IL-6/STAT3/ARF: the guardians of senescence, cancer progression and metastasis in prostate cancer
DOI: https://doi.org/10.4414/smw.2015.14215
Jan
Pencik, Robert
Wiebringhaus, Martin
Susani, Zoran
Culig, Lukas
Kenner
Summary
Prostate cancer is one of the most prevalent forms of cancer in men worldwide. It remains a clinical challenge to identify lethal metastatic prostate cancers, which escape standard therapeutic intervention. Aberrant interleukin-6 (IL-6) / signal transducer and activator of transcription-3 (STAT3) signalling and loss of p53 occur during prostate cancer progression to metastatic disease. The abnormality of the IL-6/STAT3/p53 axis is frequently accompanied by other genetic alterations; however, its potential role as an important mediator of oncogenic reprogramming, invasion and metastatic transformation remains unknown. The failure of anti-IL-6 treatments is still unexplained and may be due to an incomplete understanding of the mechanism of the in vivo role of IL-6/STAT3 in prostate cancer. The identification of the alternative reading frame protein (ARF) / murine double minute protein (MDM2) / p53 tumour suppressor pathway potentially involving the IL-6/STAT3 axis as a restricting factor in prostate cancer deficient in the tumour suppressor phosphatase and tensin homologue (PTEN) opened new avenues to currently available therapies. This review summarises the current knowledge on the role of crucial pathways driving prostate cancer progression as well as metastatic disease and discusses the potential use of novel specific target molecules and how it can be exploited to avoid overtreatment and increase quality of life.
Interleukin-6 expression in human prostate cancer and tumour-elicited microenvironment
Interleukin-6 (IL-6) is a multifunctional cytokine that is implicated in the regulation of immune responses, inflammation and cellular processes [1] in several cancers, including cancer of the prostate. IL-6 is detectable in stromal cells, but preferentially localised in the epithelium of prostate tissue [2]. The IL-6 receptor displays a highly restricted expression pattern including hepatocytes, leucocyte subsets and megakaryocytes, but is also ubiquitously expressed in prostate cancer cells [3]. In benign prostatic tissue IL-6 expression is confined to the basal cells of the epithelium. In particular, the androgen receptor-negative human prostate cancer cell lines (DU-145 and PC3) express high levels of IL-6 [4]. Whether this is a result of a direct mechanism involving the androgen receptor remains unknown. IL-6 expression is governed by nuclear factor kappa B, which is suppressed by treatment with androgenic hormones [5] and may otherwise result in an aberrant activation of the androgen receptor [6] (fig. 1). Importantly, IL-6 expression levels are high in the tissue of prostate cancer patients after radical prostatectomy, as well as in sera of patients with advanced prostate cancer that is resistant to therapy [4, 7]. IL-6 levels are upregulated by transforming growth factor-beta (TGF-β) as well, which is an important determinant of metastatic transformation [8]. Thus, IL-6 signal transduction is important for regulating cellular processes in prostate cancer. Studies with primary cells also demonstrated that there is a positive growth effect of IL-6 in such a condition [9]. Hypoxic conditions promote activation of IL-6/Notch/Jagged signalling as well as providing survival advantages and protumourigenic gene expression programmes of breast cancer cells [10]. Oxaliplatin increased IL-21 expression as well as STAT3 phosphorylation in prostate cancer B cells. More recently, Shabnam et al. showed that these B cells are immunosuppressive and also express IL-10 as well as programmed death ligand 1 (PD-L1), the appearance of which is dependent on TGF-β receptor signalling. The elimination of these B cells, which infiltrate therapy-resistant prostate cancer, helps cytotoxic T lymphocyte-dependent eradication of tumours treated with oxaliplatin [11].
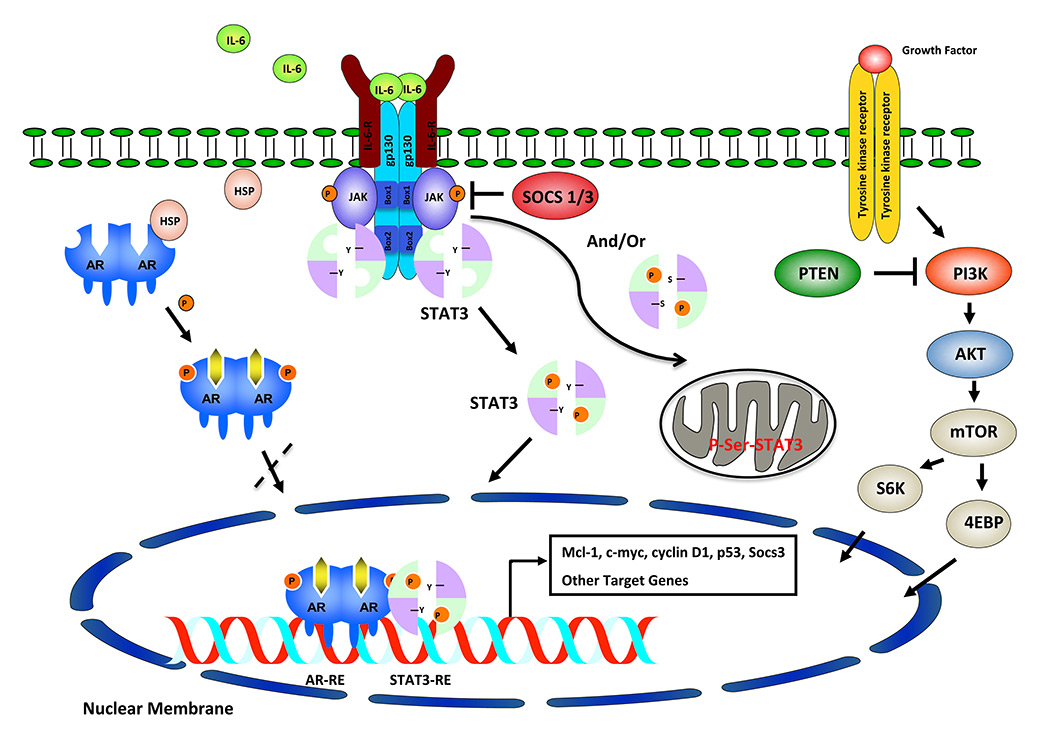
Figure 1
Overview of IL-6/STAT3/ARF and PI3K/PTEN/AKT/mTOR pathways. Activation of IL-6/STAT3 signalling leads to phosphorylation and translocation of STAT3 to the nucleus, which is associated with AR interaction regulated by HSP. JAKs serve to phosphorylate tyrosine (Y) or serine (S) residues of STAT3 and to translocate to nucleus or mitochondrial matrix. Some of these downstream signalling events (STAT3-AR-S6K) could regulate activity or expression of prostate cancer related genes.
