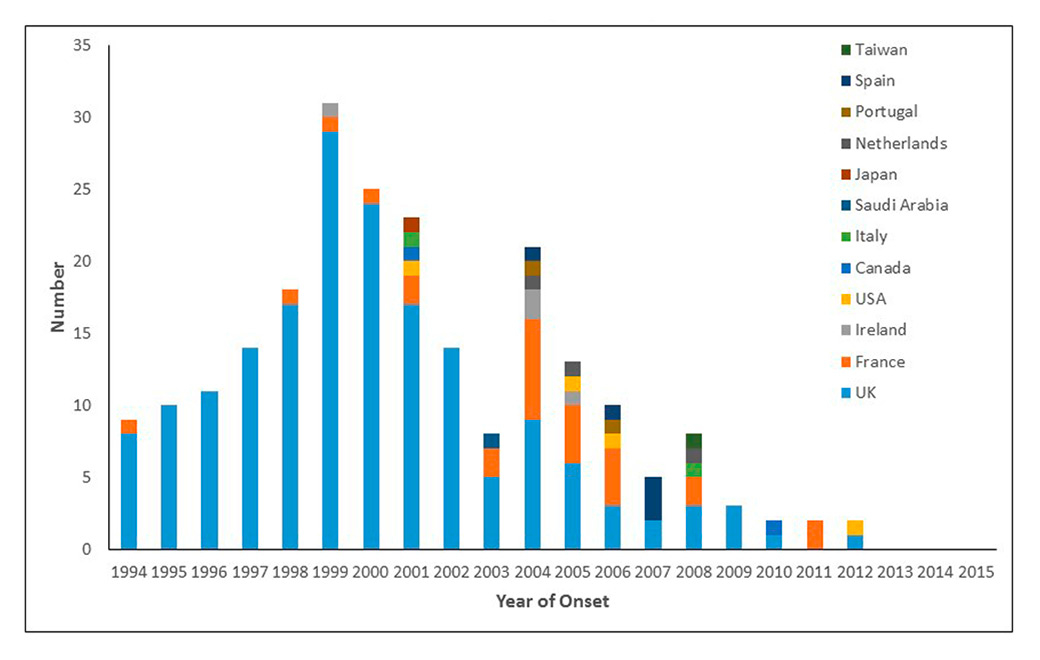
Figure 1
Variant Creutzfeldt Jakob disease cases by year and country: 1994‑2015 (n = 229).
DOI: https://doi.org/10.4414/smw.2015.14212
Abbreviations
BSE bovine spongiform encephalopathy
CJD Creutzfeldt-Jakob disease
CSF cerebrospinal fluid
EEG electroencephalogram
FFI fatal familial insomnia
GSS Gerstmann-Sträussler-Scheinker disease
gCJD genetic CJD
iCJD iatrogenic CJD
M methionine
MRI magnetic resonance imaging
PRNP prion protein gene
PrP prion protein
PrPres proteinase-resistant PrP
Rt-QuIC real-time quaking induced conversion
sCJD sporadic CJD
V valine
vCJD Variant CJD
VPSPr variably proteinase-sensitive prionopathy
WHO World Health Organization
It was a veritable news bombshell when, in March 1996, the UK Minister of Health announced the likely transmission of the epidemic of bovine spongiform encephalopathy (BSE) in cattle to humans, causing a new variant of Creutzfeldt-Jakob disease (vCJD). Frantic efforts to contain the damage already done included implementation of CJD surveillance, not only in the UK and continental EU, but in many other countries. Now, almost 20 years later, this is already history, and present-day medical students in Switzerland, and probably elsewhere, have little if any knowledge of BSE and its aftermath. While this may be partly attributed to the successful scientific and medical management of BSE and its major problems [1], the decline in concern about these diseases may invite complacency and questions whether surveillance for human prion diseases is still necessary. This article intends to explore this in more depth and, not surprisingly in an article written by two experts on surveillance of prion diseases, our answer to the question posed in the title is yes.
