The CRISPR revolution and its impact on cancer research
DOI: https://doi.org/10.4414/smw.2015.14230
Ram
Kannan, Andrea
Ventura
Summary
A revolution in cancer research is underway. Spurred by the advent of the CRISPR-Cas9 technology, new methods to probe the mammalian genomes are being developed. By providing simple, flexible, and cost-effective ways to edit and manipulate the genome of somatic cells of adult animals, these new methods present the opportunity to model cancer progression in vivowith an unprecedented degree of sophistication. Here we provide a brief overview of this exciting and fast-moving field.
An overview of the CRISPR-Cas system
Unless you have been living under a rock for the past three years, you have probably already heard of CRISPR (clustered regularly interspaced short palindromic repeats) – Cas (CRISPR-associated), the powerful new technology that allows precise manipulation of the genome of individual cells. This brief review will focus on the applications of CRISPR-based genome editing methods in cancer research, with a particular emphasis on its in vivoapplications.
The anatomy and biochemistry of CRISPR systems have been covered in depth in many excellent reviews (for example see [1–4]), but a brief description of CRISPR’s essential features is needed before focusing on its practical applications.
Twelve different CRISPR-Cas systems have been identified in bacteria and archaea, where they serve as RNA-guided antiviral defense mechanisms. Although important differences exist between the various systems, all CRISPR systems use short RNA fragments derived from the CRISPR loci in complex with one or more Cas proteins to identify and neutralize invading nucleic acids (see legend of figure 1 for additional details).
Dawn of the CRISPR revolution
Despite capturing the interest of microbiologists and molecular biologists, the CRISPR-Cas system stayed at least initially under the radar of the general biomedical research community. This suddenly changed when several groups demonstrated the Cas9 protein from Streptococcus Pyogenouscould be reprogrammed with synthetic RNAs to generate site-specific double-strand breaks (DSBs) in vitro [5] and in mammalian cells [6–8]. The CRISPR-Cas9 system is an example of a type II CRISPR-Cas system and was chosen because a single protein (Cas9) is sufficient to induce cleavage at the target site. In S. PyogenousCas9 is complexed with two RNAs: the crRNA, whose specific sequence confers target-specificity, and a 77-nucleotide transactivatingRNA (tracrRNA), which is required for target cleavage [5]. Fortunately, a single chimeric RNA – often referred to as sgRNA or gRNA – can effectively replace the two RNAs [5], thus providing a versatile binary system to introduce specific DSBs in mammalian cells [7]. The only requirement for CRISPR-Cas9 to function is the presence, on the target DNA, of a short sequence known as “protospacer adjacent sequence” (PAM) that has to be located immediately downstream of the sequence recognized by the gRNA. The PAMs recognized by the S. Pyogenous Cas9 are “NGG” and – with much reduced efficiency – “NAG” [9]. This requirement somewhat limits the sequence space that can be edited by CRISPR-Cas9. To overcome this limitation, modified versions of the Cas9 protein have been engineered that recognize different PAM sequences [10]. In addition, the Zhang group has recently shown that naturally occurring Cas proteins obtained from other microorganisms, and recognizing different PAM sequences, can also be used for genome editing in eukaryotes [11, 12].
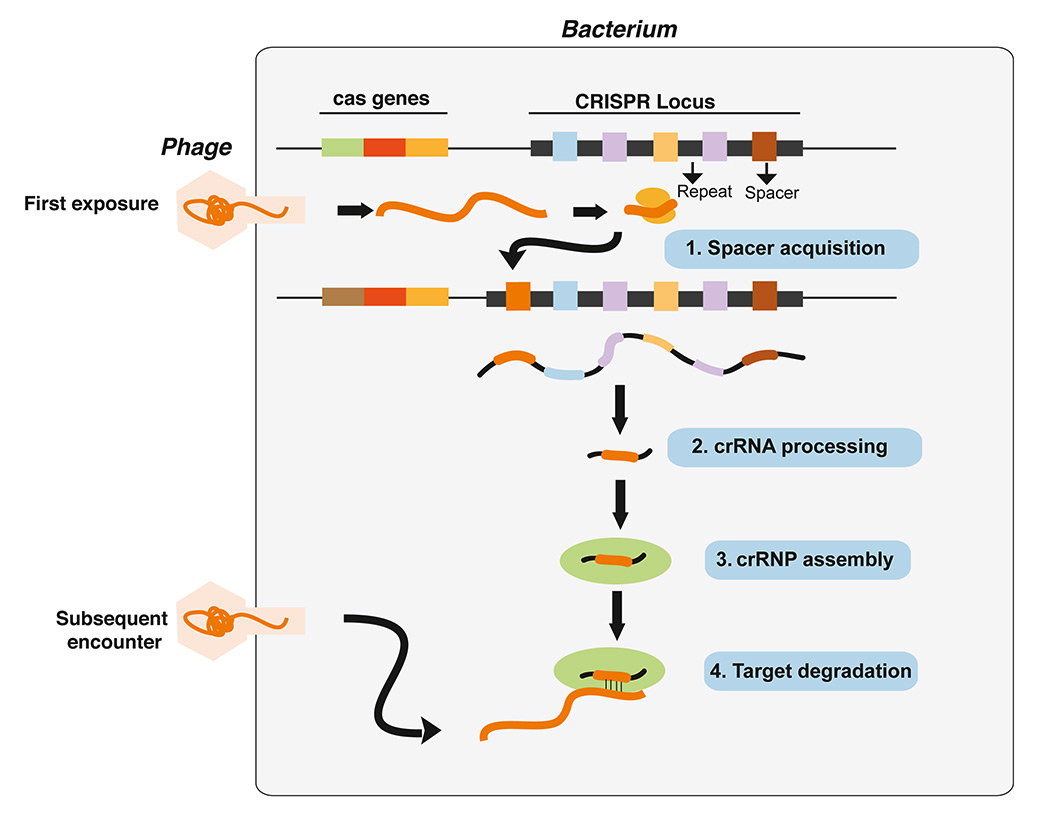
Figure 1
CRISPR directed antiviral immunity in bacteria.