ARF = alternative reading frame protein; IL-6 = interleukin-6; PI3K = phosphatidylinositol-3-kinase; PTEN = phosphatase and tensin homologue; STAT3 = signal transducer and activator of transcription-3
The role of IL-6/STAT3 signalling during transdifferentiation of prostate cancer
Studies on human-derived prostate cancer cell lines reflected considerable heterogeneity of IL-6 responses in prostate cancer. Interestingly, divergent responses to IL-6 were observed in lymph node carcinomas of the prostate (LNCaP) cells [7, 12].
In the LNCaP cell line, IL-6 induces nuclear translocation and phosphorylation of the signal transducer and activator of transcription (STAT3). STAT3 activation may lead to enhanced neuroendocrine differentiation [13]. Neuroendocrine cells are growth-arrested; however, they express high levels of neuropeptides. The neuroendocrine or endocrine-paracrine cancer cells secrete potent neurohormones, including bombesin/gastrin, calcitonin and parathyroid hormone-related peptide, which stimulate prostate growth in a paracrine manner. For this reason neuroendocrine differentiation induced by IL-6 may be considered a bad prognostic factor. There is no adequate therapy for cancer that shows a large amount of neuroendocrine differentiation. IL-6‒mediated inhibition of LNCaP cells is associated with cell cycle arrest and up-regulation of the cyclin-dependent kinase inhibitor p27 [14]. Inhibitory effects of IL-6 on in-vivogrowth of LNCaP prostate cancer xenografts have also been demonstrated [15]. Notably STAT3 is considered to play a tumour-suppressive role in murine KRas mutant lung cancer [16] and glioblastoma cells [17], questioning the value of IL-6 receptor or STAT3 inhibitors as therapeutic strategies.
However, it should be mentioned that in other laboratories growth stimulation of LNCaP cells by IL-6 has been observed [7]. This may be a result of genetic drift and differences in these cancer cells or it may result from different cell culture conditions, which make the use and conclusion from in-vitro tissue culture experiments questionable since these discrepancies have not yet been resolved. In contrast, studies from the Gao laboratory demonstrated that STAT3 inhibition in vivo causes prostate cancer cell growth retardation [18]. Pro- or anti-proliferative effects of IL-6 on prostate cancer cell lines may depend on the expression levels of the ErbB2 oncogene. In the presence of ErbB2, IL-6 may induce activation of mitogen-activated protein kinases thus leading to proliferation of prostate cancer cells [19]. Phosphorylation of STAT3 also depends on the presence of heat-shock protein 27 (HSP27), which affects prostate carcinogenesis in multiple ways and is a valid target for therapy [20].
In contrast, PC3 cells produce high levels of IL-6 (even though they carry STAT3 deletions) and are stimulated by the cytokine in an autocrine manner. IL-6 signalling in PC3 cells is accomplished through the phosphatidylinositol-3-kinase (PI3K) pathway, which is activated in prostate and other cancers as a result of the loss of the tumour suppressor phosphatase and tensin homologue (PTEN) [13] (fig. 1). Therefore, IL-6 targeting in several cell lines seems to be justified. The results of most in-vitro studies clearly showed that IL-6 is a valid target in prostate cancer, because of its proproliferative and antiapoptotic effects. IL-6 is a major regulator of the antiapoptotic myeloid cell leukaemia-1 (Mcl-1) protein. It is highly expressed in prostate cancer and induced by androgen ablation [21, 22]. IL-6 may also inhibit apoptosis through activation of the signalling pathway of the insulin-like growth factor receptor, which mediates phosphorylation and activation of AKT [23]. IL-6 may even contribute to angiogenesis through up-regulation of vascular endothelial growth factor (VEGF) in prostate cancer cells [24]. In addition to classic IL-6 signalling through the membrane receptor, it has been demonstrated that trans-signalling through the soluble receptor is important for regulation of migration, thus facilitating tumour metastasis [22].