National surveillance for human prion diseases was started in several countries prior to 1996, but after the advent of vCJD, surveillance systems were established in many EU and some other countries to determine the extent to which human exposure to BSE prions might affect national populations. Additional reasons included: assessment of the effects of costly management measures to stop entrance into, and circulation within, the human food chain of bovine prions; the need to have a human counterpart of the newly established active surveillance for animal prion diseases, which quickly showed a considerably more widespread distribution of BSE than originally assumed; exploration of the possibility that phenotypes other than vCJD might be animal-derived; and identification as early as possible of new mechanisms of iatrogenic transmission of human prion diseases. In fact, the World Health Organization (WHO) recommended global CJD surveillance at a consultation as early as May 1996 [2], and emphasised the appropriate methodologies for such surveillance at further meetings and in publications [3]. The biggest obstacle to efficient surveillance has been limited knowledge of the nature of the causative agent, which has resulted in the absence of prion-specific tests comparable in validity and speed to microbiological tests for viruses or bacteria. In fact, the gold standard for detecting the infectious agent in prion disease has been transmission to experimental animals, which is costly, laborious and takes several months to years to yield results. Thus, in practice, up to now surrogate markers have been increasingly used in diagnosis, case definitions and surveillance.
Since the first observation in the UK of BSE as a novel prion disease in cattle in November 1986, 184 511 indigenous BSE cases had been diagnosed by April 2015, with a peak in 1992 and 1993 of more than 35 000 cases annually, representing incidence rates of more than 6 000 per million in bovines over 24 months of age. Since then, the UK epidemic curve has been in spectacular decline, with single-digit case numbers since 2011, only one case in 2014, and so far two in 2015 (http://www.oie.int). A total of up to one million infected cattle were estimated to have entered the human food chain in the UK from 1980–1996. Far fewer BSE cases have been diagnosed elsewhere, with one or more annual incidence rates exceeding 10 per million bovines over 24 months of age only in Belgium, the Czech Republic, France, Germany, Ireland, Italy, Luxemburg, the Netherlands, Portugal, Slovakia, Spain, and Switzerland, where the outbreak was officially declared closed in 2011. Worldwide BSE figures, however, have to be called into question, as effective active surveillance programmes are the prerequisite for adequate case ascertainment, but have been fully implemented only in the EU, Canada, Japan, and some other countries. Despite such geographical uncertainty, it is clear that the present BSE situation is most favourable when “classical” BSE is considered, with the epidemic of BSE virtually extinct in the UK, EU, and Japan. In contrast, “atypical” BSE continues to occur consistently at very low rates, as it is likely a sporadic disease form in analogy with sporadic CJD (sCJD). Atypical BSE has been diagnosed in two forms, as H-type and L-type or bovine amyloidotic spongiform encephalopathy (BASE); according to transmission experiments to human prion protein (PrP) transgenic mice or primates, BASE in particular might have zoonotic potential [4].
There are four main types of human prion disease: sporadic CJD (sCJD), genetic or hereditary forms (gCJD), the zoonotic form vCJD and accidentally transmitted or iatrogenic CJD (iCJD). In most countries carrying out national surveillance for CJD, identification of patients depends on notification of suspected cases by targeted professional groups, including neurologists, neuropathologists and more recently radiologists. Details of the clinical features and investigations are obtained and cases are classified using internationally accepted diagnostic criteria [3], which allows valid comparisons of data on CJD between centres and countries. The identification of sCJD and vCJD rely on identification of the characteristic clinical phenotypes, while diagnosis of hereditary forms of human prion disease depends on sequencing of the prion protein gene (PRNP) to identify disease-associated mutations, and in iCJD seeking a history of specific causal exposures, e.g. human growth hormone treatment or prior human dura mater graft.
Diagnostic criteria for all forms of CJD have been formulated and partially validated [3]. These include a combination of key clinical features with the results of investigations such as electroencephalogram, MRI brain scan and cerebrospinal fluid (CSF) 14-3-3 bioassay to allow classification as “probable” or “possible” in life. A definite diagnosis requires neuropathological confirmation, usually after post-mortem examination. In general, official statistics on CJD report only cases classified as “definite” or “probable” as the true diagnosis in “possible” cases may be uncertain.