An outline of how CRISPR-mediate antiviral immunity functions in bacteria. Upon defective viral infection, short viral sequences are inserted into the CRISPR array, where they become the new “spacers” flanked by repeats. The spacers, thus provide a memory of past infection. To ensure immunity to subsequent infection, the entire CRISPR array is transcribed and processed into short crisprRNAs (crRNAs), which form complexes with other “effector” Cas proteins and guide them to degrade invading viral DNA or RNA in a sequence-specific manner.
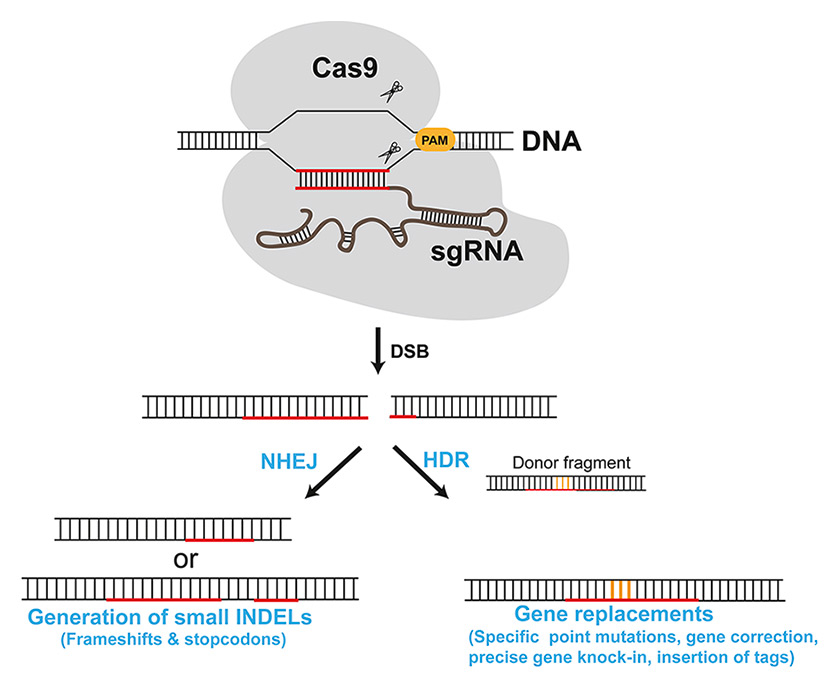
Figure 2
Genome editing by CRISPR-Cas9. Schematic of possible outcomes of CRISPR-Cas9-induced DSBs. Repairing via non-homologous end joining (NHEJ) generates indels, while repair via homologous recombination in the presence of an appropriate donor fragment can be used to introduce specific genetic changes (in orange). The sequence recognized by the sgRNA is in red. The location of the protospacer adjavent motif (PAM) in the target site is also shown.
The ability to introduce specific DSBs into the genome of mammalian cells is not entirely new: TALENs and zinc finger nucleases engineered to bind and cleave at specific sites have been used previously with success [13, 14]. However, the intrinsic simplicity of the CRISPR-Cas9 system – for which generating a new guide RNA is all that is needed to cleave at a different site – has made somatic genome editing available to virtually any laboratory equipped for basic molecular biology.
But how does introducing a site-specific DSB allow for genome editing? Two major pathways repair DSBs in eukaryotic cells: error prone non-homologous end joining (NHEJ), and homology directed repair (HDR) (fig. 2). The first, which is the predominant mechanism in cells that are in the G0 and G1 phase of the cell cycle, results in the generation of usually short indels at the repair site, and is thus ideal to cause frame-shift mutations in protein coding genes. HDR, on the other hand, is mainly active in S and G2 phases and uses homologous sequences present in the cell genome (or provided by the investigator) to repair the DSB. It can therefore be exploited to engineer precise genetic changes and, in principle, to correct genetic defects (fig. 2).