Modelling of prostate cancer progression and metastasis in mouse models
Initiation and progression of prostate cancer is driven by the stepwise accumulation of critical genetic alterations including mutations, gain-of-function of oncogenic genes or inactivation of tumour suppressor genes, such as PTEN, TP53, SMAD4, STAT3 and CDKN2A (Cancer Genome Atlas Research Network, 2015). Experimental approaches using probasin-driven Cre (Pb-Cre4) recombinase activity and ablation of PTEN in prostate epithelium [25] have been developed. Importantly, deletion of PTEN and STAT3 in prostate cancer animal models has produced inconsistent results. Pencik et al. showed that loss of IL-6 or Stat3 in this prostate cancer model leads to massive and disseminated metastases and early lethality due to complete loss of alternative reading frame protein (ARF) / murine double minute protein (MDM2) / p53 cellular senescence [26]. These data determine the tumour suppressor role of the IL-6/STAT3 pathway. In contrast, Toso et al. demonstrated that STAT3 inactivation in PTEN-deficient tumours reduced tumour size (roughly by 70%) and stromal compartment, and only 25% of age-matched PTENpc-/-STAT3pc-/- tumours developed invasive prostate cancer [27]. Moreover, loss of STAT3 in PTEN-deficient tumours restores active immunosurveillance and reprograms senescence. The main differences in these studies are that Pencik et al. generated PTENpc-/-STAT3pc
-/- mice by crossing PTENloxP/loxPmice [25] with STAT3loxP/loxPanimals [28]. Alonzi et al. observed Cre-mediated recombination through removal of the region corresponding to exons 12 to 14 accompanied by complete STAT3deletion. Toso et al. used PTENloxP/loxPmice [25] crossed with STAT3loxP/loxPanimals [29]. The original publication demonstrated Cre-mediated deletion of STAT3loxP/loxPanimals, which resulted in expression of a truncated STAT3 protein [30]. This STAT3Δ protein contained no tyrosine residue or mitogen-activated protein kinase recognition site, both of which are important for STAT3 activation; however, unphosphorylated STAT3 has an important role as transcription factor in cancers and in responses to cytokines, and activates gene expression of different target genes (cdc2, cyclin B1, mras, and E2F1) [31, 32]. The remaining mitochondrial pool of STAT3Δ has an important role in controlling energy metabolism, cell death and oncogenic transformation [33, 34].
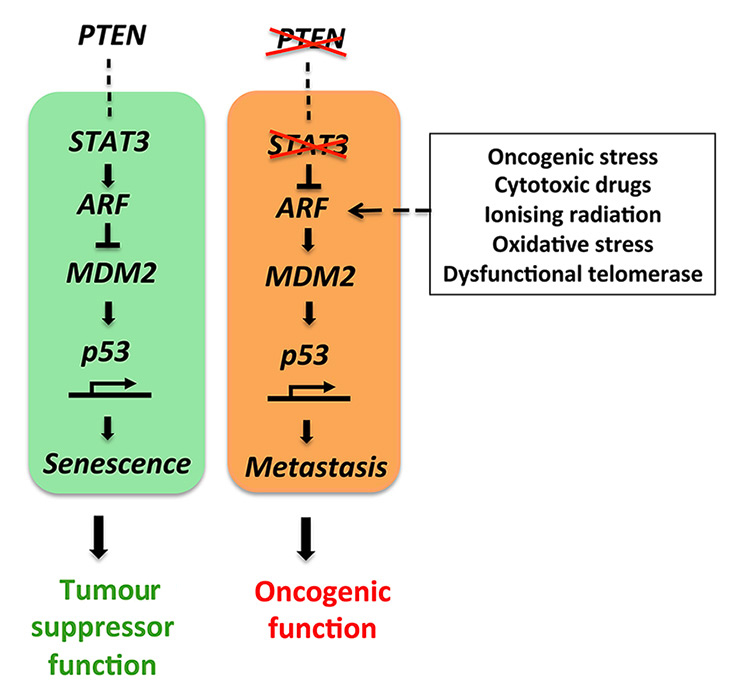
Figure 2
The STAT3-ARF tumour suppressor pathway is activated under normal conditions. Loss of PTEN and STAT3 function causes an oncogenic switch ultimately breaking the ARF-MDM2-p53 axis and inducing metastatic progression.
The findings of Pencik et al. contradict the dogma that IL-6/STAT3 signalling has an oncogenic function during prostate cancer progression, and unmasks ARF as a novel direct STAT3 target gene. The imminent clinical data identify STAT3 and ARF as key prognostic markers to stratify high from low risk prostate cancer patients. STAT3 is co-deleted with PTEN in 66% of human prostate cancer metastases and particularly STAT3 and CDKN2A deletions co-occurred in the majority of prostate cancer metastases. These data show STAT3 and ARF to be leading factors in bypassing senescence and driving metastatic progression by inhibiting p53 function (fig. 2) in prostate cancer. Importantly, Toso et al. demonstrated that docetaxel (a chemotherapeutic drug used in recurrent prostate cancer) drives a strong senescence response, but fails to activate antitumour immune response and tumour clearance. In this respect, Toso et al. identified the tyrosine phosphatase SRC-homology 2 domain-containing phosphate 2 (SHP2) as a critical factor favouring escape from immune surveillance. However, genetic ablation or pharmacological inhibition of SHP2 was recently shown to induce senescence together with increased levels of p53, p27 and H3K9me3 [35]. SHP2 inhibition could therefore particularly reprogram senescence antitumour immune responses and elicit senescence to activate tumour clearance.
