Gains and losses on the road to understanding Alzheimer’s disease
DOI: https://doi.org/10.4414/smw.2015.14233
Summary
Alzheimer’s disease (AD) is a neurodegenerative disorder and the most common cause for dementia, which affects approximately 120 thousand people in Switzerland and 35 million worldwide. Aging is a major risk factor for developing AD and thus, as our societies are growing older, we face great challenges to find treatment strategies. The disease is characterised by loss of memory, deposition of extracellular amyloid plaques containing Aβ peptides and intraneuronal tangles of the tau protein. To date, there is no effective treatment and the cause of the disease is still debated.
The Schweizerische Alzheimervereinigung states that we need “continuous manifold research” into all possible causes of AD to find a cure for this disease. Fitting this proposition, a recent publication by Xia et al. (2015) described a novel mouse model that for the first time reproduces cortical neuron death as observed in human AD cases. At the same time, this publication questions the major theory of AD pathogenesis and points towards different treatment avenues that should be followed to find a cure for AD.
Key words: Alzheimer’s disease; amyloid precursor protein (APP); nuclear signaling; presenilin; AICD; neurodegeneration
Gain-of-toxicity
Although familial Alzheimer’s disease (FAD) represents less than 1% of all AD cases, it has been instrumental in shaping the hypotheses on the pathogenesis of this disease. FAD is caused by mutations in the amyloid precursor protein (APP) as well as presenilin 1 (PS1) or PS2, catalytic subunits of the γ-secretase complex. A major breakthrough was the discovery that Aβ peptides are generated by sequential proteolytic cleavage of APP, with the final intramembraneous γ-secretase cleavage releasing the Aβ peptide (fig. 1). The majority of Aβ peptides are 40 amino acids long, but γ-secretase also generates Aβ species of different length, for example Aβ42, which is more prone to aggregation because the additional hydrophobic amino acids derive from the transmembrane region of APP [1].
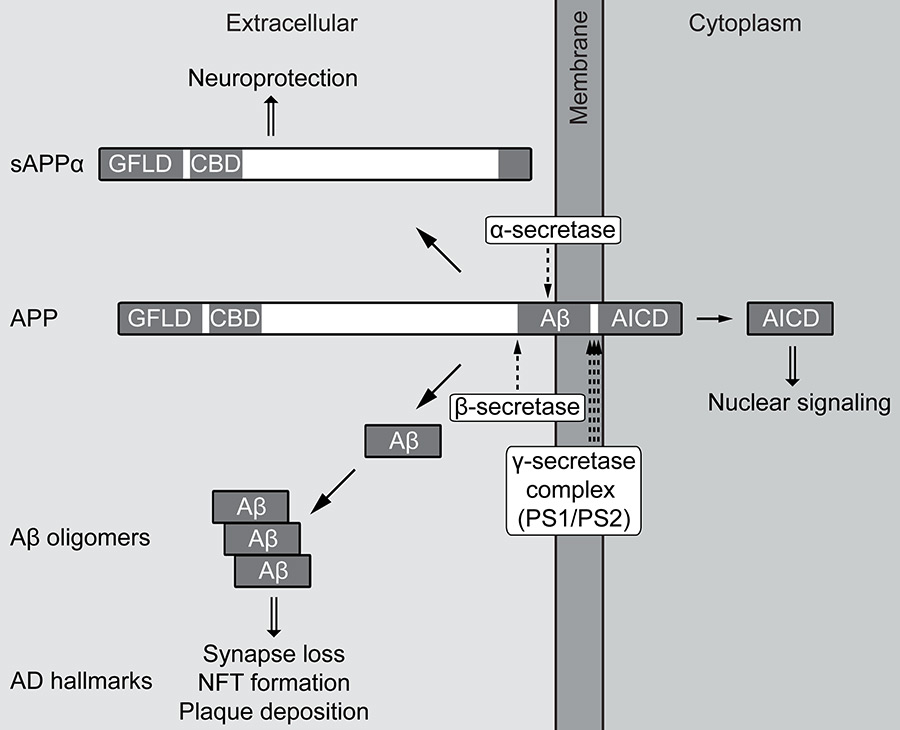
Figure 1
Proteolytic processing of the amyloid precursor protein (APP).