In vivosomatic genome editing: building better mouse models of human cancer
For the past two decades, gene targeting by homologous recombination in embryonic stem cells has been the gold standard to develop genetically engineered mouse models of human cancers [15]. When used in conjunction with site-specific recombinases such as Cre or Flpe, this technology enables the generation mice harboring loss-of-function and gain-of-function alleles of tumor suppressors and oncogenes whose expression can be induced in a temporally and spatially controlled manner. These models have proven essential in dissecting the complex molecular mechanisms underlying tumor initiation and progression in vivo and are indispensable to study tumor-host interactions in a physiologic context.
However, making a new genetically engineered mouse model is a technically challenging, time-consuming, and costly process. Murine embryonic stem cells engineered by homologous recombination are injected into blastocysts to generate chimeric mice, which are further bred to achieve germ-line transmission of the mutant alleles. If the allele is inducible, as is often desirable, mice have to be crossed to the appropriate Cre- or Flpe-expressing strain. Altogether, the process can easily take more than a year before the phenotypic consequences of the mutation introduced can be evaluated. These limitations are overcome by CRISPR-based genome editing, which not only provides a more efficient and accessible platform to introduce specific genetic changes in ES cells or in the zygote, but also offers the unprecedented opportunity to modify directly the genome of somatic cells of adult animals [16–20].
In addition to the obvious therapeutic potential of in vivo somatic genome editing as a way to correct genetic defects [21, 22], its applications to cancer modeling have already been convincingly demonstrated (table 1). For example, several groups have shown that direct in vivodelivery of Cas9 and the relevant gRNA to the tissue of interest using naked DNA or viral vectors can be used to inactivate somatically tumor-suppressor genes and initiate tumorigenesis [18, 19, 23–26]. This strategy has been successfully employed to inactivate the tumor suppressors Tp53, Lkb1, Apc, and Pten in the lungs of adult mice [18, 24]. Notably, available data indicate that the tumors obtained in these experiments are histologically very similar to the tumors that are observed in mice in which the same tumor-suppressor genes are inactivated using conventional gene targeting. For example, CRISPR-mediated Nkx2-1 inactivation in a KRasG12D; p53-null background was shown to induce lung cancers with a characteristic mucinous histology [24], identical to what had been previously described using a floxed Nkx2-1 allele [27]
The lung is not the only organ in which somatic gene inactivation has been successfully used to induce tumors in mice. For example, hydrodynamic injection of plasmids expressing Cas9 and sgRNAs against Pten and Tp53 lead to the formation of hepatocellular carcinomas [19], while retrograde pancreatic ductal injection of lentiviral vectors expressing sgRNAs against Lkb1in KRas-G12D-expressing mice resulted in ductal pancreatic adenocarcinomas [23]. Gene editing in mouse pancreas has also been recently employed to demonstrate a key role for the cyclin-dependent kinase inhibitor p57 in mediating the therapeutic response of pancreatic ductal adenocarcinoma to JQ1 and SAHA [28]. Finally, postnatal and in utero CRISPR-mediated inactivation of Ptch1, Pten, Nf1, and Tp53 has been achieved in the brain, where it has been employed to model medulloblastomas (Ptch1) and glioblastomas (concomitant inactivation of Pten, Nf1, and Tp53) [26].
Equally appealing is the possibility of using CRISPR-Cas9 to generate somatically specific point mutations in adult mice. Although technically more challenging and less efficient because it requires the concomitant delivery of a “donor” DNA fragment carrying the desired mutation, the feasibility of this approach has been recently demonstrated with the generation of activating mutations of K-Ras in the lung [18] and of beta-catenin in the liver [19].
A major limitation of CRISPR-based somatic genome editing is the need to simultaneously deliver the guide RNA and the Cas9 enzyme to the tissue of interest. To overcome this limitation, three groups have recently generated transgenic mice expressing Cas9 in a Cre- [18, 23] or tetracycline- dependent [29] manner. Because the Cas9 nuclease is encoded by the animal genome, the investigator need only to introduce the desired gRNAs and (for gene replacement experiments) the donor template. This allows the use of recombinant adeno-associated viruses, for example, which can infect a wide array of cell types but whose genome would not be able to accommodate the large cDNA encoding Cas9. Moreover, because the guide RNAs cannot cleave in the absence of Cas9, the use of these animals virtually eliminates potential biohazards for the investigators.