Previously, other prostate cancer mouse models based on conditional deletion of PTEN and other genes involved in metastatic progression were established. Inactivation of PTEN and SMAD4 generated a metastatic prostate cancer mouse model with a 100% penetrance. Molecular and histopathological analysis revealed highly proliferative and invasive tumours functionally driven by prometastatic CCND1 and SPP1 expression [36]. Importantly, in other prostate cancer mouse models harbouring specific inactivation of PTEN and p53 in the prostate led to rapid prostate cancer formation, but failed to trigger a prostate cancer metastatic phenotype [37]. Ding et al. employed a prostate-specific p53/PTEN double null model with telomere dysfunctional (G3/G4 LSL-mTert) named G3/G4 LSL-mTert PB-PTEN/p53(third and fourth generation LSL-mTert) mouse model [36]. The telomere dysfunction together with loss of PTEN and p53 led to rapid and progressive adenocarcinomas with lumbar spine metastases (in 25% of cases). Validation of genes that are capable of driving this bone metastatic progression suggested involvement of TGF-β signalling and specifically SMAD4 as a key driver. The genetic validation using a prostate-specific p53/PTEN/Smad4 knockout mouse model confirmed a very aggressive tumour phenotype with short overall survival and spontaneous bone metastasis (in 12.5% cases). Further in-vivo genetic studies will be essential to draw the complete picture of the cascade of the drivers of prostate cancer initiation, progression and metastatic spread. Prostate cancer mouse models will be essential to elucidate these cancer-relevant genes. This will turn out to be essential for patient management and therapy.
Patient-derived xenograft models and organoid-derived cultures of prostate cancer patients
Another preclinical prostate cancer model is based on transplantation of fresh cancer tissue specimens, patient-derived xenografts (PDX), which are engrafted into immunodeficient mice (e.g. NOD-SCID, NSG mice) [39]. At the histopathological level, the PDX models retain the cellular heterogeneity, architecture and stromal components of the original tumour tissue microenvironment [40]. The PDX model has the advantage that the original prostate cancer tissue can be serially propagatedin vivo. However, the PDX model shows low success rates and reduced vascularisation. The de-novo generation of three-dimensional in-vivo human prostate organoids recapitulates the diversity and molecular subtypes of human prostate cancer [41, 42]. The organoid-derived culture technology supports long-term expansion and is genetically stable (showing loss of p53 and RB tumour suppressor pathways commonly found in advanced prostate cancer). This method enables a robust long-term culture of human prostate cancer, covering multiple subgroups or subtypes of prostate cancer, and is one of the most appropriate models to address genetic and pharmacological studies.
Mutational landscape of high-risk prostate cancer patients
The major advances in whole exome sequencing technology enable the complete DNA sequences of prostate cancer patients to be determined with high-sensitivity and provide ground for complete mutational landscape of prostate cancer. Certain gene mutations are predominant e.g. TP53 and PTEN. Recent discoveries of recurrent mutations STAT3 and CDKN2A [26], SPOP, MED12 and FOXA1 [43] or MLL2 and RB1 [44] suggested that prostate cancer patients could be subdivided on the basis of mutational status. New aspects of preclinical mouse models of prostate cancer highlighted the link between STAT3-ARF [26], HER2 [45] or ERG [46] and metastatic prostate cancer. Accumulation of genetic alterations, including amplifications and deletions of genomic segments in metastatic prostate cancer, call for exploration of future insights into biological function and identification of exclusive subgroups of high-risk prostate cancer patients for specific management strategies or targeted therapies.
IL-6-androgen receptor signalling in prostate carcinogenesis and progression
Interaction between IL-6 and androgen receptor in prostate cancer is of special importance in prostate cancer. The androgen receptor is a therapeutic target whose effectiveness has been improved by use of abiraterone acetate and enzalutamide. The androgen receptor is a central regulator of proliferation, apoptosis, differentiation and angiogenesis. Activation of the androgen receptor was detectable in cells that express endogenous androgen receptor as well as in those transfected with androgen receptor cDNA [47]. IL-6 stimulates the expression of PSA in LNCaP cells, which was shown to be blocked by the nonsteroidal antiandrogen bicalutamide. Another mechanism includes IL-6/STAT3 signalling and p53 in the context of a metastatic niche during prostate cancer progression. IL-6 regulates prostate cancer autophagy in a paracrine manner by shifting cytoplasmatic and nuclear STAT3 with involvement of direct p53, ER stress-Ca2+ signalling [48‒50]. IL-6 also exerts a growth effect in vitroand in vivo on MDA prostate cancer 2b cells which contain a double-mutated androgen receptor [51]. Thus, IL-6/androgen receptor interaction is clinically relevant. It is known that IL-6 may enhance androgen-independent growth of LNCaP cells [3]. Furthermore, it is likely that ligand-independent activation of the androgen receptor may have a role in this cellular process [52].