APP is a single-pass transmembrane protein with a large extracellular domain and the short APP intracellular domain (AICD) facing the cytoplasm. Sequential proteolytic cleavage by the secretases occurs via two opposing pathways, both initiated by the release of the extracellular domain. Non-amyloidogenic cleavage by α-secretase destroys the Aβ peptide and releases secreted APPα (sAPPα). This APP fragment contains a growth factor-like domain (GFLD) and a copper-binding domain (CBD) and displays neuroprotective and neuroproliferative properties. In the amyloidogenic pathway, cleavage by β-secretase generates the Aβ N-terminus and sAPPβ, which lacks the properties of sAPPα. After release of the extracellular domain, the remaining APP transmembrane C-terminal fragments (CTFs) can enter the γ-secretase complex to undergo regulated intramembrane proteolysis (RIP), which consists of consecutive cleavages denoted by three arrows. This initially releases AICD from the membrane, which in the case of the amyloidogenic pathway can signal to the nucleus. In the same pathway, further cleavages by γ-secretase shape the Aβ C-terminus that is critical for determining aggregation propensity. Aβ aggregates into different oligomeric species that can induce synapse loss and neurofibrillary tangle (NFT) formation. Finally, Aβ deposits in large extracellular structures known as amyloid plaques – besides NFTs, a hallmark of Alzheimer’s disease (AD).
FAD mutations in the presenilins cause a shift in cleavage, resulting in generation of more Aβ42, thereby shifting the Ab42/Aβ40 ratio that is a crucial determinant for the deposition of amyloid. This is exemplified by transgenic mice expressing solely Aβ40 or Aβ42 in the absence of APP; mice with high levels of Aβ40 exhibited no plaque deposition, whereas mice expressing low levels of Aβ42 had massive amyloid deposits [2]. Crossing these two mouse lines abolished plaque formation, i.e. Aβ40 was able to prevent the aggregation and deposition of Aβ42, demonstrating the importance of the Aβ42/Aβ40 ratio [3].
These findings pointed toward a gain-of-toxic-function of FAD mutations and led to the formulation of the amyloid cascade hypothesis. The cause of the disease was thought to be the aggregation of Aβ into amyloid plaques, resulting in cell death [4]. Subsequently, this hypothesis was refined to state that the disease is initiated not by plaques, but by smaller Aβ oligomers directly influencing synaptic function, which over time changes neuronal homeostasis, leading to tangle formation, cell death and dementia [5].The initiating event is still thought to be a gain-of-toxic-function of the Aβ peptide.
Aβ peptides can be isolated from affected brains, cell cultures expressing FAD mutant APP or synthesised chemically. With these different preparations, myriad downstream effects on cellular processes have been described. However, it remains to be determined what is essentially the cause of neurodegeneration or if all processes work in parallel with different impacts [1, 6]. Especially noteworthy is the connection of Aβ to tau. Besides Aβ deposition, the second hallmark of AD is the accumulation of intracellular neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau protein. Tau phosphorylation and aggregation seems to occur downstream of Aβ oligomerisation [7, 8], with Aβ activating several tau kinases [9, 10]. Deletion of tau in FAD-APP transgenic mice ameliorates many of the cognitive deficits, axonal transport deficits and premature mortality, together demonstrating a central role of tau in neuronal homeostasis [11].
Unresolved issues
Although a great deal of data in favour of the amyloid cascade has been reported, there are still some contradictions that need to be resolved. Findings from post mortem studies revealed persons with an abundance of amyloid plaques, yet showing no mental deterioration until death [12]. The advent of imaging techniques for amyloid deposition in brain has resulted in frequent reports of cognitively healthy elderly with brains full of amyloid plaques [13–15]. The lack of correlation between dementia severity and plaque load is opposed by the strong correlation of synapse loss with dementia [16]. The expression of FAD mutant APP and PS1 in various transgenic mouse models has reproduced human amyloid plaque deposition and memory problems, but has not resulted in widespread neurodegeneration [17]. Even more, mice that solely express the Aβ42 peptide, without the rest of the APP molecule, develop strong plaque deposition as described above, but their cognitive performance is intact [18]. Shutting down transgenic APP expression in inducible mouse models corrects abnormal hypersynchronous network activity and restores cognitive performance with no effect on existing plaque load [19, 20]. Together, these reports show that amyloid plaques alone are not sufficient to mimic the neuropathology of AD.