|
Table 1: Cancer mouse models developed by CRISPR–Cas9 based genome editing |
|
Cancer type
|
Mouse strain and genotype
|
Delivery
|
Mutations/ genomic alterations
|
References
|
|
In vivo
|
| Lung adenocarcinoma |
CD1 and C57BL/6J (B6) |
Adenoviral |
Eml4-‐Alktranslocation in lung |
Maddalo D, et al. Nature. 2014 [17] |
| Lung adenocarcinoma |
p53+/-− or p53-‐/-‐
|
Lentiviral |
Eml4-‐Alktranslocation in lung |
Blasco RB, et al. Cell Reports. 2014 [37] |
| Lung adenocarcinoma |
KrasLSL-G12D/+
|
Lentiviral |
Nkx, Pten, Apc
|
Sanchez-Rivera FJ, et al. Nature. 2014 [24] |
| Liver cancer |
FVB/NJ mice |
Hydrodynamic injection |
p53, Pten,Ctnb1
|
Xue W, et al. Nature. 2014 [19] |
| Burkitt lymphoma |
Arf/- EμMyc |
Lentiviral and retroviral |
p53
|
Malina A, et al. Genes and Dev. 2013 [60] |
| Pancreatic ductal adenocarcinoma |
KrasLSL-‐G12D/+; R26LSL-Tom; H11LSL- Cas9/+ |
Retrograde ductal injection of adeno/lentivirus |
Lkb1
|
Chiou, et al. Genes and Dev. 2015 [23] |
| Pancreatic ductal adenocarcinoma |
Kras
+/LSL-‐G12D; Trp53
loxP/loxP
|
Lentiviral |
p53, Kras G12D, and p57
|
Mazur, et al. Nature Medicine. 2015 [28] |
| Medulloblastoma |
C57BL/6N mice |
PEI-mediated transfection and inutero electroporation |
Ptch1
|
Zuckermann M , et al. Nature Communications. 2015 [26] |
| Glioblastoma |
Crl:CD1(ICR) mice |
PEI-mediated transfection and inutero electroporation |
Trp53,Pten,Nf1
|
Zuckermann M , et al. Nature Communications. 2015 [26] |
|
Ex vivo
|
| Acute myeloid leukemia |
C57Bl/6 mice or heterozygous Flt3-‐ITD knock-‐in mice |
Lentiviral |
Dnmt3a, Ezh2,Runx1, Smc3 and Nf1
|
Heckl, et al. Nature Biotech. 2014 [16] |
| Acute myeloid leukemia |
p53null HSPC |
Lentiviral |
Mll3
|
Chen, et al. Cancer Cell. 2014 [61] |
| Burkitt lymphoma |
HSPC from Eμ-‐Myc mice |
Lentiviral |
p53
|
Aubrey, et al. Cell Reports. 2015 [62] |
|
Xenograft
|
| Tumor regression |
Nude-‐Foxn1
numice |
Lipid transfection |
Correction of PKCβA509T |
Antal C, et al. Cell. 2015 [63] |
|
Cas9 knock-in
|
| Lung adenocarcinoma
|
Cre-‐dependent Cas9
|
Adenoviral delivery of guide RNA
|
p53, Lkb1 and Kras G12D
|
Platt, et al. Cell. 2014 [18]
|
| Intestinal hyperplastic polyps |
Doxycycline-inducible Cas9 |
ESC targeting |
Apc, Trp53
|
Dow, et al. Nature Biotech. 2015 [29] |
| Pancreatic ductal adenocarcinoma |
KrasLSL-‐G12D/+;R26LSL-‐Tom;H11LSL-‐Cas9/+ |
Retrograde ductal injection of adeno/lentivirus |
Lkb1
|
Chiou, et al. Genes and Dev. 2015 [23] |
|
In vivo metastases screen
|
| Lung metastases |
Kras
G12D/+;p53
−/−;Dicer1
+/−
|
Lentiviral |
Multiple hits from screen |
Chen, et al. Cell. 2015 [14] |
Modeling chromosomal rearrangements using CRISPR-Cas9
The CRISPR technology is also uniquely suited to engineer chromosomal rearrangements, a class of mutations that are particularly challenging to model using conventional gene-targeting strategies. Commonly observed in human cancers, chromosomal rearrangements can contribute to tumorigenesis through a variety of mechanisms, including inactivation of tumor-suppressor genes, transcriptional activation of oncogenes, and the generation of oncogenic gene fusions. Engineering this class of mutations using gene targeting by homologous recombination requires the generation of mice harboring loxP sites at the desired breakpoints and the expression of the Cre-recombinase to induce the rearrangement [30, 31]. With CRISPR, cells harboring a specific chromosomal rearrangement can be generated much more rapidly because only the transient expression of Cas9 and two gRNAs is required [32, 33]. This approach is very effective in murine ES cells and has already been used to generate mouse models of human diseases caused by structural chromosomal variants [34, 35]. In principle the same approach can also be used to revert a disease-causing chromosomal rearrangements, as has been recently reported for hemophilia type A [36].
Of relevance to cancer modeling, this strategy can be used to engineer specific rearrangements directly in adult animals, a process that entirely bypasses the need to modify the germline and allows for the rapid generation of preclinical models of human cancers driven by structural chromosomal aberrations.
Our group and the Chiarle group have recently provided proof of concept for this approach by modeling EML4-ALK driven non-small cell lung cancer in mice [17, 37]. In humans, the EML4-ALK gene fusion is generated by an inversion on the short arm of chromosome 2 that leads to the juxtaposition of the echinoderm microtubule-associated protein like 4 (EML4) and the anaplastic lymphoma kinase (ALK) genes [38]. This rearrangement characterizes a subset (4–5%) of non-small cell lung cancers and is clinically relevant because it confers strong sensitivity to targeted therapy with ALK inhibitors [39].