The paradigm features of sCJD are rapidly progressive dementia and myoclonic involuntary movements, associated with focal neurological features such as ataxia. The mean survival is short, which distinguishes sCJD from more common forms of dementia. Terminally, sCJD is characterised by a state of akinetic mutism with death usually due to intercurrent illness such as pneumonia. In vCJD the clinical course is more protracted with a mean survival of 14 months and, although the terminal stages are similar to sCJD, with dementia, ataxia and involuntary limb movements, there is a prodrome of psychiatric symptoms such as depression and anxiety for a mean of 6 months before cognitive and focal neurological features develop.
The clinical features of gCJD are heterogeneous and this is determined largely by the specific associated PRNPmutation. Some cases develop a syndrome very similar to sCJD, while other cases present with nonspecific neurological features such as a progressive ataxic syndrome or isolated progressive dementia. In iCJD due to peripheral exposure to infection, for example human growth hormone treatment, there is a progressive ataxic syndrome, with little evidence of cognitive impairment. With a central exposure, for example to an infected human dura mater graft, the clinical features are similar to sCJD.
There is no diagnostic blood test for human prions and, although specialist investigations are nonspecific in CJD, they are nonetheless invaluable to diagnosis. The electroencephalogram (EEG) shows periodic triphasic complexes in a majority of cases of sCJD, but has largely been replaced by the 14-3-3 cerebrospinal fluid (CSF) assay [5] and magnetic resonance imaging (MRI) brain scan, which shows high signal in caudate, putamen and/or cortical regions in about 80% of cases. The 14-3-3 assay is nonspecific, as it is a marker of neuronal damage, and it may be replaced in time with CSF real-time quaking induced conversion (Rt-QuIC), which amplifies prion protein, but not infectivity, to produce a fluorescent signal, and has a very high sensitivity and specificity in sCJD [6].
In vCJD the most helpful test is the MRI brain scan, which shows high signal in the posterior thalamus, the “pulvinar sign”, in about 90% of cases by use of fluid-attenuated inversion recovery (FLAIR) and diffusion-weighted imaging (DWI) sequences. Tonsil biopsy to identify disease-associated prion protein can be a useful investigation in cases of vCJD in which there is diagnostic uncertainty, but this investigation has risks, as does brain biopsy, which is rarely carried out in CJD. The RT-QuIC is negative in vCJD using current methodologies and the 14-3-3 assay is positive in only about 50% of cases.
The MRI brain scan and 14-3-3 CSF assays can also be helpful in the diagnosis of both gCJD and iCJD, and should be considered in all CJD suspect cases. Most hospitals now have an MRI scanner and there is no evidence of a risk from carrying out CSF examination.
A definite diagnosis of human prion disease requires neuropathological confirmation and/or confirmation of protease-resistant prion protein (PrPres) by immunocytochemistry or Western blot [3]. Thus neuropathology is an essential element of any surveillance system. Paramount histological features are spongiform change, neuronal loss, astro- and microgliosis and deposition of disease-associated PrP in affected CNS tissue [7]. However, each of these features may be variably present and distinctly distributed, giving rise to neuropathological heterogeneity that mirrors that seen in clinical presentation. In fact, for sporadic CJD (sCJD), such heterogeneity has been attributed to distinct “strains” defined by two molecular determinants, the PRNP polymorphism (methionine[M] vs valine [V]) at codon 129 on one hand, and to one of two types of PrPres demonstrable by immunoblotting on the other. Thus neuropathology includes distinct profiles that distinguish at least six major sCJD “histotypes” (MM or MV/1, VV1, MM2 cortical, MM2 thalamic, MV2, VV2) and also mixed types [8, 9]. Immunohistochemical demonstration of disease-associated PrP in sections of affected brain tissue is part of this heterogeneity, with distinct deposition patterns described as synaptic, patchy-perivacuolar, pericellular, plaque-like or with Kuru-type plaques [7].
Western blotting to identify PrPres in affected brain tissue usually complements the full diagnostic spectrum applicable in autopsy cases of human prion diseases. A brain tissue homogenate is digested with proteinase K and immunoblotted with an anti-PrP antibody (mostly 3F4), revealing three distinct PrPres bands representing di-, mono- and unglycosylated protein fragments. However, banding patterns of proteinase K digestion products do not strictly correspond to differences in glycosylation degree but also to chain length differences as shown with the POM antibody family [10]. According to the size of the unglycosylated fragment, PrPres is classified as type 1 (with a molecular size of 21 kDa) or type 2 (19 kDa), as the proteinase K cleavage site differs between both types [9]. In vCJD, a peculiar pattern, denoted as type 2B, has predominance of the diglycosylated fragment in addition to an unglycosylated fragment of 19 kDa, in contrast with type 2A seen in sCJD with predominantly monoglycosylated PrPres. In genetic prion diseases, however, the blotting patterns may be more variable and reveal also lower size bands.