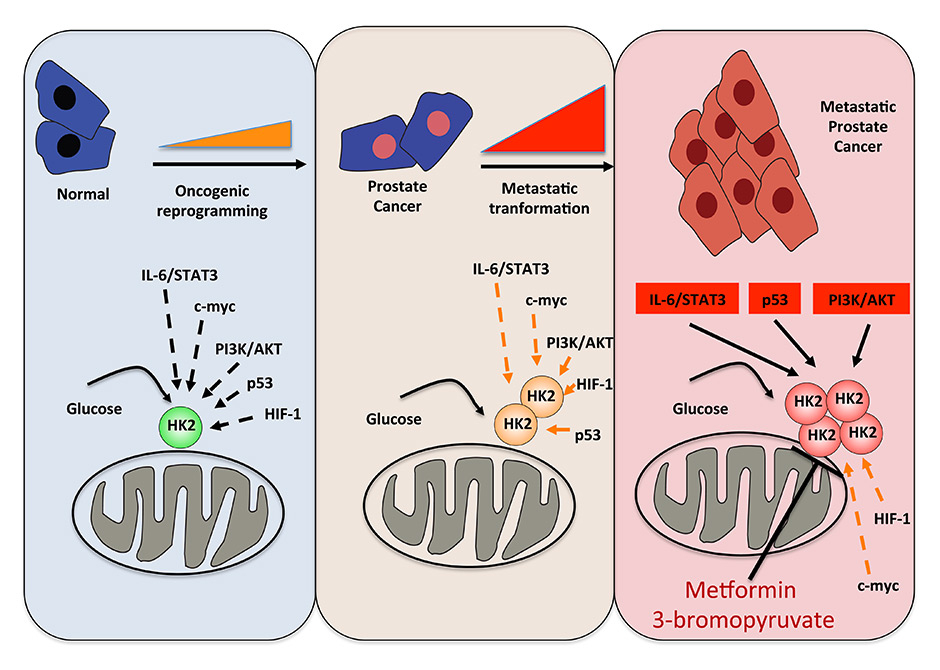
Figure 3
Targeting IL-6/STAT3-p53-PI3K/AKT signalling inducing metastatic prostate cancer metabolism by HK2 inhibitors (metformin or 3-bromopyruvate).
In addition to regulation of proliferation through androgen receptor signalling, other pathways may be activated. In androgen-insensitive cells, autocrine production of IL-6 activates the pathway of PI3K, thus inhibiting apoptosis [13]. For ligand-independent activation of the androgen receptor by IL-6, specific coactivators such as SRC-1 and p300 are of importance [53, 54]. These coactivators facilitate enhancement of androgen receptor activity through interaction with the N-terminal of the receptor. Importantly, these coactivators are highly expressed in prostate cancer tissue, particularly during tumour progression. Silencing of p300 could completely abrogate the enhancement of androgen receptor activity by IL-6. Interestingly, the ability of p300 to induce the expression of proteins that are normally androgen receptor-regulated in a cellular compartment with low androgen receptor expression has been demonstrated [55]. This mechanism may be important for progression of prostate cancer in which the androgen receptor is expressed at a lower level owing to epigenetic modifications.
IL-6/STAT3/p53 in prostate cancer therapy
IL-6 may be of particular importance for the level of tumour stem cells. The presence of prostate cancer stem cells is associated with a resistance to endocrine and cytotoxic therapy. Current cancer therapies fail in most instances to eradicate cancer stem cells efficiently. Blocking the Janus kinase (JAK) / STAT pathway in cancer stem cells results in inhibition of tumour initiation, which is in fact of utmost importance in the design of new therapies for advanced prostate cancer [56].
As expected, continuous treatment with IL-6 has some measurable effects. IL-6 resistance may occur in prostate cancer. An example of such a cell line is LNCaP-IL-6+. This cell line shows a more aggressive behaviour in vitro and in vivo than parental cells [2, 24]. Although the cells do not respond to exogenous IL-6 after continuous treatment, they produce large amounts of endogenous IL-6. Increased growth of LNCaP-IL-6+ cells is associated with a loss of the retinoblastoma tumour suppressor Rb. Whether transition from paracrine growth inhibition to autocrine growth stimulation by IL-6 occurs in a larger number of prostate cancer models remains to be determined.
As mentioned before, IL-6 is a valid target in prostate cancer therapy. Experimental therapy with a monoclonal anti-IL-6 antibody yielded unexpected results. For example, growth of PC3 cells was inhibited by the IL-6 receptor blocking antibody CNTO 328 in vitro and in vivo[57]. Growth inhibition is mainly a consequence of induced apoptosis in these cells. However, a limited effect of the antibody was observed in LNCaP-IL-6+ cells or in a highly metastatic derivative of PC3 cells, PC3-M [24, 58]. In that in-vivo model, anti-IL-6 treatment was associated with inhibition of cachexia. An important effect of the CNTO 328 antibody on delaying the progression of prostate cancer was demonstrated in the LNCaP 35 xenograft model [59]. Anti-IL-6 treatment also decreased the levels of the coactivators p300 and CBP, which are clearly implicated in prostate cancer progression. It is important to state the dependence of anti-IL-6 treatment on the presence of Mcl-1 in vivo. If Mcl-1 was downregulated by specific small interfering RNA, no effect of CNTO 328 was observed [21]. IL-6 levels were also inhibited by vitamin D in vitro, but vitamin D clinical trials in prostate cancer did not show sufficient efficacy [60]. Terakawa and associates reported that prostate cancer cells which also expressed IL-6 showed a greater sensitivity to androgen withdrawal, which in fact may be of interest for future combination therapies in prostate cancer [61].
Moreover, treatment of patients with an IL-6R blocking antibody alone did not result in a survival advantage in patients with advanced prostate cancer. Clinical studies with CNTO 328 as a monotherapy did not show any significant effect in castration-resistant prostate cancer [62, 63]. One should keep in mind that advanced prostate cancer is a very heterogeneous disease for which any monotherapy is unlikely to yield a significant long-lasting effect.
