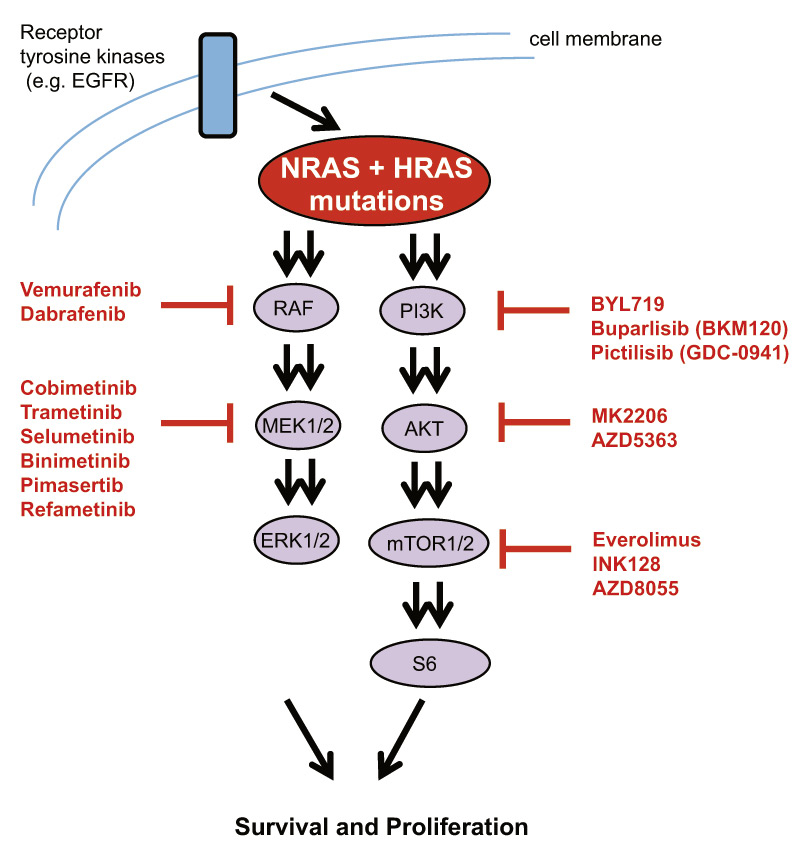
Figure 1
Schematic representation of the RAS signalling pathway. RAS activates both the MAPK and the PI3K/mTOR pathways downstream. Inhibitors for both pathways are shown. Vemurafenib, dabrafenib and cobimetinib are approved in Switzerland, whereas others are mostly in early and some in advanced clinical development.
Approved MEK inhibitors and MEK inhibitors in advanced clinical studies
Cobimetinib (GDC-0973, XL-518, Cotellic®)
Cobimetinib is the first MEK inhibitor approved by Swissmedic in Switzerland [28]. It is approved for treatment of BRAF V600 mutant metastatic melanoma in combination with vemurafenib (Zelboraf®). The approval of Swissmedic came even before the approval by the US Food and Drug Administration (FDA), which was approved by FDA in November 2015. Approval was based on the phase III CoBRIM trial with 495 patients and its recent update [29, 30]. Patients received 960 mg vemurafenib twice daily for 28 days in combination with either placebo or 60 mg cobimetinib once a day from days 1 to 21 (in a 28-day cycle) (29). In the follow up report, the overall response rate was 70% for the combination versus 50% for vemurafenib alone [30]. Progression-free survival was significantly higher for the combination at 12.3 versus 7.2 months whereas median overall survival was not reached yet [30]. Risk for death seemed to be significantly lower in the combination arm. Cobimetinib showed an acceptable safety profile but grade 3 and 4 events were reported for 63% of patients in the combination group and 58% in the vemurafenib arm [30]. The majority of adverse events included diarrhoea, nausea, rash, arthralgia, fatique and increased creatine phosphokinase levels (higher in the combination arm). As expected, keratoacanthomas and cutaneous squamous-cell carcinoma were significantly less frequent in the combination arm [30]. In conclusion, the combination of vermurafenib and cobimetinib represents a novel treatment option for BRAF mutant melanoma patients in Switzerland.