Loss-of-function
The γ-secretase complex has many more substrates in addition to APP [21]. A result of the intramembraneous cleavage of transmembrane proteins by γ-secretase is the release of the intracellular domain (ICD) from the membrane, which in several cases has been shown to signal to the nucleus to regulate transcription. Many FAD mutations in the presenilin genes have been shown to lower Aβ40 as well as APP ICD (AICD) and Notch ICD (NICD) production, when analysed by means of in vitro assays [22]. With highly purified γ-secretase complexes, different FAD mutations in the PS protein lead to a complete loss of AICD production [23]. In the light of this loss-of-function, the presenilin hypothesis was put forward to explain neuronal death in AD [22].
Mice lacking the presenilin 1 gene are not viable [24]. Therefore, mice with a conditional forebrain knockout of PS1 (cPS1) were generated, to study the effects of loss of presenilin function in the adult brain [25]. PS1 reduction was accompanied by accumulation of transmembrane C-terminal fragments of APP (APP-CTFs = γ-secretase substrates) and reduction of Aβ peptides (γ-secretase products). Crossing of cPS1 mice to PS2 knockout (KO) mice, to exclude compensatory effects by this homologue, resulted in viable mice with intact brain morphology until 6 months of age. This was followed by progressive loss of grey and white matter and thinning of cortical layers, accompanied by reduced dendrite complexity and spine density, as well as disruption of synaptic plasticity and severe memory impairment. Similar findings of massive cortical and hippocampal atrophy due to progressive neuron loss have been reported in independently generated cPS1xPS2KO mice [26]. The loss of presenilin function in brain thus results in many phenotypes that are seen in AD.
New mouse models knock in
A caveat of most mouse models engineered to express FAD mutant PS1 is that the transgene is overexpressed. If PS FAD mutations cause a loss-of-function, this might be compensated for by the increased expression. To circumvent this, Xia et al. created knockin (KI) mice, introducing two different human PS1 FAD mutations (L435F and C410Y) into the mouse presenilin 1 gene locus [27].
Both mouse lines show normal levels of PS1 mRNA, but a drastic reduction of endoproteolysis: PS1 holoprotein increases and N- and C-terminal fragments decrease in a KI allele dosage-dependent manner. Analysing three γ-secretase substrates – APP, N-Cadherin and Notch – the authors report a strong increase in their CTFs in both homozygous mouse lines, equivalent to the increase in homozygous PS1 KO mice. NICD generation is reduced in a dosage-dependent manner, being virtually abolished in homozygous KI lines. At the same time, the generation of Aβ40 and Aβ42 species is absent in homozygous mice. Thus, both presenilin FAD mutations display a clear loss-of-function phenotype, resulting in severe developmental deficits, including decreased proliferation of progenitors in the ventricular zone and thinning of the cortical plate, closely resembling the phenotype of homozygous PS1 KO mice. In heterozygous KI mice the de novo production of Aβ40 and Aβ42 is reduced by approximately 50% in 3-month-old animals, again pointing to a complete loss-of-function due to the FAD mutation. In addition, insoluble brain fractions displayed an increased Aβ42/Aβ40 ratio, a finding also seen in FAD patients [28]. The catalytic core of γ-secretase probably consists of a presenilin dimer [29], although this is debated [30]. In heterozygous animals the dimer can be a mix of wild type and FAD mutant presenilin. FAD mutations alter the conformation of presenilin, thereby affecting how APP is presented to the active site [31]. Homozygous FAD mutant mice have only mutated presenilin in the dimer, inhibiting γ-secretase activity. In heterozygous animals, the mutant presenilin obviously influences the conformation of the active wild-type version, leading to APP cleavage at different positions that result in increased Aβ42/Aβ40 ratios.
Crossing the KI mice with mice overexpressing mutant human APP allowed analysis of plaque deposition. As with endogenous mouse Aβ, the insoluble fraction showed a greater decrease of Aβ40, leading to an increased Aβ42/Aβ40 ratio. Despite the decrease in total Aβ levels, the plaque area in cortex at 9 months of age was 5-fold greater. This is in line with data from the mouse models described earlier. Expression of only Aβ without the rest of APP revealed that the determining factor for plaque deposition is the Aβ42/Aβ40 ratio, not the absolute amount of Aβ [3].