For treatment of metastatic disease, combination therapies to reactivate the IL-6/STAT3-p53 tumour suppressive pathway should be tested. Moreover, IL-6/STAT3 signalling enhances glycolysis and regulates the glycolytic enzyme hexokinase-2 [64]. Hexokinase-2 overexpression has been observed in cancer cell lines as well as in primary tumour tissue [65, 66]. Administration of potent hexokinase-2 inhibitors (metformin or 3-bromopyruvate) showed significantly reduced tumour growth [67, 68] (fig. 3). Concomitant loss of PTEN and p53 in a preclinical prostate cancer mouse model showed robustly enhanced hexokinase-2 expression [69]. Therefore, PTEN and p53-deficient human prostate cancer are relevant for the treatment of metastatic prostate cancer currently untreatable with pharmacotherapy. Therefore, hexokinase-2 inhibitors accompanied by abiraterone acetate or enzalutamide could be a promising novel therapeutic strategy for prostate cancer patients carrying PTEN and TP53 mutations or similar genomic alterations.
Authors’ contribution: JP and RW contributed equally to this work.
References
1 Schaper F, Rose-John S. Interleukin-6: Biology, signaling and strategies of blockade. Cytokine Growth Factor Rev. 2015;26(5):475–87.
2 Hobisch A, Rogatsch H, Hittmair A, Fuchs D, Bartsch G, Jr., Klocker H, et al. Immunohistochemical localization of interleukin-6 and its receptor in benign, premalignant and malignant prostate tissue. J Pathol. 2000;191:239–44.
3 Lee SO, Lou W, Hou M, de Miguel F, Gerber L, Gao AC. Interleukin-6 promotes androgen-independent growth in LNCaP human prostate cancer cells. Clin Cancer Res. 2003;9:370–6.
4 Twillie DA, Eisenberger MA, Carducci MA, Hseih WS, Kim WY, Simons JW. Interleukin-6: a candidate mediator of human prostate cancer morbidity. Urology. 1995;45:542–9.
5 Keller ET, Chang C, Ershler WB. Inhibition of NFkappaB activity through maintenance of IkappaBalpha levels contributes to dihydrotestosterone-mediated repression of the interleukin-6 promoter. J Biol Chem. 1996;271:26267–75.
6 Nadiminty N, Lou W, Sun M, Chen J, Yue J, Kung HJ, et al. Aberrant activation of the androgen receptor by NF-kappaB2/p52 in prostate cancer cells. Cancer Res. 2010;70:3309–19.
7 Giri D, Ozen M, Ittmann M. Interleukin-6 is an autocrine growth factor in human prostate cancer. Am J Pathol. 2001;159:2159–65.
8 Park JI, Lee MG, Cho K, Park BJ, Chae KS, Byun DS, et al. Transforming growth factor-beta1 activates interleukin-6 expression in prostate cancer cells through the synergistic collaboration of the Smad2, p38-NF-kappaB, JNK, and Ras signaling pathways. Oncogene. 2003;22:4314–32.
9 Culig Z, Steiner H, Bartsch G, Hobisch A. Interleukin-6 regulation of prostate cancer cell growth. J Cell Biochem. 2005;95:497–505.
10 Sansone P, Storci G, Tavolari S, Guarnieri T, Giovannini C, Taffurelli M, et al. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J Clin Invest. 2007;117:3988–4002.
11 Shalapour S, Font-Burgada J, Di Caro G, Zhong Z, Sanchez-Lopez E, et al. Immunosupressive plasma cells impede T-Cell dependent immunogenic chemotherapy. Nature 2015;521(7550):94–8.
12 Degeorges A, Tatoud R, Fauvel-Lafeve F, Podgorniak MP, Millot G, de Cremoux P, Calvo F. Stromal cells from human benign prostate hyperplasia produce a growth-inhibitory factor for LNCaP prostate cancer cells, identified as interleukin-6. Int J Cancer. 1996;68:207–14.
13 Chung TD, Yu JJ, Kong TA, Spiotto MT, Lin JM. Interleukin-6 activates phosphatidylinositol-3 kinase, which inhibits apoptosis in human prostate cancer cell lines. Prostate. 2000;42:1–7.
14 Mori S, Murakami-Mori K, Bonavida B. Interleukin-6 induces G1 arrest through induction of p27(Kip1), a cyclin-dependent kinase inhibitor, and neuron-like morphology in LNCaP prostate tumor cells. Biochem Biophys Res Commun. 1999;257:609–14.
15 Wang Q, Horiatis D, Pinski J. Interleukin-6 inhibits the growth of prostate cancer xenografts in mice by the process of neuroendocrine differentiation. Int J Cancer. 2004;111:508–13.
16 Grabner B, Schramek D, Mueller KM, Moll HP, Svinka J, Hoffmann T, et al. Disruption of STAT3 signalling promotes KRAS-induced lung tumorigenesis. Nat Commun. 2015;6:6285.
17 McFarland BC, Gray GK, Nozell SE, Hong SW, Benveniste EN. Activation of the NF-kappaB pathway by the STAT3 inhibitor JSI-124 in human glioblastoma cells. Mol Cancer Res. 2013;11:494–505.
18 Ni Z, Lou W, Leman ES, Gao AC. Inhibition of constitutively activated Stat3 signaling pathway suppresses growth of prostate cancer cells. Cancer Res. 2000;60:1225–8.