There are occasional cases in which such immunoblotting is negative or equivocal at first hand. One condition more recently described as variably proteinase-sensitive prionopathy (VPSPr) [11] may yield immunoblots that may be negative when the anti-PrP 3F4 antibody is used, but show a ladder-like pattern with other antibodies such as 1E4, which is directed against PrP epitope 97-15. Moreover, clinically probable or possible CJD cases found recently by surveillance in Austria did not demonstrate any PrPres at all by immunoblotting, but at autopsy thalamic degeneration with a peculiar immunohistochemical pattern of PrP was found[12]; however, the nosological position of this entity still needs to be determined.
More than 30 PRNP mutations or insertions have been found associated with clinical manifestations that may be similar to, or strikingly different from, sCJD [13]. For surveillance, it is important to recognise the absence of a positive family history in about half of all genetic cases [14]. Thus genetic testing may be useful in all CJD-suspected patients, as a disease-associated aberration would dismiss further diagnostic considerations. Moreover, knowledge of the PRNP 129 M/V polymorphism is useful for further subtyping of sCJD [15].
Sporadic CJD is by far the most common human prion disease, but is overall rare. Most established surveillance systems find sCJD at annual mortality rates between 1 and 2 per million [16]. However, there are considerable differences between countries, and a positive correlation between incidence and surveillance intensity has been demonstrated [17]. While some countries report differences in gender distribution, this has not been consistent in international studies, and only a slight increase of deaths in men over age 70 has been found [16].
Sporadic CJD is a disease mainly of the elderly, with a peak between ages 60 to 79 years [16]. A decline over age 80 has been described [16], but it is unclear whether this might be an artefact of previously incomplete case ascertainment in the elderly [18]. Duration of disease is usually short, with a median of 5 months [19], as reflected by the early term of “subacute” spongiform encephalopathy. However, longer survival has been found to correlate with younger age at onset, female gender, heterozygosity at PRNP 129, presence of CSF 14-3-3 protein and type 2A PrPres [19].
In countries on different continents with well-established surveillance, sCJD has been shown to occur at similar rates [16]. Thus it seems that globally it is relatively evenly distributed.
Case-control studies in sCJD have produced varying results but no consistent risk factor for the development of disease has been identified. Notably, there is no evidence of direct spread of infection from person to person or of an increased risk through occupation, for example in medical or paramedical professions [20]. An increased risk through prior surgery has been found in one study [21], but not others, and the evidence overall, including look-back studies, does not suggest that having received a blood transfusion increases the risk of the disease.[22] Although many of the studies of risk factors in sCJD may be subject to bias or confounding [23], the findings are consistent with the hypothesis that sCJD is not an environmentally acquired disease.
VPSPr was recently described as a new but rare type of sporadic prion disease that occurs in all genotypes of the PRNP 129 polymorphism, although V129V is most common [11]. Most cases have occurred in the USA, with a few cases most recently found also in Europe. For surveillance, VPSPr might pose a problem because of the peculiar properties of its disease-associated PrP (see above) when diagnosis relies on immunoblotting only. However, neuropathology is able to recognise a prion disease because of some spongiform change and positive immunohistochemistry for PrP, although the presence of microplaques is peculiar [11].
Variant CJD was identified in the UK in 1996 and has an overall mortality rate of 0.15/million. Cases of vCJD have been identified in a number of other countries, mainly in Europe (table 1), most likely through exposure to BSE infection through UK exports rather than indigenous BSE. It is of note that there is international agreement that cases of vCJD are classified according to the country of normal residence at the time of disease onset and there are, therefore, cases in which the country of exposure to BSE in not the same as the country of attribution, for example the cases in the USA.