Besides the presenilin 1 gene, the homologous presenilin 2 gene is also affected by FAD mutations, albeit at lower frequency. PS2 expression is upregulated in the absence of PS1 and might then compensate for behavioural and synaptic deficits caused by PS1 deficiency [32]. Therefore, Xia et al. crossed the L435F KI mice onto a PS2 KO background. Heterozygous KI mice on this background showed diverse synaptic deficits. The impaired synaptic plasticity resulted in memory impairment, although the effects were rather subtle. Heterozygous KI mice on a PS2 KO background, with additional deletion of the second wild type presenilin 1 allele, showed the most dramatic effects. Animals aged twelve months showed increased activation of astroglia and microglia in cortex and hippocampus, a cellular reaction that classically accompanies AD. Most stunningly, 12- and 18-month-old animals had reduced cortical volume and neuron numbers with an increase in apoptotic cells. These results are striking in the sense that this is the first mouse model encompassing human FAD mutations that shows massive age-dependent neurodegeneration in the cortex. Nevertheless, one has to bear in mind that the genotype of human FAD cases is very different. These patients still have one wild-type copy of the presenilin 1 and both presenilin 2 alleles, in contrast to the mice showing neurodegeneration that express only the FAD mutant copy of presenilin 1. Still, this kind of genetic exaggeration is common in mouse models – such as the overexpression of FAD-APP – and is used to accelerate processes that take 10–20 years in humans, in order to be able to detect effects during the short lifespan of mice. The findings in the new mouse models show that FAD mutations in PS1 lead to a clear loss-of-function – the question is: which function is lost?
Loss-of-what?
APP is known to play a role in various cellular processes, with the full-length protein itself, as well as the different fragments produced by the secretase cleavages, being involved in distinct functions, including cell adhesion, cell proliferation, cell migration, neurite outgrowth, synaptogenesis, nuclear signalling and regulation of copper, calcium and mitochondrial homeostasis [33, 34]. The extracellular domain is shed through cleavage by α- or β-secretase, with α-cleavage generating a secreted extracellular domain (sAPPα) that has neuroprotective functions and promotes neural progenitor proliferation. But, because α-cleavage precedes APP cleavage by γ-secretase, sAPPα levels are not expected to be directly affected by PS FAD mutations, although this was not analysed by Xia et al. In addition to reducing Aβ levels, loss-of-presenilin-function also reduces AICD production. AICD has been shown to translocate to the nucleus and regulate transcription [35, 36], with approximately 30 target genes described to date [21, 37]. Deregulated expression of these target genes might contribute to the cellular processes that ultimately result in neurodegeneration [38]. Target genes encompass proteins regulating lipid, mitochondrial and APP metabolism, cell cycle, proliferation, apoptosis, tumour suppression, cytoskeleton and synaptic functions – all of which are reported to be altered in AD.
In addition to the effects on APP, loss-of-function of presenilin will affect many other γ-secretase substrates, as shown for Notch and N-Cadherin by Xia et al. Notch has important roles in development, but also in adulthood. Consequently, the recent clinical trials targeting AD by means of γ-secretase inhibition showed severe Notch-related side effects [39, 40]. Altered processing of these additional substrates might also impact the degenerative processes and could be involved in the earlier onset of the disease in presenilin mutant carriers. Nevertheless, there are nearly 50 different FAD mutations in APP and in these cases there is no described effect on the other substrates. Furthermore, in sporadic AD the major hallmark is amyloid plaques derived from processing of APP. In the following section, I have therefore restricted my discussion to APP.
Speculating beyond the cascade
What if Aβ were not the sole or major cause of neurodegeneration in AD? Toxicity has been observed with high concentrations of synthetic Aβ preparations in vitro. But, using naturally secreted Aβ oligomers or those isolated from AD brain, the described effects are restricted to synapses, with impairment of synaptic plasticity and memory [41, 42]. Aβ generation is induced by neuronal activity and it is therefore conceivable that Aβ oligomers are physiologic regulators of synaptic plasticity and memory, with synapse loss being reversible after removal of excess Aβ [43–45].