As discussed for other MEK inhibitors in this review, responses in early clinical trials were observed mostly exclusively in patients with mutations in the RAS pathway. In a phase I study, for example, treatment with cobimetinib induced 3 partial responses among the 46 patients with various advanced solid tumors. The 3 partial responses were observed in patients with BRAF mutant melanoma, BRAF mutant pancreatic cancer and in a KRAS-mutant endometrial cancer [31]. This demonstrates the importance of a selected patient population based on mutations and other biomarkers for successful outcome of clinical trials.
Trametinib (GSK1120212, Mekinist®)
Trametinib first showed clinical activity in a phase I study including 206 patients with pancreas cancer, colorectal cancer and melanoma [32]. In unselected populations, 2 pancreas cancer patients out of 21 showed a partial response and 2 out of 26 patients with NSCLC had a partial response [32]. The most sensitive population had BRAF mutant and NRAS mutant melanoma patients, with a response rate of 33%, demonstrating the importance of highly selected populations [32].
Thus, trametinib was further studied in BRAF mutant melanoma. In patients with metastatic melanoma harbouring a BRAF V600E/K mutation, single-agent trametinib compared with dacarbazine showed significantly longer progression-free survival of 4.8 months in the trametinib group compared with 1.5 months in the standard chemotherapy group [23]. Based on these results trametinib was approved by the FDA in 2013, and by the European Medicines Agency (EMA) in 2014. Trametinib has not been approved by Swissmedic.
Combinations of trametinib and dabrafenib were found to be even more effective. Progression-free survival and overall response rate were higher for the combination of trametinib and dabrafenib in metastatic melanoma, which led to approval of the combination therapy by the FDA in 2014 [30]. These findings were supported by more mature data from a phase III study. The combination of trametinib and dabrafenib increased median overall survival by 6 months [21, 22]. Thus, two different combinations of BRAF and MEK inhibitors have by now been shown to induce clinically meaningfull responses.
Selumetinib (AZD6244)
Selumetinib is another prominent example of a clinically successful MEK inhibitor. Though selumetinib is not approved yet, the FDA has granted it orphan drug status for the treatment of uveal melanoma. Phase III data are expected by the end of this year. Further, selumetinib is being investigated in phase III studies in KRAS mutant lung cancer and thyroid cancer.
In preclinical studies, selumetinib effectively blocked cell growth of melanoma and hepatocellular cell lines in cell culture and in in-vivo models [34, 35]. Selumetinib decreased ERK phosphorylation in tumour cell lines and also in 80% of tumour biopsies in a phase I study [36]. Clinical activity was first demonstrated with three cases of prolonged stable disease in patients with medullary thyroid cancer or uveal melanoma or renal cancer (one case each) [36]. In addition, several phase II studies proved the efficacy of single-agent selumetinib. Selumetinib caused partial remission in 3 (12%) and stable disease in 14 (50%) of 28 patients with metastatic biliary cancers, whereas no response was detected in papillary thyroid carcinoma or hepatocellular carcinoma [37]. In an unselected population of melanoma patients, selumetinib and temozolomid had comparable response rates of 10% [38]. Of note, patients responding to selumetinib showed a BRAF mutation in five out of six cases [38]. In a study focusing on BRAF mutated melanoma, 3 of 15 patients had partial responses [39].
However, inhibition of MEK did not result in a clinical response in acute myeloid leukaemia, which might be owing to greater heterogeneity of the disease [40]. Furthermore, single-agent selumetinib did not demonstrate superiority over temozolomide in melanoma, over pemetrexed in NSCLC, over capecitabine in pancreatic cancer and over capecitabine in colorectal cancer.
Interestingly, selumetinib has been extensively studied in combination treatments with other drugs. One recent early trial investigated the combined effect of selumetinib with the AKT inhibitor MK2206. Twenty-nine patients with various KRAS mutant cancers were included; 3 of 13 (23%) patients with NSCLC had a partial response as did one of two (50%) patients with ovarian cancer [41]. One of the NSCLC patients had a durable) response for 15 months [41]. This, again, nicely demonstrates the importance of carefully selecting patient populations on the basis of histology and molecular biomarkers.
The combination of selumetinib and cetuximab showed promising first results in a phase I study with solid tumours including KRAS mutated colorectal cancer [42]. Two patients with partial responses and two with with stable disease out of 13 patients were observed in patients with KRAS mutated colorectal cancer and one stable disease in a patient with tonsillar squamous cell carcinoma and one with NSCLC [42].