19 Qiu Y, Ravi L, Kung HJ. Requirement of ErbB2 for signalling by interleukin-6 in prostate carcinoma cells. Nature. 1998;393:83–5.
20 Shiota M, Bishop JL, Nip KM, Zardan A, Takeuchi A, Cordonnier T, et al. Hsp27 regulates epithelial mesenchymal transition, metastasis, and circulating tumor cells in prostate cancer. Cancer Res. 2013;73:3109–19.
21 Cavarretta IT, Neuwirt H, Untergasser G, Moser PL, Zaki MH, Steiner H, et al. The antiapoptotic effect of IL-6 autocrine loop in a cellular model of advanced prostate cancer is mediated by Mcl-1. Oncogene. 2007;26:2822–32.
22 Santer FR, Malinowska K, Culig Z, Cavarretta IT. Interleukin-6 trans-signalling differentially regulates proliferation, migration, adhesion and maspin expression in human prostate cancer cells. Endocr Relat Cancer. 2010;17:241–53.
23 Rojas A, Liu G, Coleman I, Nelson PS, Zhang M, Dash R, et al. IL-6 promotes prostate tumorigenesis and progression through autocrine cross-activation of IGF-IR. Oncogene. 2011;30:2345–55.
24 Steiner H, Berger AP, Godoy-Tundidor S, Bjartell A, Lilja H, Bartsch G, et al. An autocrine loop for vascular endothelial growth factor is established in prostate cancer cells generated after prolonged treatment with interleukin 6. Eur J Cancer. 2004;40:1066–72.
25 Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003;4:209–21.
26 Pencik J, Schlederer M, Gruber W, Unger C, Walker SM, Chalaris A, et al. STAT3 regulated ARF expression suppresses prostate cancer metastasis. Nat Commun. 2015;6:7736.
27 Toso A, Revandkar A, Di Mitri D, Guccini I, Proietti M, Sarti M, et al. Enhancing chemotherapy efficacy in pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 2014;9:75–89.
28 Alonzi T, Maritano D, Gorgoni B, Rizzuto G, Libert C, Poli V. Essential role of STAT3 in the control of the acute-phase response as revealed by inducible gene inactivation [correction of activation] in the liver. Mol Cell Biol. 2001;21:1621–32.
29 Akira S. Roles of STAT3 defined by tissue-specific gene targeting. Oncogene. 2000;19:2607–11.
30 Takeda K, Kaisho T, Yoshida N, Takeda J, Kishimoto T, Akira S. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J Immunol. 1998;161:4652–60.
31 Yang J, Chatterjee-Kishore M, Staugaitis SM, Nguyen H, Schlessinger K, Levy DE, et al. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 2005;65:939–47.
32 Yang J, Stark GR. Roles of unphosphorylated STATs in signaling. Cell Res. 2008;18:443–51.
33 Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner C, Levy DE. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009;324:1713–6.
34 Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, et al. Function of mitochondrial Stat3 in cellular respiration. Science. 2009;323:793–7.
35 Lan L, Holland JD, Qi J, Grosskopf S, Vogel R, Gyorffy B, et al. Shp2 signaling suppresses senescence in PyMT-induced mammary gland cancer in mice. EMBO J. 2015;34:1493–508.
36 Ding Z, Wu CJ, Chu GC, Xiao Y, Ho D, Zhang J, et al. SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature. 2011;470:269–73.
37 Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–30.
38 Ding Z, Wu CJ, Jaskelioff M, Ivanova E, Kost-Alimova M, Protopopov A, et al. Telomerase reactivation following telomere dysfunction yields murine prostate tumors with bone metastases. Cell. 2012;148:896–907.
39 Lin D, Wyatt AW, Xue H, Wang Y, Dong X, Haegert A, et al. High fidelity patient-derived xenografts for accelerating prostate cancer discovery and drug development. Cancer Res. 2014;74:1272–83.
40 Russell PJ, Russell P, Rudduck C, Tse BW, Williams ED, Raghavan D. Establishing prostate cancer patient derived xenografts: lessons learned from older studies. Prostate. 2015;75:628–36.
41 Chua CW, Shibata M, Lei M, Toivanen R, Barlow LJ, Bergren SK, et al. Single luminal epithelial progenitors can generate prostate organoids in culture. Nat Cell Biol. 2014;16:951–61, 951–4.
42 Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, et al. Organoid cultures derived from patients with advanced prostate cancer. Cell. 2014;159:176–87.
43 Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44:685–9.
44 Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–43.
45 Ahmad I, Patel R, Singh LB, Nixon C, Seywright M, Barnetson RJ, et al. HER2 overcomes PTEN (loss)-induced senescence to cause aggressive prostate cancer. Proc Natl Acad Sci U S A. 2011);108:16392–7.
46 Chen Y, Chi P, Rockowitz S, Iaquinta PJ, Shamu T, Shukla S, et al. ETS factors reprogram the androgen receptor cistrome and prime prostate tumorigenesis in response to PTEN loss. Nat Med. 2013;19:1023–9.
47 Hobisch A, Culig Z, Radmayr C, Bartsch G, Klocker H, Hittmair A. Distant metastases from prostatic carcinoma express androgen receptor protein. Cancer Res. 1995;55:3068–72.