The lowering of Aβ levels with concomitant changes in the Aβ42/Aβ40 ratio induced by PS FAD mutations or the increased Aβ production due to higher β-secretase expression in sporadic AD [46], will probably lead to alterations of synaptic plasticity that could manifest in memory problems. Over decades, this altered plasticity – leading to a net loss of synapses – could result in failure of neurotrophic support that relies on functioning synapses [47], resulting in neuronal death. Amyloid plaques might be generated to sequester excess amounts of Aβ that would disrupt memory and thus, rather subserve a protective function. Accordingly, amyloid plaques would only tell us that something is wrong with APP metabolism, leading potentially to the development of AD.
At the same time that Aβ generation is altered in familial and sporadic AD, FAD PS mutations reduce AICD production. What about FAD mutations in APP? Most of them also lead to increased Aβ42/Aβ40 ratios. Processing of APP CTFs by γ-secretase occurs in consecutive cleavages along different product lines [48]. Whereas PS FAD mutations block the fourth enzymatic cleavage, leading to accumulation of Aβ42 and Aβ43, APP FAD mutations cause a shift towards the product line producing Aβ42 [49]. In this product line, the initial ε-cleavage releases a one amino acid longer AICD species that starts with a leucine, instead of the N-terminal valine exposed in the product line leading to Aβ40. AICD is suggested to be degraded via the proteasome by an N-end rule-mediated pathway [50]. In this pathway, the N-terminal amino acid determines the stability of the protein, with valine being a stabilising and leucine a destabilising residue. Together, these data imply that the product line leading to higher Aβ42/Aβ40 ratios is inherently coupled to the production of less stable AICD species. Thus, both PS and APP FAD mutations lead to reduced AICD levels. The Swedish APP FAD mutation stands out in that it does not change the Aβ42/Aβ40 ratio, but increases the total level of Aβ, because this mutation renders APP a better β-secretase substrate. Similarly, the increased β-secretase activity in sporadic AD enhances Aβ production [46]. In both cases, higher Aβ levels are accompanied by higher levels of AICD nuclear signalling, as this depends on the β-secretase-mediated cleavage pathway [51]. In addition to enhanced β-secretase activity, there is also evidence for γ-secretase dysfunction in sporadic AD [52] that might again alter AICD production. The resulting deregulation of AICD target genes could further disrupt synaptic function, adding to the effects of Aβ. Increases in β-secretase activity or reduction of γ-secretase activity will also lead to the accumulation of βCTFs that can disrupt neuronal functions, for instance by inducing mitochondrial dysfunction, enhancing endocytosis or disrupting axonal transport of growth factors [53–55].
But what is finally responsible for neurodegeneration in human AD patients? Excess Aβ can be captured in amyloid plaques, whereas the deregulation of AICD-mediated transcription might be more deleterious for neuronal survival. As stated in the introduction, we need “continuous manifold research” into all possible causes of AD to find a cure for this disease.
Acknowledgements:I am grateful to Dr. Stephan Mertens from the Deutsche Ärzteblatt and Dr. Manuel Gersbacher from the Division of Psychiatry Research for a critical review of this manuscript.
References
1 Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8(2):101–12.
2 McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, et al. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005;47(2):191–9.
3 Kim J, Onstead L, Randle S, Price R, Smithson L, Zwizinski C, et al. Abeta40 inhibits amyloid deposition in vivo. J Neurosci. 2007;27(3):627–33.
4 Hardy JA and Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–5.
5 Hardy J and Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–6.
6 Benilova I, Karran E, De Strooper B. The toxic Abeta oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci. 2012;15(3):349–57.
7 Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293(5534):1487–91.
8 Gotz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293(5534):1491–5.
9 Yu W, Polepalli J, Wagh D, Rajadas J, Malenka R, Lu B. A critical role for the PAR-1/MARK-tau axis in mediating the toxic effects of Abeta on synapses and dendritic spines. Hum Mol Genet. 2012;21(6):1384–90.
10 Hoshi M, Sato M, Matsumoto S, Noguchi A, Yasutake K, Yoshida N, et al. Spherical aggregates of beta-amyloid (amylospheroid) show high neurotoxicity and activate tau protein kinase I/glycogen synthase kinase-3beta. Proc Natl Acad Sci U S A. 2003;100(11):6370–5.
11 Morris M, Maeda S, Vossel K, Mucke L. The many faces of tau. Neuron. 2011;70(3):410–26.
12 Snowdon DA. Healthy aging and dementia: findings from the Nun Study. Ann Intern Med. 2003;139(5 Pt 2):450–4.
13 Mormino EC, Kluth JT, Madison CM, Rabinovici GD, Baker SL, Miller BL, et al. Episodic memory loss is related to hippocampal-mediated beta-amyloid deposition in elderly subjects. Brain. 2009;132(Pt 5):1310–23.