Several combinations of selumetinib with vandetanib, cixutumumab, docetaxel, gemcitabine, and irinotecan are currently being tested in clinical trials.
MEK inhibitors in early clinical development
CI-1040/PD-0325901
CI-1040 was the first MEK inhibitor which made it to clinical trials but did not produce an objective response in an unselected population of 67 patients including NSCLC, breast, colon and pancreatic cancers [43]. Its predecessor PD-0325901 has excellent oral bioavailability and has higher potency to block MEK1/2. However, PD-0325901 caused unacceptable side effects, such as retinal vein occlusion, and clinical development was stopped. Currently, only two clinical studies aim to investigate PD-0325901 in combination with (1) a CDK4/6 inhibitor in KRAS mutant NSCLC and (2) an endothelial growth factor receptor (EGFR) inhibitor in KRAS mutant colon cancer (clinicaltrials.gov).
Pimasertib (AS703026, MSC1936369B)
The efficacy of pimasertib was studied in a phase I study of patients with advanced solid tumours including melanoma and colorectal cancer [44]. As seen with other MEK inhibitors, pimasertib caused tumour shrinkage mostly in patients with BRAF or NRAS mutations. Now it is being studied in a phase II study of pimasertib versus dacarbazine in patients with NRAS mutated metastatic melanoma. In a setting of 88 KRAS mutated pancreatic cancer patients, pimasertib did not improve overall response rate or overall survival in combination with gemcitabine versus gemcitabine alone [45]. The finding suggests that KRAS mutations alone are not a sufficient predictive marker for treatment with MEK inhibitors (see molecular markers for KEK inhibitors, below). Combination of pimasertib with 5-fluorouracil, leucovorin and irinotecan (FOLFIRI) in KRAS-mutated metastatic colorectal cancer showed high toxicity and is therefore not a treatment option [46]. Several combination studies of pimasertib with the PI3K/mTOR inhibitor SAR245409, or with the mTOR inhibitor temsirolimus, or with gemcitabine are ongoing.
Refametinib (BAY 86-9766, RDEA119)
Refametinib is under early clinical investigation with only one partial response in 53 unselected patients with various cancers [47]. The one responding patient had colorectal cancer and was considered “wild-type” according to RAS and PI3K mutation status [47]. Of note, two BRAF mutant melanoma patients showed progression on refametinib. The combination of refametinib with sorafenib led to partial response in three and stable diseases in 25 of 69 Asian patients with hepatocellular carcinoma [48]. However, toxicity was high, with four grade 5 treatment-related adverse events and dose modifications for all patients. Combination studies of refametinib with gemcitabine in patients with metastatic pancreatic cancer and with the PI3K inhibitor BAY80-6946 are ongoing.
More MEK inhibitors under clinical evaluation
Several MEK inhibitors are currently under early clinical evaluation including AZD8330 (ARRY-424704), RO4987655 (CH4987655), WX-554, E6201, and TAK-733. Further, RO5126766, which is a novel dual RAF/MEK inhibitor, is also currently investigated in phase I clinical trials.
Toxicity of MEK inhibitors
Most toxicity from MEK inhibitors observed so far includes unspecific events that are common to most small-molecule kinase inhibitors. These toxicities include rash, fatigue and diarrhoea [26]. However, ocular toxicities are specific to MEK inhibitors and occur as blurred and reduced vision. Retinal vein occlusion stopped development of PD-0325901, but has not been observed with other inhibitors of MEK. More commonly described is central serous retinopathy [26]. Toxicities are temporary and can be reversed by reduction of dose or drug interruption. Side effects with low impact on patients’ status are periorbital oedema and elevation of creatine phosphokinase. Overall, toxicities from MEK inhibitors are similar to toxicities known from other small-molecule inhibitors and readily manageable.