48 Chang GS, Chen XA, Park B, Rhee HS, Li P, Han KH, et al. A comprehensive and high-resolution genome-wide response of p53 to stress. Cell Rep. 2014;8:514–27.
49 Delk NA, Farach-Carson MC. Interleukin-6: a bone marrow stromal cell paracrine signal that induces neuroendocrine differentiation and modulates autophagy in bone metastatic PCa cells. Autophagy. 2012;8:650–63.
50 Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D’Amelio M, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10:676–87.
51 Malinowska K, Neuwirt H, Cavarretta IT, Bektic J, Steiner H, Dietrich H, et al. Interleukin-6 stimulation of growth of prostate cancer in vitro and in vivo through activation of the androgen receptor. Endocr Relat Cancer. 2009;16:155–69.
52 Liu C, Lou W, Armstrong C, Zhu Y, Evans CP, Gao AC. (2015). Niclosamide suppresses cell migration and invasion in enzalutamide resistant prostate cancer cells via Stat3-AR axis inhibition. Prostate.
53 Debes JD, Schmidt LJ, Huang H, Tindall DJ p300 mediates androgen-independent transactivation of the androgen receptor by interleukin 6. Cancer Res. 2002;62:5632–6.
54 Ueda T, Mawji NR, Bruchovsky N, Sadar MD. Ligand-independent activation of the androgen receptor by interleukin-6 and the role of steroid receptor coactivator-1 in prostate cancer cells. J Biol Chem. 2002;277:38087–94.
55 Heemers HV, Sebo TJ, Debes JD, Regan KM, Raclaw KA, Murphy LM, et al. Androgen deprivation increases p300 expression in prostate cancer cells. Cancer Res. 2007;67:3422–30.
56 Kroon P, Berry PA, Stower MJ, Rodrigues G, Mann VM, Simms M, et al. JAK-STAT blockade inhibits tumor initiation and clonogenic recovery of prostate cancer stem-like cells. Cancer Res. 2013;73:5288–98.
57 Azevedo A, Cunha V, Teixeira AL, Medeiros R. IL-6/IL-6R as a potential key signaling pathway in prostate cancer development. World J Clin Oncol. 2011;2:384–96.
58 Zaki MH, Nemeth JA, Trikha M. CNTO 328, a monoclonal antibody to IL-6, inhibits human tumor-induced cachexia in nude mice. Int J Cancer. 2004;111:592–5.
59 Wallner L, Dai J, Escara-Wilke J, Zhang J, Yao Z, Lu Y, et al. Inhibition of interleukin-6 with CNTO328, an anti-interleukin-6 monoclonal antibody, inhibits conversion of androgen-dependent prostate cancer to an androgen-independent phenotype in orchiectomized mice. Cancer Res. 2006;66:3087–95.
60 Nonn L, Peng L, Feldman D, Peehl DM. Inhibition of p38 by vitamin D reduces interleukin-6 production in normal prostate cells via mitogen-activated protein kinase phosphatase 5: implications for prostate cancer prevention by vitamin D. Cancer Res. 2006;66:4516–24.
61 Terakawa T, Miyake H, Furukawa J, Ettinger SL, Gleave ME, Fujisawa M. Enhanced sensitivity to androgen withdrawal due to overexpression of interleukin-6 in androgen-dependent human prostate cancer LNCaP cells. Br J Cancer. 2009;101:1731–9.
62 Dorff TB, Goldman B, Pinski JK, Mack PC, Lara PN, Jr., Van Veldhuizen PJ, Jr., et al. Clinical and correlative results of SWOG S0354: a phase II trial of CNTO328 (siltuximab), a monoclonal antibody against interleukin-6, in chemotherapy-pretreated patients with castration-resistant prostate cancer. Clin Cancer Res. 2010;16:3028–34.
63 Fizazi K, De Bono JS, Flechon A, Heidenreich A, Voog E, Davis NB, et al. Randomised phase II study of siltuximab (CNTO 328), an anti-IL-6 monoclonal antibody, in combination with mitoxantrone/prednisone versus mitoxantrone/prednisone alone in metastatic castration-resistant prostate cancer. Eur J Cancer. 2012;48:85–93.
64 Ando M, Uehara I, Kogure K, Asano Y, Nakajima W, Abe Y, et al. Interleukin 6 enhances glycolysis through expression of the glycolytic enzymes hexokinase 2 and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3. J Nippon Med Sch. 2010;77:97–105.
65 Patra KC, Wang Q, Bhaskar PT, Miller L, Wang Z, Wheaton W, et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell. 2013;24:213–28.
66 Wolf A, Agnihotri S, Micallef J, Mukherjee J, Sabha N, Cairns R, et al. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J Exp Med. 2011;208:313–26.
67 Cardaci S, Desideri E, Ciriolo MR. Targeting aerobic glycolysis: 3-bromopyruvate as a promising anticancer drug. J Bioenerg Biomembr. 2012;44:17–29.
68 Kato H, Sekine Y, Furuya Y, Miyazawa Y, Koike H, Suzuki K. Metformin inhibits the proliferation of human prostate cancer PC-3 cells via the downregulation of insulin-like growth factor 1 receptor. Biochem Biophys Res Commun. 2015;461:115–21.
69 Wang L, Xiong H, Wu F, Zhang Y, Wang J, Zhao L, et al. Hexokinase 2-mediated Warburg effect is required for PTEN- and p53-deficiency-driven prostate cancer growth. Cell Rep. 2014;8:1461–74.