14 Villemagne VL, Pike KE, Chetelat G, Ellis KA, Mulligan RS, Bourgeat P, et al. Longitudinal assessment of Abeta and cognition in aging and Alzheimer disease. Ann Neurol. 2011;69(1):181–92.
15 Jansen WJ, Ossenkoppele R, Knol DL, Tijms BM, Scheltens P, Verhey FR, et al. Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. JAMA. 2015;313(19):1924–38.
16 Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30(4):572–80.
17 Braidy N, Munoz P, Palacios AG, Castellano-Gonzalez G, Inestrosa NC, Chung RS, et al. Recent rodent models for Alzheimer’s disease: clinical implications and basic research. Journal of neural transmission. 2012;119(2):173–95.
18 Kim J, Chakrabarty P, Hanna A, March A, Dickson DW, Borchelt DR, et al. Normal cognition in transgenic BRI2-Abeta mice. Mol Neurodegener. 2013;8:15.
19 Born HA, Kim JY, Savjani RR, Das P, Dabaghian YA, Guo Q, et al. Genetic suppression of transgenic APP rescues Hypersynchronous network activity in a mouse model of Alzeimer's disease. J Neurosci. 2014;34(11):3826–40.
20 Fowler SW, Chiang AC, Savjani RR, Larson ME, Sherman MA, Schuler DR, et al. Genetic modulation of soluble Abeta rescues cognitive and synaptic impairment in a mouse model of Alzheimer’s disease. J Neurosci. 2014;34(23):7871–85.
21 Pardossi-Piquard R and Checler F. The physiology of the beta-amyloid precursor protein intracellular domain AICD. J Neurochem. 2012;120(Suppl 1):109–24.
22 Shen J and Kelleher RJ, 3rd. The presenilin hypothesis of Alzheimer's disease: evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci U S A. 2007;104(2):403–9.
23 Cacquevel M, Aeschbach L, Houacine J, Fraering PC. Alzheimer’s Disease-Linked Mutations in Presenilin-1 Result in a Drastic Loss of Activity in Purified gamma-Secretase Complexes. PloS one. 2012;7(4):e35133.
24 Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell. 1997;89(4):629–39.
25 Saura CA, Choi SY, Beglopoulos V, Malkani S, Zhang D, Shankaranarayana Rao BS, et al. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron. 2004;42(1):23–36.
26 Feng R, Wang H, Wang J, Shrom D, Zeng X, Tsien JZ. Forebrain degeneration and ventricle enlargement caused by double knockout of Alzheimer’s presenilin-1 and presenilin-2. Proc Natl Acad Sci U S A. 2004;101(21):8162–7.
27 Xia D, Watanabe H, Wu B, Lee SH, Li Y, Tsvetkov E, et al. Presenilin-1 knockin mice reveal loss-of-function mechanism for familial Alzheimer’s disease. Neuron. 2015;85(5):967–81.
28 Kumar-Singh S, Theuns J, Van Broeck B, Pirici D, Vennekens K, Corsmit E, et al. Mean age-of-onset of familial alzheimer disease caused by presenilin mutations correlates with both increased Abeta42 and decreased Abeta40. Hum Mutat. 2006;27(7):686–95.
29 Schroeter EH, Ilagan MX, Brunkan AL, Hecimovic S, Li YM, Xu M, et al. A presenilin dimer at the core of the gamma-secretase enzyme: insights from parallel analysis of Notch 1 and APP proteolysis. Proc Natl Acad Sci U S A. 2003;100(22):13075–80.
30 Sato T, Diehl TS, Narayanan S, Funamoto S, Ihara Y, De Strooper B, et al. Active gamma-secretase complexes contain only one of each component. J Biol Chem. 2007;282(47):33985–93.
31 Berezovska O, Lleo A, Herl LD, Frosch MP, Stern EA, Bacskai BJ, et al. Familial Alzheimer's disease presenilin 1 mutations cause alterations in the conformation of presenilin and interactions with amyloid precursor protein. J Neurosci. 2005;25(11):3009–17.