Resistance to MEK inhibitors
Cobimetinib and trametinib are the only MEK inhibitors approved so far. Trametinib and cobimetinib are mostly used in combination with BRAF inhibitors and rarely as single agent. Thus, knowledge gained on resistance is based on combined therapy and for a limited number of patients. Despite impressive clinical efficacy and few reports of long-term responding patients, half of patients under combined combination treatment ultimately relapse, after about 10 months. Whereas acquired resistance to BRAF inhibitors is driven by activating alterations in the RAS pathway (MAPK activation, increased BRAF copy numbers, altered splicing of BRAF, mutations in NRAS or MEK1/2 or upregulation of other receptor tyrosine kinases), it was commonly believed that mechanisms independent of the RAS pathway would occur [49]. Surprisingly, a study from last year identified MEK2 Q60P and MEK2 C125S mutations, BRAF amplification and BRAF splice variants as resistance mechanism for combined treatment of BRAF and MEK inhibitors [49, 50]. This show that resistance arises within the RAS pathway, which renders it more difficult to overcome resistance. How to best treat patients with tumour progression on MEK inhibitors (in combination with RAF inhibitors) is currently unknown. One group which investigated the MEK2 Q60P mutation found that resistant cells could be resensitised by a triple combination of dabrafenib, trametinib, and a PI3K/mTOR inhibitor [51]. Hence, resistance might require novel combination therapies to overcome it.
Molecular markers for MEK inhibitors
The RAS pathway is a prominent example of successful targeted therapy. BRAF mutations are a strong predictive marker for treatment with BRAF inhibitors in clinical studies, as described above. Nonmutant BRAF tumours are not responding to BRAF inhibitors in preclinical studies, on the contrary; tumours even grow as a result of a paradoxical activation of the RAS pathway [52, 53]. RAS mutations are, however, not yet established and whether KRAS, NRAS and HRAS mutation can be used as reliable predictive positive markers for MEK inhibitors is currently under investigation and will be discussed in the next paragraphs.
KRAS mutation is a poor predictive marker for single MEK inhibitor treatment
Based on preclinical and early clinical studies, KRAS seems to be a poor predictive marker for response to MEK inhibitors in vitro and in vivo. A preclinical study evaluating the inhibitory concentrations of selumetinib showed strong sensitivity of BRAF mutant cell lines whereas about 60% of KRAS mutant cell lines were insensitive towards selumetinib with EC50 concentrations often far above 1 μM [54]. Concentrations of higher than 1 μM are hardly achieved in patients and cell lines are considered sensitive towards inhibitors if the EC50 concentration is well below 1 μM; usually concentrations are within the nanomolar range and BRAF mutant cell lines, for example, show sensitivities lower than 0.1 μM [24]. In a phase I study of trametinib, three patients with NSCLC responded to treatment [32]. Only one of the three was KRAS mutated and the two other responders had EGFR mutation, whereas most nonresponders were KRAS mutated [32]. Thus, this study shows that (1) clinical data correlate with in-vitro studies in cell lines and (2) that KRAS mutations alone are a poor predictive marker for single-agent therapy with MEK inhibitors. However, a recent phase II study showed that the combination of selumetinib with docetaxel has promising efficacy in second-line treatment for KRAS mutant NSCLC [55]. Although, treatment was not compared between KRAS wildtype and KRAS mutant patients in this study, clinical activity might be higher for KRAS mutant patients if results are compared with other phase II studies of selumetinib in unselected patients [56]. Therefore, outcomes from the phase III combination study of selumetinib and docetaxel in KRAS mutant NSCLC patients are awaited. In conclusion, KRAS mutations have so far failed to prove a powerful predictive marker for treatment with MEK inhibitors and KRAS mutations might require combination treatments to result in clinical meaningful effects.
NRAS is a promising predictive marker
Interestingly, NRAS as a predictive target for MEK inhibitors is rarely studied. Although, a plethora of recent preclinical data clearly shows that NRAS mutations predict for sensitivity to MEK inhibitors in cell line studies, clinical proof-of-concept data are rare. NRAS mutations were detected in 2% of high-risk cutaneous T-cell lymphomas (CTCL), and a CTCL cell line harbouring a NRAS Q61K mutation was highly sensitive to three different MEK inhibitors at low nanomolar concentrations [56]. NRAS mutations occur in 0.5–1% of NSCLC patients, mostly of adenocarcinoma histology and five of six NRAS mutant lung cancer cell lines were sensitive to the MEK inhibitors selumetinib and trametinib [56]. NRAS is mutated in 15–25% of melanoma patients and NRAS mutant melanoma cell lines are very sensitive to MEK inhibitors [3, 59].