Variant CJD affects a younger age group than sCJD with a mean age at death of 28 years in comparison with 66 years in sCJD. The reason for the young age in vCJD is unknown, but possibilities include age-related susceptibility to infection or variation in exposure to BSE in the food chain according to age group.
Initial fears of a massive epidemic of vCJD have not materialised; the outbreak, having peaked in 2000, is now in decline and there have been no new cases in the UK since 2013. Cases of vCJD outside the UK are also in decline (fig. 1). There was an extensive exposure of the UK population to BSE in the food chain and the likely explanation of the limited epidemic of vCJD is that there is a substantial species barrier between bovines and humans [24].
The major risk factor for vCJD is a history of residence in the UK, which is consistent with BSE as the cause of this disease. The other important determinant is the PRNPcodon 129 polymorphism, as all tested probable and definite clinical cases have been methionine homozygotes at this locus (the distribution of PRNP 129 genotypes in the normal population is about 51% MV, 37% MM, and 12% VV). There are concerns that further outbreaks may appear at a later date in the alternative codon 129 genotypes. A case-control study in vCJD, which showed an increased risk through consumption of food potentially containing high levels of BSE infectivity, is consistent with the hypothesis of food born transmission of infection [25].
Figure 1
Variant Creutzfeldt Jakob disease cases by year and country: 1994‑2015 (n = 229).
There is a significantly higher level of disease-associated prion protein in peripheral tissues such as the lymphoreticular system in vCJD as compared with sCJD. This has led to pathological studies in the UK of routine tonsil and appendix specimens to determine whether members of the general population may be infected with BSE, but showing no symptoms. In prion disease there is a silent incubation period between exposure to infection and the development of symptoms, which is estimated to be around a mean of 15 years in vCJD. A recent study found 16 positive appendix specimens in over 32 000 samples, leading to an estimated prevalence of infection of 1/2 000 [26], suggesting a significant fraction of the UK population with a carrier state.
The significant peripheral pathogenesis in vCJD led to concerns that this condition might be transmissible through blood transfusion and a look-back study was established in the UK in 1996. In 2003 a case of vCJD was identified with a history of having received a blood transfusion donated by an individual who themselves later developed vCJD. Since then two further clinical cases have been linked to a blood transfusion derived from another case, providing strong evidence that this condition can be transmitted via blood transfusion [23, 27]. In addition one recipient who died of a non-neurological illness was found to have abnormal prion protein in the spleen and a lymph node, and a patient with haemophilia, treated with UK plasma products, had a positive Western blot in a spleen sample.
| Table 1: Variant Creutzfeldt-Jakob disease - current data: August 2015. | |||
| Country | Total number of primary cases (number alive) | Total number of secondary cases (number alive) | Residence in the UK >6 months during period 1980–1996 |
| UK | 174 (0) | 3 (0) | 177 |
| France | 27 (0) | – | 1 |
| Republic of Ireland | 4 (0) | – | 2 |
| Italy | 2 (0) | – | 0 |
| USA | 42 (0) | – | 2 |
| Canada | 2 (0) | – | 1 |
| Saudi Arabia | 1 (0) | – | 0 |
| Japan | 11 (0) | – | 0 |
| Netherlands | 3 (0) | – | 0 |
| Portugal | 2 (0) | – | 0 |
| Spain | 5 (0) | – | 0 |
| Taiwan | 1 (0) | – | 1 |
| 1 The case from Japan had resided in the UK for 24 days in the period 1980–1996. 2 The third US patient with vCJD was born and raised in Saudi Arabia and has lived permanently in the United States since late 2005. According to the US case-report, the patient was most likely infected as a child when living in Saudi Arabia. In the fourth US patient the history indicated that exposure to infection most likely occurred prior to moving to the USA. | |||
About 3% of all cases of CJD are iatrogenic in origin, but the incidence varies markedly from country to country in relation to differences in medical and surgical practice and the source of medical treatments such as human pituitary derived hormones.
The main types of iCJD numerically are cases caused by surgical treatment with human dura mater grafts, particularly the brand Lyodura® (n = >220 cases) and cases caused by treatment in childhood with human pituitary growth hormone (n = >220). The presumption is that tissues derived from cases of sCJD were included in the production process, contaminating the final product with infectivity. Rarer causes of iCJD include six cases caused by infected neurosurgical instruments or EEG depth electrodes, two linked to corneal grafts and four linked to human pituitary gonadotrophin treatment [28]. Transfusion transmission of vCJD is described above.