To model this rearrangement in vivo, we used recombinant adenoviral vectors to deliver Cas9 and two gRNAs targeting intron 14 of Eml4 and intron 20 of ALK (corresponding to the most common breakpoints observed in EML4-ALK+ lung cancers) to the lungs of adult mice. Intratracheal injection of the virus in wild-type animals resulted in the development of multiple bilateral lung adenocarcinomas within 8 weeks. Importantly, every analyzed tumor harbored the desired Eml4-Alk inversion, and the lesions rapidly regressed upon treatment with the ALK inhibitor crizotinib [17]. Chiarle and colleagues obtained analogous results using two independent lentiviral vectors to deliver the two gRNAs rather than a adenoviral vector [37].
In addition to its speed and simplicity, this novel CRISPR-based approach offers other important advantages compared with conventional transgenic strategies. By inducing the rearrangement in only a subset of somatic cells, the resulting lesions more closely recapitulate the stochastic nature of tumor formation in humans. In addition, by engineering the specific chromosomal rearrangement, transcriptional and epigenetic regulations of the gene fusion are faithfully reproduced, as are the reduced dosage of the wild-type alleles and the expression of the reciprocal product of the translocation/inversion. Finally, tumor initiation and tumor load can be easily modulated by controlling the timing of infection and the viral titer.
Whether this general strategy can be adapted to model other types of cancers remains to be determined, but preliminary studies from our lab indicate that a wide range of rearrangements – including duplications and reciprocal translocations – can be efficiently engineered. Provided that a feasible way to infect the relevant cell type is available, it seems likely that somatic genome editing will substantially increase the number of human cancers that can be modeled in mice.
CRISPR-based screens
An appealing aspect of the CRISPR technology is the ease with which large libraries can be generated for functional screens. Because the short RNA sequence determines the site of cleavage, pooled libraries against hundreds or thousands of genes can be created in a relatively inexpensive way. In fact, targeted and genome-wide CRISPR-libraries have already been successfully used for positive selection screens [40–42]. The power of this approach was recently demonstrated in a study in which a genome-wide library containing more than 65,000 gRNAs was used to identify drivers of cancer metastasis [40].
Compared to positive selection screens, negative selection screens are more challenging because, on average, one in three indels will not generate a frameshift and therefore bi-allelic gene disruption can be expected in only approximately 45% of cells. This is a significant problem for drop-out screens because it results in unacceptably high background noise. A clever way around this limitation is to design the gRNAs against essential protein domains rather than against the first coding exons, so that even non-frameshift mutations are likely to result in a functionally null allele. Vakoc and colleagues demonstrated the feasibility of this approach by using a gRNA library designed against 192 protein domains involved in chromatin regulation to identify novel therapeutic targets in an acute myeloid leukemia cell line [43].
CRISPR-based screens are also likely to have a major impact on the study of DNA regulatory elements and non-coding RNAs. Because the actual DNA sequence is irreversibly modified, the CRISPR technology has clear advantages compared to RNAi screens as it allows screening for functionally relevant DNA regulatory elements. Analogously, using paired gRNA vectors designed to generate targeted deletions of non-coding RNA loci should make it possible to systematically explore the functions of this largely uncharacterized class of genes [44].
CRISPR-based approaches to modulate gene expression
Although genome editing is the most obvious application of the CRISPR-Cas system in cancer research, it is not the only one. Cong et al. have generated a Cas9 mutant that can still be recruited to specific sites in a gRNA-dependent manner even though it has lost the ability to introduce DSB [6]. When fused to chromatin-modifying enzymes [45, 46], transcriptional activators [47], or repressor domains [48], dCas9 can be used to modulate the expression of endogenous loci in a selective and powerful manner. Additional flexibility can be achieved by indirectly tethering the second protein to Cas9d using the guide RNA itself [45, 50]. It is easy to predict that these new ways of using CRISPR will lead to a better understanding of gene regulation and chromatin dynamics in cancer. These systems could be adapted to reversibly control the expression of endogenous oncogenic or tumor-suppressive loci directly in vivo, especially when combined with transgenic mice expressing the mutant Cas9 in a temporally and spatially controlled manner [48, 51].
The road ahead
The pace at which new applications of CRISPR-Cas9 are being reported is astounding. Although predicting the future is notoriously risky, it is probably safe to say that the application of CRISPR-based somatic genome editing to model cancer in mice and other organism will greatly accelerate the pace of discovery.