Since NRAS mutations are most abundant in melanoma, the MEK inhibitor binimetinib (MEK162) is so far the sole MEK inhibitor studied specifically in a selected NRAS mutant patient population. In a phase I study with biliary cancer and colorectal cancer patients, 1 complete response and 1 partial response out of 26 patients were observed [60]. Consequently, a phase II study investigated the effect of binimetinib in NRAS and BRAF mutant melanoma patients. Objective responses were found in 6 out of 30 (20%) patients with NRAS mutant melanoma and in 8 out of 41 (20%) with BRAF mutant melanoma [61]. Patients with previous BRAF inhibitor treatment were allowed in this trial, which may explain the lower response rates of 20%. Nevertheless, this study clearly shows that NRAS can be a predictive marker for MEK inhibitors in clinical therapy. These encouraging data led to the initiation of a phase III study of the MEK inhibitor binimetinib. This study is called NEMO trial for “NRAS mElanoma and MEK inhibitOr” and compares binimetinib with dacarbazine in patients with metastatic NRAS mutant melanoma.
There have been fewer studies of MEK inhibitors in haematological malignancies. In acute myeloid leukaemia (AML), a phase II trial of selumetinib was performed including 3 of 47 patients harbouring a NRAS mutation. However, selumetinib resulted in only modest response, independent of the NRAS or KRAS mutation status [40]. This indicates that either more AML patients with NRAS mutations have to be included in clinical trials or that the potency of NRAS as a predictive marker is dependent on tissue background and lineage.
Targeting mutant HRAS: preclinical and clinical data
Even though HRAS mutations can be found in a significant number of cancers, including squamous cell cancer of the lung (2.8%), head and neck cancer (3.9%) and bladder cancer (5.1%), little is known about targeting mutant HRAS [62]. We have investigated several HRAS mutant cancer cell lines and observed strong sensitivity to MEK inhibitors in vitro and in vivo compared with HRAS wildtype cell lines [63]. These data showed that, at least in a preclinical setting, HRAS mutations predict sensitivity to MEK inhibitors.
Clinical characteristics and behaviour of HRAS mutant cancer patients have been described rarely. One recent report described an adenocarcinoma of the lung with HRAS Q61L mutation associated with from rapid progression and deterioration, suggesting that HRAS mutations in NSCLC tumours could be fast growing and associated with poor prognosis [64]. Interestingly, one phase I trial for the novel MEK inhibitor RO5126766 reported a tumour patient with HRAS mutation who had 20% tumour shrinkage due to MEK inhibitor treatment [65]. This is the first hint that HRAS mutant cancer patients might benefit clinically from MEK inhibitor treatment.
Outlook to the future
Despite the high number of mutations found in the RAS pathway only a few patients seem to be eligible for treatment with specific RAF and MEK inhibitors. Whereas BRAF mutations are a crucial predictive marker for treatment with RAF and MEK inhibitors in melanoma patients, the role of NRAS, KRAS and HRAS as markers has to be further studied. However, striking activity has been demonstrated for MEK inhibitors in selected RAS mutant patient populations and several phase II and phase III studies in RAS mutant patients are currently ongoing. The activity of MEK inhibitors might be further enhanced by combination with other small molecule inhibitors (e.g., AKT inhibitors and PI3K inhibitors) or classical cytotoxic agents. The results of ongoing trials are awaited with high interest.
References
1 Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nature reviews Drug discovery. 2014;13(12):928–42.
2 Yordy JS, Muise-Helmericks RC. Signal transduction and the Ets family of transcription factors. Oncogene. 2000;19(55):6503–13.
3 Posch C, Moslehi H, Feeney L, Green GA, Ebaee A, Feichtenschlager V, et al. Combined targeting of MEK and PI3K/mTOR effector pathways is necessary to effectively inhibit NRAS mutant melanoma in vitro and in vivo. Proc Natl Acad Sci U S A. 2013;110(10):4015–20.
4 Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, et al. A landscape of driver mutations in melanoma. Cell. 2012;150(2):251‒63.
5 Scheffzek K, Ahmadian MR, Kabsch W, Wiesmuller L, Lautwein A, Schmitz F, et al. The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science. 1997;277(5324):333–8.
6 Shimizu K, Goldfarb M, Suard Y, Perucho M, Li Y, Kamata T, et al. Three human transforming genes are related to the viral ras oncogenes. Proc Natl Acad Sci U S A. 1983;80(8):2112–6.