The interval between exposure to a treatment and the development of clinical symptoms can be accurately determined in iCJD cases linked to specific surgical procedures: with human dura mater grafts the mean incubation period is 12 years and with surgical instruments 1–2 years. In human growth hormone cases the incubation period estimates are made in relation to the mid-point of treatment, which sometimes extended to many years, and the mean incubation period in these cases is estimated to be 17 years. In the more common forms of iCJD there is a wide spread of incubation periods, which can range from 1 to more than 40 years. In transfusion transmitted vCJD the incubation period ranges from 3–8 years.
Cases of iCJD have been identified in 21 countries, but there is a clear excess of cases linked to human dura mater grafts in Japan, probably because these grafts were used more frequently in Japan than other countries. More than half the total human growth hormone cases have been in France, possibly because of a higher level of contamination in the production process.
About 10 to 15% of prion diseases in Caucasians have a genetic origin. In the series of the EuroCJD consortium, 10.2% of all cases were genetic, but with considerable differences between countries [14]. The responsible PRNP aberrations and their clinicopathological correlates have been amply described, e.g. in [13]. In addition to gCJD (with E200K as most frequent) that usually mimics sCJD, distinct phenotypes include Gerstmann-Sträussler-Scheinker disease (GSS, with P102L as the most frequent mutation), classically with a slowly evolving spinoataxic syndrome and neuropathologically pathognomonic multicentric PrP plaques; fatal familial insomnia (FFI, with D178N M129M as the most frequent constellation and predominantly thalamic degeneration); and a novel systemic PrP amyloidosis with Y163X [13]. Sporadic fatal insomnia (SFI) is a very rare peculiar clinical-pathological phenotype that is indistinguishable from FFI, but the FFI-associated PRNP aberration is absent.
GSS and FFI usually manifest earlier than gCJD or sCJD and have a longer disease duration [14]. Insert mutations may have a gCJD or GSS phenotype and are variable in age at onset and duration of disease [14].
Israel (mainly Sephardic Jews emigrating from Libya) and Slovakia (preferentially the Orava and Lucenec provinces) have a high prevalence of the E200K mutation that manifests mostly indistinguishably from sCJD [14]. In Italy, the number of genetic cases was higher in Campania and Calabria regions than in other regions [29].
As with classical BSE, Kuru is now largely a matter of medical history. This epidemic of the 20th century in the Fore linguistic group of Papua-New Guinea was caused by ritual cannibalism that was stopped in the late 1950s. Cases are, however, still occurring with incubation periods exceeding 40 years [30]. The clinical presentation was with an ataxic syndrome and death followed in 12–18 months. Kuru has taught the medical and scientific community several lessons about external prion infection in humans, including the length of incubation time, the absence of vertical transmission from mother to child and the role of the genetic background, with the recent finding of a human PRNP polymorphism that completely protects against infectious prions [31]. It seems that Kuru acted as a selection factor for a resistance genotype in Kuru-affected regions. Kuru was the first prion disease to be experimentally transmitted in a laboratory setting, a finding that led to successful experimental transmission of sCJD.
It is obvious that the epidemic of classical BSE is coming to an end. However, atypical BSE is expected to persist, possibly as a sporadic disease, and recent experimental data on the transmission of classical sheep scrapie strains to humanised mice [32] and primates [33] are questioning the traditional belief that prion diseases of small ruminants do not have zoonotic potential. Thus, even in the absence of BSE, the possibility of a remaining animal source for human prion diseases cannot be excluded and this should stimulate continued research and analysis. Moreover, the threat of circulation of prions between humans, from external sources or by iatrogenic procedures including blood/blood product therapies, must also be considered, an issue highlighted by the prevalence data on vCJD infection in the UK that suggest a significant fraction with a carrier state of the population. Finally, prion diseases have more recently served as the gold standard paradigm to compare with the “prion-like” spread of other aggregated proteins in neurodegenerative diseases. It remains to be seen whether a novel public health hazard is emerging from diseases that are several logs more frequent in the population. Continuation of detailed surveillance of human prion disorders would be prudent in view of all these issues that deserve clarification, together with research into the potential transmissibility of other neurodegenerative diseases. Moreover, it would be wise to maintain well-organized surveillance structures for human prion diseases: it is easy to destroy functioning structures, but it is extremely difficult and expensive to build up novel structures.