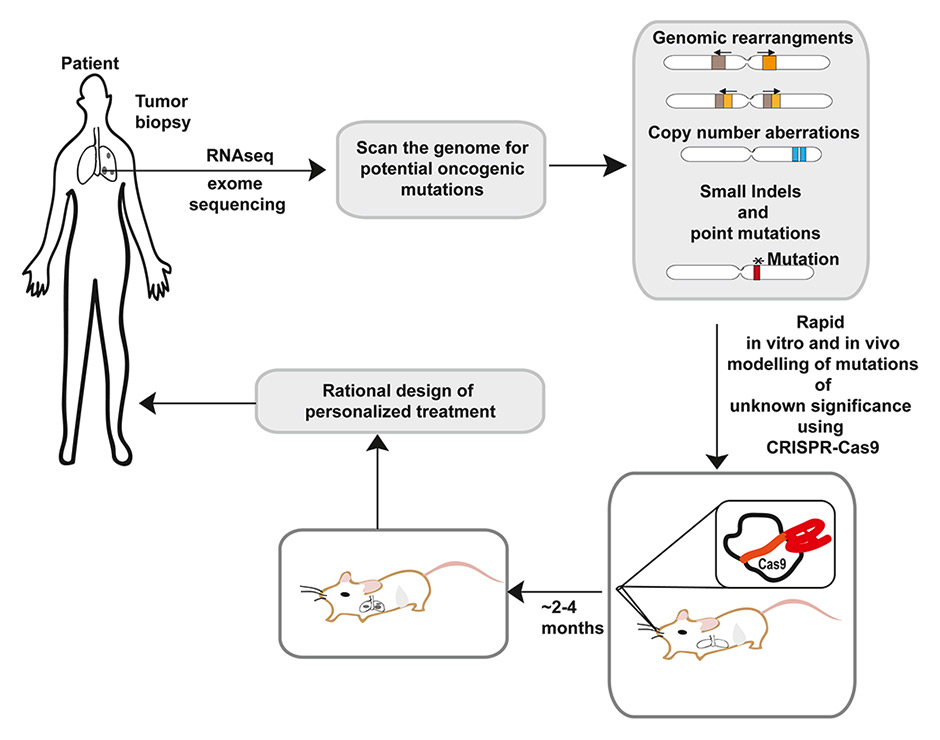
Figure 3
Applications of CRISPR-Cas9 in personalized medicine.
A possible scenario for the use of CRISPR-Cas9 in personalized medicine. Genetic information derived from high-throughput analysis of the patient’s tumor samples often results in the identification of mutations of unclear pathogenetic significance in known oncogenes and tumor suppressors. CRISPR could be used to model such mutations in vitro and in vivo and rapidly determine their functional consequences. Such information can then be used to design an optimized personalized treatment.
Furthermore, combining somatic genome editing with the vast array of already available genetically engineered strains to sequentially introduce specific genetic lesions will provide new insights into tumor evolution and metastasis. As already mentioned, specific areas that will likely benefit the most from these new approaches include the investigation of the non-coding fraction of the human genome and the functional characterization of recurrent chromosomal rearrangements. Beyond the commonly used animals to model cancer like Mus musculus,the relative ease of using CRISPR-Cas9 technologies should allow cancer modeling in other model organisms, including pigs [52], non-human primates [53], and other vertebrates [54].
An important benefit of the CRISPR-Cas system is that it provides an experimental pipeline to rapidly model and functionally test newly identified mutations in oncogenes and tumor suppressor genes. This could have important implication in personalized medicine where it could be used to guide the choice of the optimal targeted therapy for individual cancer patients (fig. 3).
Of even more direct translational value is the use of CRISPR-based genome editing to, for example, facilitate the engineering of patient-derived chimeric antigen receptor (CAR) T-cells. In this setting, CRISPR could be used to mediate the insertion of the desired T cell receptor gene at a specific locus in the T cell genome, thus ensuring optimal expression and eliminating undesired effects.
As with any new technology, the initial phase of excitement is invariably followed by a more careful evaluation of its limitations. In the case of CRISPR, reducing or at least taking into account off-target effects is clearly of great importance. The use of the Cas9-nickase together with two offset gRNAs has been shown to greatly reduce the chance of undesired DSBs and is an encouraging step in the right direction. The recent development of methods to rapidly generate large pooled libraries of paired gRNA vectors should allow the use of Cas9-nickase-based approaches also for large scale screens [55].
Equally important will be the creation of Cas enzymes with intrinsically lower tendency to cleave off-target sites, and the development of improved algorithms to identify gRNAs with reduced off-target potential.
In addition to improving specificity, other important limitations need to be overcome before the full potential of CRISPR-based genome editing is realized. For example, although currently available viral vectors have been used with some success, more effective methods to deliver the Cas enzyme and the gRNAs to somatic cells of adult animals are needed. Encouraging steps in this direction are the discovery of more compact Cas enzymes [11], as well as the development of new non-viral methods to transfer proteins and nucleic acids to tissues and cells [56, 57].
It will also be important to improve the efficiency of CRISPR-mediated gene replacement in order to be able to introduce specific genetic changes – including gain-of-function mutations in oncogenes – in vivo. This result could be achieved by temporarily suppressing NHEJ to enhance HDR, as recently reported in cells [58, 59].
Finally, the widespread availability of Cas9-expressing viral vectors, together with the ease with which tumor-suppressor genes can be inactivated or oncogenes induced, raises concerns about possible biohazards. Clear guidelines will have to be implemented and strictly followed to minimize such risks and to take full advantage of the incredible potential of somatic genome editing.
Despite all these caveats and limitations, the CRISPR revolution is here to stay and will be seen by future generations as a watershed moment in cancer research. A bright future lays ahead, filled with opportunities and exciting discoveries.