32 Watanabe H, Iqbal M, Zheng J, Wines-Samuelson M, Shen J. Partial loss of presenilin impairs age-dependent neuronal survival in the cerebral cortex. J Neurosci. 2014;34(48):15912–22.
33 Zheng H and Koo EH. Biology and pathophysiology of the amyloid precursor protein. Mol Neurodegener. 2011;6(1):27.
34 Dawkins E and Small DH. Insights into the physiological function of the beta-amyloid precursor protein: beyond Alzheimer's disease. J Neurochem. 2014;129(5):756–69.
35 von Rotz RC, Kohli BM, Bosset J, Meier M, Suzuki T, Nitsch RM, et al. The APP intracellular domain forms nuclear multiprotein complexes and regulates the transcription of its own precursor. Journal of cell science. 2004;117(Pt 19):4435–48.
36 Cao X and Sudhof TC. A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 2001;293(5527):115–20.
37 Grimm MO, Mett J, Stahlmann CP, Haupenthal VJ, Zimmer VC, Hartmann T. Neprilysin and Abeta Clearance: Impact of the APP Intracellular Domain in NEP Regulation and Implications in Alzheimer’s Disease. Front Aging Neurosci. 2013;5:98.
38 Konietzko U. AICD nuclear signaling and its possible contribution to Alzheimer's disease. Current Alzheimer research. 2012;9(2):200–16.
39 Coric V, Salloway S, van Dyck CH, Dubois B, Andreasen N, Brody M, et al. Targeting Prodromal Alzheimer Disease With Avagacestat: A Randomized Clinical Trial. JAMA neurology. 20151–10.
40 Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. The New England journal of medicine. 2013;369(4):341–50.
41 Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416(6880):535–9.
42 Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14(8):837–42.
43 Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27(11):2866–75.
44 Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, et al. APP processing and synaptic function. Neuron. 2003;37(6):925–37.
45 Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, et al. AMPAR Removal Underlies Abeta-Induced Synaptic Depression and Dendritic Spine Loss. Neuron. 2006;52(5):831–43.
46 Yang LB, Lindholm K, Yan R, Citron M, Xia W, Yang XL, et al. Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med. 2003;9(1):3–4.
47 Howe CL and Mobley WC. Long-distance retrograde neurotrophic signaling. Curr Opin Neurobiol. 2005;15(1):40–8.
48 Kakuda N, Funamoto S, Yagishita S, Takami M, Osawa S, Dohmae N, et al. Equimolar production of amyloid beta-protein and amyloid precursor protein intracellular domain from beta-carboxyl-terminal fragment by gamma-secretase. J Biol Chem. 2006;281(21):14776–86.
49 Chavez-Gutierrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, et al. The mechanism of gamma-Secretase dysfunction in familial Alzheimer disease. The EMBO journal. 2012
50 Gersbacher MT, Goodger ZV, Trutzel A, Bundschuh D, Nitsch RM, Konietzko U. Turnover of amyloid precursor protein family members determines their nuclear signaling capability. PLoS ONE. 2013;8(7):e69363.
51 Goodger ZV, Rajendran L, Trutzel A, Kohli BM, Nitsch RM, Konietzko U. Nuclear signaling by the APP intracellular domain occurs predominantly through the amyloidogenic processing pathway. Journal of cell science. 2009;122(Pt 20):3703–14.
52 Hata S, Fujishige S, Araki Y, Taniguchi M, Urakami K, Peskind E, et al. Alternative processing of gamma-secretase substrates in common forms of mild cognitive impairment and Alzheimer’s disease: evidence for gamma-secretase dysfunction. Ann Neurol. 2011;69(6):1026–31.
53 Kim S, Sato Y, Mohan PS, Peterhoff C, Pensalfini A, Rigoglioso A, et al. Evidence that the rab5 effector APPL1 mediates APP-betaCTF-induced dysfunction of endosomes in Down syndrome and Alzheimer's disease. Mol Psychiatry. 2015
54 Devi L and Ohno M. Mitochondrial dysfunction and accumulation of the beta-secretase-cleaved C-terminal fragment of APP in Alzheimer’s disease transgenic mice. Neurobiol Dis. 2012;45(1):417–24.
55 Salehi A, Delcroix JD, Belichenko PV, Zhan K, Wu C, Valletta JS, et al. Increased App expression in a mouse model of Down’s syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron. 2006;51(1):29–42.