1 Budka H. Editorial: The European Response to BSE: A Success Story. EFSA Journal 2011;9(9):e991 http://www.efsa.europa.eu/en/efsajournal/pub/e991.htm.
2 WHO: Public health issues and clinical and neurological characteristics of the new variant of Creutzfeldt-Jakob disease and other human and animal transmissible spongiform encephalopathies: Memorandum from two WHO meetings. Bull WHO. 1996;74(5):453–63.
3 WHO: WHO manual for surveillance of human transmissible spongiform encephalopathies including variant Creutzfeldt-Jakob disease. WHO Communicable Disease Surveillance and Response, WHO Geneva 2003.
4 European Food Safety Authority (EFSA), European Centre for Disease Prevention and Control (ECDC): Joint Scientific Opinion on any possible epidemiological or molecular association between TSEs in animals and humans. EFSA Journal 2011;9(1):1945 [1111 pp.] http://www.efsa.europa.eu/en/efsajournal/doc/1945.pdf.
5 Green AJE, Ramljak S, Müller WEG, Knight RSG, Schröder HC. 14-3-3 in the cerebrospinal fluid of patients with variant and sporadic Creutzfeldt-Jakob disease measured using capture assay able to detect low levels of 14-3-3 protein. Neurosci Lett. 2002;324(1):57–60.
6 McGuire LI, Peden AH, Orrú CD, Wilham JM, Appleford NE, Mallinson G, et al. Real time quaking-induced conversion analysis of cerebrospinal fluid in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 2012;72(2):278–85.
7 Budka H. Neuropathology of prion diseases. Br Med Bull. 2003;66(1):121–30.
8 Parchi P, de Boni L, Saverioni D, Cohen ML, Ferrer I, Gambetti P, et al. Consensus classification of human prion disease histotypes allows reliable identification of molecular subtypes: an inter-rater study among surveillance centres in Europe and USA. Acta Neuropathol. 2012;124(4):517–29.
9 Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46:224–33.
10 Polymenidou M, Moos R, Scott M, Sigurdson C, Shi Y-z, Yajima B, et al. The POM Monoclonals: A Comprehensive Set of Antibodies to Non-Overlapping Prion Protein Epitopes. PLoS ONE 2008;3(12):e3872.
11 Zou W-Q, Puoti G, Xiao X, Yuan J, Qing L, Cali I, et al. Variably protease-sensitive prionopathy: A new sporadic disease of the prion protein. Ann Neurol. 2011;68(2):162–72.
12 Kovacs GG, Peden A, Weis S, Höftberger R, Berghoff A, Yull H, et al. Rapidly progressive dementia with thalamic degeneration and peculiar cortical prion protein immunoreactivity, but absence of proteinase K resistant PrP: a new disease entity? Acta Neuropathol Commun. 2013;1(1):72.
13 Gelpi E, Colom-Cadena M, Budka H. Molecular Genetics of Creutzfeldt-Jakob Disease and Gerstmann- Sträussler-Scheinker Disease. In: eLS. John Wiley & Sons, LTD: Chichester. DOI: 10.1002/9780470015902.a0024442; 2015: 1–11.
14 Kovacs GG, Puopolo M, Ladogana A, Pocchiari M, Budka H, van Duijn C, et al. Genetic prion disease: the EUROCJD experience. Hum Genet. 2005;118(2):166–74.
15 Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG, et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 1996,39:767–78.
16 Ladogana A, Puopolo M, Croes EA, Budka H, Jarius C, Collins S, Klug GM, et al. Mortality from Creutzfeldt-Jakob disease and related disorders in Europe, Australia, and Canada. Neurology. 2005;64(9):1586–91.
17 Klug GM, Wand H, Simpson M, Boyd A, Law M, Masters CL, et al. Intensity of human prion disease surveillance predicts observed disease incidence. J Neurol Neurosurg Psychiatry. 2013;84:1372–7.