7 COSMIC G. Catalogue of somatic mutations in cancer. http://cancer.sanger.ac.uk/cancergenome/projects/studies/. 2015. Accessed August 2015.
8 Atreya CE, Corcoran RB, Kopetz S. Expanded RAS: refining the patient population. J Clin Oncol. 2015;33(7):682–5.
9 To MD, Wong CE, Karnezis AN, Del Rosario R, Di Lauro R, Balmain A. Kras regulatory elements and exon 4A determine mutation specificity in lung cancer. Nat Genet. 2008;40(10):1240–4.
10 Haigis KM, Kendall KR, Wang Y, Cheung A, Haigis MC, Glickman JN, et al. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat Genet. 2008;40(5):600–8.
11 Li Q, Haigis KM, McDaniel A, Harding-Theobald E, Kogan SC, Akagi K, et al. Hematopoiesis and leukemogenesis in mice expressing oncogenic NrasG12D from the endogenous locus. Blood. 2011;117(6):2022–32.
12 Burd CE, Liu W, Huynh MV, Waqas MA, Gillahan JE, Clark KS, et al. Mutation-specific RAS oncogenicity explains NRAS codon 61 selection in melanoma. Cancer discovery. 2014;4(12):1418–29.
13 Badalian-Very G, Vergilio JA, Degar BA, MacConaill LE, Brandner B, Calicchio ML, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116(11):1919–23.
14 Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med.364(26):2507–16.
15 Mao M, Tian F, Mariadason JM, Tsao CC, Lemos R, Jr., Dayyani F, et al. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin Cancer Res. 2013;19(3):657–67.
16 Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483(7387):100–3.
17 Rao S, Cunningham D, de Gramont A, Scheithauer W, Smakal M, Humblet Y, et al. Phase III double-blind placebo-controlled study of farnesyl transferase inhibitor R115777 in patients with refractory advanced colorectal cancer. J Clin Oncol. 2004;22(19):3950–7.
18 Van Cutsem E, van de Velde H, Karasek P, Oettle H, Vervenne WL, Szawlowski A, et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol. 2004;22(8):1430–8.
19 Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–65.
20 Rizos H, Menzies AM, Pupo GM, Carlino MS, Fung C, Hyman J, et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin Cancer Res. 2014;20(7):1965–77.
21 Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372(1):30–9.
22 Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet. 2015;386(9992):444–51.
23 Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367(2):107–14.
24 Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439(7074):358–62.
25 Ohren JF, Chen H, Pavlovsky A, Whitehead C, Zhang E, Kuffa P, et al. Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nature structural & molecular biology. 2004;11(12):1192–7.
26 Adjei A, Zhao, Y. Inhibiting MEK for Cancer Therapy. ASCO Annual Meeting. 2015.
27 Bissonauth V, Roy S, Gravel M, Guillemette S, Charron J. Requirement for Map2k1 (Mek1) in extra-embryonic ectoderm during placentogenesis. Development. 2006;133(17):3429–40.
28 Swissmedic. Cotellic®, Filmtabletten (Cobimetinibum). https://www.swissmedic.ch/zulassungen/00153/00189/00200/02938/index.html?lang=de. 2015. Accessed 07.11.2015.
29 Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371(20):1867–76.
30 Larkin JMG, Yan Y, McArthur GA, Ascierto PA, Liszkay G, Maio M, et al. Update of progression-free survival (PFS) and correlative biomarker analysis from coBRIM: Phase III study of cobimetinib (cobi) plus vemurafenib (vem) in advanced BRAF-mutated melanoma. J Clin Oncol 33, 2015 (suppl; abstr 9006). 2015.
31 LoRusso PGS, Pandya SS, Kwak EL, Jones C, Belvin M, Musib LC, et al. A first-in-human phase Ib study to evaluate the MEK inhibitor GDC-0973, combined with the pan-PI3K inhibitor GDC-0941, in patients with advanced solid tumors. J Clin Oncol 30, 2012 (suppl; abstr 2566). 2012;2012 ASCO Annual Meeting (Abstract #2566).
32 Infante JR, Fecher LA, Falchook GS, Nallapareddy S, Gordon MS, Becerra C, et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13(8):773–81.
33 Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367(18):1694–703.
34 Haass NK, Sproesser K, Nguyen TK, Contractor R, Medina CA, Nathanson KL, et al. The mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor AZD6244 (ARRY-142886) induces growth arrest in melanoma cells and tumor regression when combined with docetaxel. Clin Cancer Res. 2008;14(1):230–9.