Acknowledgements: We thank Jennifer Hollenstein for editing the manuscript, and members of the Ventura lab for comments and suggestions. We apologize to our colleagues whose work we couldn’t cite due to space limitation. Work on genome editing in the Ventura lab is supported by the MSK Cancer Center Support Grant/Core Grant (P30 CA008748) and by grants from the Geoffrey Beene Cancer Research Foundation, the Uniting Against Lung Cancer Foundation, and the Cycle for Survival Foundation.
References
1 Barrangou R, Marraffini LA. CRISPR-Cas systems: Prokaryotes upgrade to adaptive immunity. Mol Cell. 2014;54:234–44.
2 Bondy-Denomy J, Davidson AR. To acquire or resist: the complex biological effects of CRISPR-Cas systems. Trends Microbiol. 2014;22:218–25.
3 Plagens A, Richter H, Charpentier E, Randau L. (2015). DNA and RNA interference mechanisms by CRISPR-Cas surveillance complexes. FEMS Microbiol Rev.
4 van der Oost J, Westra ER, Jackson RN, Wiedenheft B. Unravelling the structural and mechanistic basis of CRISPR-Cas systems. Nat Rev Microbiol. 12:479-92.
5 Jinek M, Chylinski K, Fonfara I, HauerM, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–21.
6 Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013; 339:819–23.
7 Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. RNA-programmed genome editing in human cells. Elife. 2013;2:e00471.
8 Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–6.
9 Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013; 31:827–32.
10 Kleinstiver BP, Prew MS, Tsai SQ, Topkar VV, Nguyen NT, Zheng Z, et al. (2015). Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature.
11 Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–91.
12 Zetsche B, Gootenberg Jonathan S, Abudayyeh Omar O, Slaymaker Ian M, Makarova Kira S, Essletzbichler P, et al. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell.
13 Kim H, Kim JS. A guide to genome engineering with programmable nucleases. Nat Rev Genet. 2014;15;321–34.
14 Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. 2010; 11:636–46.
15 Tuveson DA, Jacks T. Technologically advanced cancer modeling in mice. Current opinion in genetics & development. 2002;12:105–10.
16 Heckl D, Kowalczyk MS, Yudovich D, Belizaire R, Puram RV, McConkey ME, et al. Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat Biotechnol. 2014;32:941–6.
17 Maddalo D, Manchado E, Concepcion CP, Bonetti C, Vidigal JA, Han YC, et al. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature. 2014;516:423–7.
18 Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014;159:440–55.
19 Xue W, Chen S, Yin H, Tammela T, Papagiannakopoulos T, Joshi NS, et al. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature. 2014;514:380–4.
20 Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 2013;154:1370–9.
21 Long C, McAnally JR, Shelton JM, Mireault AA, Bassel-Duby R, Olson EN. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science. 2014;345:1184–8.
22 Yin H, Xue W, Chen S, Bogorad RL, Benedetti E, Grompe M, et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotech. 2014;32:551–3.
23 Chiou SH, Winters IP, Wang J, Naranjo S, Dudgeon C, Tamburini FB, et al. Pancreatic cancer modeling using retrograde viral vector delivery and in vivo CRISPR/Cas9-mediated somatic genome editing. Genes & development. 2015;29:1576–85.
24 Sanchez-Rivera FJ, Papagiannakopoulos T, Romero R, Tammela T, Bauer MR, Bhutkar A, et al. Rapid modelling of cooperating genetic events in cancer through somatic genome editing. Nature. 2014;516:428–31.
25 Wang D, Mou H, Li S, Li Y, Hough S, Tran K, et al. (2015). Adenovirus-mediated somatic genome editing of Pten by CRISPR/Cas9 in mouse liver in spite of Cas9-specific immune responses. Hum Gene Ther.
26 Zuckermann M, Hovestadt V, Knobbe-Thomsen CB, Zapatka M, Northcott PA, Schramm K, et al. Somatic CRISPR/Cas9-mediated tumour suppressor disruption enables versatile brain tumour modelling. Nature communications. 2015;6:7391.
27 Snyder EL, Watanabe H, Magendantz M, Hoersch S, Chen TA, Wang DG, et al. Nkx2-1 Represses a Latent Gastric Differentiation Program in Lung Adenocarcinoma. Mol Cell. 2013;50:185–99.
28 Mazur PK, Herner A, Mello SS, Wirth M, Hausmann S, Sanchez-Rivera FJ, et al. (2015). Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nature Medicine.
29 Dow LE, Fisher J, O’Rourke KP, Muley A, Kastenhuber ER, Livshits G, et al. Inducible in vivo genome editing with CRISPR-Cas9. Nat Biotechnol. 2015;33:390–4.
30 Collins EC, Pannell R, Simpson EM, Forster A, Rabbitts TH. Inter-chromosomal recombination of Mll and Af9 genes mediated by cre-loxP in mouse development. EMBO reports. 2000;1:127–32.
31 Smith AJ, De Sousa MA, Kwabi-Addo B, Heppell-Parton A, Impey H, Rabbitts P. A site-directed chromosomal translocation induced in embryonic stem cells by Cre-loxP recombination. Nature Genetics. 1995;9:376–85.