18 Gelpi E, Heinzl H, Höftberger R, Unterberger U, Ströbel T, Voigtländer T, et al. Creutzfeldt-Jakob Disease in Austria: An Autopsy-Controlled Study. Neuroepidemiology. 2008;30(4):215–21.
19 Pocchiari M, Puopolo M, Croes EA, Budka H, Gelpi E, Collins S, et al. Predictors of survival in sporadic Creutzfeldt-Jakob disease and other human transmissible spongiform encephalopathies. Brain. 2004;127(10):2348–59.
20 Alcalde-Cabero E, Almazán-Isla J, Brandel J-P, Breithaupt M, et al. Health professions and risk of sporadic Creutzfeldt–Jakob disease, 1965 to 2010. Euro Surveill. 2012;17(15):13–22.
21 Mahillo-Fernandez I, de Pedro-Cuesta J, Bleda MJ, Cruz M, Mølbak K, Laursen H, et al. Surgery and Risk of Sporadic Creutzfeldt-Jakob Disease in Denmark and Sweden: Registry-Based Case-Control Studies. Neuroepidemiology. 2008;31(4):229–40.
22 Dorsey K, Zou S, Schonberger LB, Sullivan M, Kessler D, Notari E, et al. Lack of evidence of transfusion transmission of Creutzfeldt-Jakob disease in a US surveillance study. Transfusion. 2009;49(5):977–84.
23 de Pedro Cuesta J, Ruiz Tovar M, Ward H, Calero M, Smith A, Verduras CA, et al. Sensitivity to Biases of Case-Control Studies on Medical Procedures, Particularly Surgery and Blood Transfusion, and Risk of Creutzfeldt-Jakob Disease. Neuroepidemiology. 2012;39(1):1–18.
24 Chen C-C, Wang Y-H. Estimation of the Exposure of the UK Population to the Bovine Spongiform Encephalopathy Agent through Dietary Intake During the Period 1980 to 1996. PLoS ONE 2014;9(4):e94020.
25 Ward HJT, Everington D, Cousens SN, Smith-Bathgate B, Leitch M, Cooper S, et al. Risk factors for variant Creutzfeldt-Jakob disease: A case-control study. Ann Neurol. 2006;59(1):111–20.
26 Gill ON, Spencer Y, Richard-Loendt A, Kelly C, Dabaghian R, Boyes L, et al. Prevalent abnormal prion protein in human appendixes after bovine spongiform encephalopathy epizootic: large scale survey. Br Med J. 2013;347:f5675.
27 Hewitt PE, Llewelyn CA, Mackenzie J, Will RG. Creutzfeldt-Jakob disease and blood transfusion: results of the UK Transfusion Medicine Epidemiological Review study. Vox Sanguinis. 2006;91(3):221–30.
28 Brown P, Brandel J-P, Sato T, Nakamura Y, MacKenzie J, Will RG, et al. Iatrogenic Creutzfeldt-Jakob Disease, Final Assessment. Emerging Infectious Diseases. 2012;18(6):901–7.
29 Ladogana A, Puopolo M, Poleggi A, Almonti S, Mellina V, Equestre M, et al. High incidence of genetic human transmissible spongiform encephalopathies in Italy. Neurology. 2005;64(9):1592–7.
30 Alpers MP. The epidemiology of kuru: monitoring the epidemic from its peak to its end. Philosophical Transactions of the Royal Society B: Biological Sciences. 2008;363(1510):3707–13.
31 Asante EA, Smidak M, Grimshaw A, Houghton R, Tomlinson A, Jeelani A, et al. A naturally occurring variant of the human prion protein completely prevents prion disease. Nature. 2015;522(7557):478–81.
32 Cassard H, Torres J-M, Lacroux C, Douet J-Y, Benestad SL, Lantier Fdr, et al. Evidence for zoonotic potential of ovine scrapie prions. Nat Commun. 2014;5:5821.
33 Comoy EE, Mikol J, Luccantoni-Freire S, Correia E, Lescoutra-Etchegaray N, Durand Vr, et al. Transmission of scrapie prions to primate after an extended silent incubation period. Sci Rep. 2015;5:11573.
Disclosure statement: No financial support and no other potential conflict of interest relevant to this article was reported.