35 Huynh H, Soo KC, Chow PK, Tran E. Targeted inhibition of the extracellular signal-regulated kinase kinase pathway with AZD6244 (ARRY-142886) in the treatment of hepatocellular carcinoma. Mol Cancer Ther. 2007;6(1):138–46.
36 Widemann Brigitte C. LJM, Michael J. Fisher, Brian D. Weiss, AeRang Kim, Eva Dombi, Andrea Baldwin, Patricia Whitcomb, Staci Martin, Andrea Gillespie, Austin Doyle. Phase I study of the MEK1/2 inhibitor selumetinib (AZD6244) hydrogen sulfate in children and young adults with neurofibromatosis type 1 (NF1) and inoperable plexiform neurofibromas. 2014 ASCO Annual Meeting 2014;J Clin Oncol 32:5s, 2014 (suppl; abstr 10018) (Abstract number: 10018).
37 Banerji U, Camidge DR, Verheul HM, Agarwal R, Sarker D, Kaye SB, et al. The first-in-human study of the hydrogen sulfate (Hyd-sulfate) capsule of the MEK1/2 inhibitor AZD6244 (ARRY-142886): a phase I open-label multicenter trial in patients with advanced cancer. Clin Cancer Res. 2010;16(5):1613–23.
38 Kirkwood JM, Bastholt L, Robert C, Sosman J, Larkin J, Hersey P, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res. 2012;18(2):555–67.
39 Catalanotti F, Solit DB, Pulitzer MP, Berger MF, Scott SN, Iyriboz T, et al. Phase II trial of MEK inhibitor selumetinib (AZD6244, ARRY-142886) in patients with BRAFV600E/K-mutated melanoma. Clin Cancer Res. 2013;19(8):2257–64.
40 Jain N, Curran E, Iyengar NM, Diaz-Flores E, Kunnavakkam R, Popplewell L, et al. Phase II study of the oral MEK inhibitor selumetinib in advanced acute myelogenous leukemia: a University of Chicago phase II consortium trial. Clin Cancer Res. 2014;20(2):490–8.
41 Tolcher AW, Khan K, Ong M, Banerji U, Papadimitrakopoulou V, Gandara DR, et al. Antitumor activity in RAS-driven tumors by blocking AKT and MEK. Clin Cancer Res. 2015;21(4):739–48.
42 Deming D.A. WRS, Sam Joseph Lubner, Daniel Mulkerin, Noelle K. LoConte, Suzanne Fioravanti, Tim Greten, et al. A phase I study of selumetinib (AZD6244/ARRY-142866) in combination with cetuximab (cet) in refractory solid tumors and KRAS mutant colorectal cancer (CRC). J Clin Oncol. 30, 2012 (suppl; abstr 3103). 2012.
43 Rinehart J, Adjei AA, Lorusso PM, Waterhouse D, Hecht JR, Natale RB, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol. 2004;22(22):4456–62.
44 Delord J, N. Houede, A. Awada, A. Taamma, S. J. Faivre, T. Besse-Hammer, A. Italiano, C. Vignaud, M. Donica, E. Raymond. First-in-human phase I safety, pharmacokinetic (PK), and pharmacodynamic (PD) analysis of the oral MEK-inhibitor AS703026 (two regimens [R]) in patients (pts) with advanced solid tumors. J Clin Oncol 28:15s, 2010 (suppl; abstr 2504). 2010.
45 Van Cutsem E, Manuel Hidalgo, Igor Bazin, Jean-Luc Canon, Elena Poddubskaya, Nebojsa Manojlovic, et al. Phase II randomized trial of MEK inhibitor pimasertib or placebo combined with gemcitabine in the first-line treatment of metastatic pancreatic cancer. J Clin Oncol. 33, 2015 (suppl 3; abstr 344). 2015.
46 Macarulla T, Cervantes A, Tabernero J, Rosello S, Van Cutsem E, Tejpar S, et al. Phase I study of FOLFIRI plus pimasertib as second-line treatment for KRAS-mutated metastatic colorectal cancer. Br J Cancer. 2015.