32 Choi PS, Meyerson M. Targeted genomic rearrangements using CRISPR/Cas technology. Nature communications. 2014;5:3728.
33 Torres R, Martin MC, Garcia A, Cigudosa JC, Ramirez JC, Rodriguez-Perales S. Engineering human tumour-associated chromosomal translocations with the RNA-guided CRISPR-Cas9 system. Nature communications. 2014;5:3964.
34 Kraft K, Geuer S, Will AJ, Chan WL, Paliou C, Borschiwer M, et al. (2015). Deletions, Inversions, Duplications: Engineering of Structural Variants using CRISPR/Cas in Mice. Cell reports.
35 Lupianez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015;161:1012–25.
36 Park CY, Kim DH, Son JS, Sung JJ, Lee J, Bae S, et al. Functional Correction of Large Factor VIII Gene Chromosomal Inversions in Hemophilia A Patient-Derived iPSCs Using CRISPR-Cas9. Cell stem cell. 2015;17:213–20.
37 Blasco RB, Karaca E, Ambrogio C, Cheong TC, Karayol E, Minero VG, et al. Simple and rapid in vivo generation of chromosomal rearrangements using CRISPR/Cas9 technology. Cell reports. 2014;9:1219–27.
38 Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–6.
39 Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–703.
40 Chen S, Sanjana NE, Zheng K, Shalem O, Lee K, Shi X, et al. Genome-wide CRISPR Screen in a Mouse Model of Tumor Growth and Metastasis. Cell. 2015;160:1246–60.
41 Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–7.
42 Shalem O, Sanjana NE, Zhang F. High-throughput functional genomics using CRISPR-Cas9. Nat Rev Genet. 2015;16:299–311.
43 Shi J, Wang E, Milazzo JP, Wang Z, Kinney JB, Vakoc CR. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat Biotechnol. 2015;33:661–7.
44 Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154:26–46.
45 Hilton IB, D’Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol. 2015;33:510–7.
46 Kearns NA, Pham H, Tabak B, Genga RM, Silverstein NJ, Garber M, Maehr R. Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat Methods. 2015;12;401–3.
47 Perez-Pinera P, Kocak DD, Vockley CM, Adler AF, Kabadi AM, Polstein LR, et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat Methods. 2013;10:973–6.
48 Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–51.
49 Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, et al. (2014). Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature advance online publication.
50 Zalatan JG, Lee ME, Almeida R, Gilbert LA, Whitehead EH, La Russa M, et al. Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell. 2015;160:339–50.
51 Tanenbaum ME, Gilbert LA, Qi LS, Weissman JS, Vale RD. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell. 2014;159:635–46.
52 Whitworth KM, Lee K, Benne JA, Beaton BP, Spate LD, Murphy SL, et al. Use of the CRISPR/Cas9 system to produce genetically engineered pigs from in vitro-derived oocytes and embryos. Biol Reprod. 2014;91:78.
53 Niu Y, Shen B, Cui Y, Chen Y, Wang J, Wang L, et al. Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell. 2014;156:836–43.
54 Harel I, Benayoun BA, Machado B, Singh PP, Hu CK, Pech MF, et al. A platform for rapid exploration of aging and diseases in a naturally short-lived vertebrate. Cell. 2015;160:1013–26.
55 Vidigal JA, Ventura A. (2015). Rapid and efficient one-step generation of paired gRNA CRISPR-Cas9 libraries. Nature communications. 2014;6:8083.
56 D’Astolfo DS, Pagliero RJ, Pras A, Karthaus WR, Clevers H, Prasad V, et al. Efficient intracellular delivery of native proteins. Cell. 2015; 161:674–90.
57 Zuris JA, Thompson DB, Shu Y, Guilinger JP, Bessen JL, Hu JH, et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat Biotechnol. 2015;33:73–80.
58 Chu VT, Weber T, Wefers B, Wurst W, Sander S, Rajewsky K, Kuhn R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat Biotechnol. 2015;33:543–8.
59 Maruyama T, Dougan SK, Truttmann MC, Bilate AM, Ingram JR, Ploegh HL. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat Biotechnol. 2015;33:538–42.
60 Malina A, Mills JR, Cencic R, Yan Y, Fraser J, Schippers LM, et al. Repurposing CRISPR/Cas9 for in situ functional assays. Genes Dev. 2013;27(23):2602–14.
61 Chen C, Liu Y, Rappaport AR, Kitzing T, Schultz N, Zhao Z, et al. MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell. 2014;25(5):652–65.
62 Aubrey BJ, Kelly GL, Kueh AJ, Brennan MS, O'Connor L, Milla L, et al. An inducible lentiviral guide RNA platform enables the identification of tumor-essential genes and tumor-promoting mutations in vivo. Cell Rep. 2015;10(8):1422–32.
63 Antal CE, Hudson AM, Kang E, Zanca C, Wirth C, Stephenson NL, et al. Cancer-associated protein kinase C mutations reveal kinase's role as tumor suppressor. Cell. 2015;160(3):489–502.