47 Weekes CD, Von Hoff DD, Adjei AA, Leffingwell DP, Eckhardt SG, Gore L, et al. Multicenter phase I trial of the mitogen-activated protein kinase 1/2 inhibitor BAY 86-9766 in patients with advanced cancer. Clin Cancer Res. 2013;19(5):1232–43.
48 Lim HY, Heo J, Choi HJ, Lin CY, Yoon JH, Hsu C, et al. A phase II study of the efficacy and safety of the combination therapy of the MEK inhibitor refametinib (BAY 86-9766) plus sorafenib for Asian patients with unresectable hepatocellular carcinoma. Clin Cancer Res. 2014;20(23):5976–85.
49 Long GV, Fung C, Menzies AM, Pupo GM, Carlino MS, Hyman J, et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat Commun. 2014;5:5694.
50 Wagle N, Van Allen EM, Treacy DJ, Frederick DT, Cooper ZA, Taylor-Weiner A, et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer discovery. 2014;4(1):61–8.
51 Villanueva J, Infante JR, Krepler C, Reyes-Uribe P, Samanta M, Chen HY, et al. Concurrent MEK2 mutation and BRAF amplification confer resistance to BRAF and MEK inhibitors in melanoma. Cell Rep. 2013;4(6):1090–9.
52 Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464(7287):431–5.
53 Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140(2):209–21.
54 Davies Barry R AL, Jennifer S. McKay PM, Samantha Steele RJ, Mark Cockerill SC, and Paul D. Smith. AZD6244 (ARRY-142886), a potent inhibitor of mitogenactivated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol Cancer Ther. 2007;2007(6(8).
55 Janne PA, Shaw AT, Pereira JR, Jeannin G, Vansteenkiste J, Barrios C, et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. 2013;14(1):38–47.
56 Hainsworth JD, Cebotaru CL, Kanarev V, Ciuleanu TE, Damyanov D, Stella P, et al. A phase II, open-label, randomized study to assess the efficacy and safety of AZD6244 (ARRY-142886) versus pemetrexed in patients with non-small cell lung cancer who have failed one or two prior chemotherapeutic regimens. J Thorac Oncol. 2010;5(10):1630–6.
57 Kiessling MK, Oberholzer PA, Mondal C, Karpova MB, Zipser MC, Lin WM, et al. High-throughput mutation profiling of CTCL samples reveals KRAS and NRAS mutations sensitizing tumors toward inhibition of the RAS/RAF/MEK signaling cascade. Blood. 2011;117(8):2433–40.
58 Ohashi K, Sequist LV, Arcila ME, Lovly CM, Chen X, Rudin CM, et al. Characteristics of lung cancers harboring NRAS mutations. Clin Cancer Res. 2013;19(9):2584–91.
59 Vujic I, Posch C, Sanlorenzo M, Yen AJ, Tsumura A, Kwong A, et al. Mutant NRASQ61 shares signaling similarities across various cancer types--potential implications for future therapies. Oncotarget. 2014;5(17):7936–44.
60 Finn R.S. MMJ, B.R. Tan Jr., C.C Weekes, J.C. Bendell, A. Patnaik, G.N. Khan, et al. A Phase 1 Study of MEK Inhibitor MEK162 (ARRY-438162) in Patients with Biliary Tract Cancer. ASCO GI, Jan 19–21, 2012, San Francisco, CA. 2012.
61 Ascierto PA, Schadendorf D, Berking C, Agarwala SS, van Herpen CM, Queirolo P, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 2013;14(3):249–56.
62 Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505(7484):495–501.
63 Kiessling MK C-FA, Samaras P, Atrott K, Cosin-Roger J, Lang S, Scharl M, Gerhard Rogler. Mutant HRAS as novel target for MEK and mTOR inhibitors. Oncotarget. 2015;published online, accessed 06.11.2015(DOI: 10.18632/oncotarget.5619).
64 Cathcart-Rake E, Corless C, Sauer D, Lopez-Chavez A. Elderly former smoker with HRAS mutant non-small-cell lung cancer. J Thorac Oncol. 2014;9(10):e75–8.
65 Martinez-Garcia M, Banerji U, Albanell J, Bahleda R, Dolly S, Kraeber-Bodere F, et al. First-in-human, phase I dose-escalation study of the safety, pharmacokinetics, and pharmacodynamics of RO5126766, a first-in-class dual MEK/RAF inhibitor in patients with solid tumors. Clin Cancer Res. 2012;18(17):4806–19.