Astonishing advances in mouse genetic tools for biomedical research
DOI: https://doi.org/10.4414/smw.2015.14186
Walker Scot
Jackson, Lech
Kaczmarczyk
Summary
The humble house mouse has long been a workhorse model system in biomedical research. The technology for introducing site-specific genome modifications led to Nobel Prizes for its pioneers and opened a new era of mouse genetics. However, this technology was very time-consuming and technically demanding. As a result, many investigators continued to employ easier genome manipulation methods, though resulting models can suffer from overlooked or underestimated consequences. Another breakthrough, invaluable for the molecular dissection of disease mechanisms, was the invention of high-throughput methods to measure the expression of a plethora of genes in parallel. However, the use of samples containing material from multiple cell types could obfuscate data, and thus interpretations. In this review we highlight some important issues in experimental approaches using mouse models for biomedical research. We then discuss recent technological advances in mouse genetics that are revolutionising human disease research. Mouse genomes are now easily manipulated at precise locations thanks to guided endonucleases, such as transcription activator-like effector nucleases (TALENs) or the CRISPR/Cas9 system, both also having the potential to turn the dream of human gene therapy into reality. Newly developed methods of cell type-specific isolation of transcriptomes from crude tissue homogenates, followed by detection with next generation sequencing (NGS), are vastly improving gene regulation studies. Taken together, these amazing tools simplify the creation of much more accurate mouse models of human disease, and enable the extraction of hitherto unobtainable data.
Abbreviations
4-TU 4-thiouracil
Ago2 Argonaute 2 protein
BAC bacterial artificial chromosome
Cas9 CRISPR-associated system nuclease 9
CRISPR clustered regularly interspaced short palindromic repeats
DSB double strand break
ESCs embryonic stem cells
Floxed flanked by LoxP sites
HR homologous recombination
LCM laser capture microdissection
NGS next generation sequencing
NHEJ nonhomologous end joining
Prnp prion protein gene
RGENs RNA-guided endonucleases
RISC RNA-induced silencing complex
RIT random integration transgenic
Rpl22 ribosomal protein L22
sgRNA synthetic guide RNA
TALENs transcription activator-like effector nucleases
TALEs transcription activator-like effectors
ZFN zinc finger nuclease
Introduction
Despite their diminutive size, and our lack of body fur and a tail, there are deep, genetically encoded, incontrovertible parallels in human and mouse biology. Like humans, mice have colour vision and close their eyes during sleep, and their four-chambered hearts pump warm blood, which carries gases via haemoglobin proteins. The mouse brain has the same major regions present in humans, including those most severely affected in neurodegenerative diseases. Human and mouse behaviour, metabolism and bodies change with aging [1–3]. Importantly, genomes of mice and humans have a similar linear length and gene content, both are typically methylated on silenced regions and hydroxymethylated on transcribed regions [4, 5], and many features of genome organisation and control are conserved [6–9]. All these similarities, together with their small size and fast life-cycle, have made the mouse a favourite model system for studying human diseases.
The utility of mouse models was dramatically augmented with the advent of recombinant DNA technologies, enabling the modelling of genetic alterations linked to human diseases, and the manipulation of gene networks hypothesised to be involved in disease mechanisms. An important example involves a mouse model of one of the most notorious genetic diseases: sickle cell anaemia. The mice were genetically engineered to express the human sickle cell mutation and developed blood phenotypes that closely resembled those in humans [10]. Remarkably, the mice were experimentally “cured” by a combined gene therapy / stem cell therapy approach, much like what is envisioned to be commonly used in humans one day [10, 11]. The importance of the mouse’s role in medical research has been thoroughly articulated by others [12–14] (see also http://ec.europa.eu/research/health/pdf/summary-report-25082010_en.pdf). Herein we review several cutting edge technologies that are revolutionising the utility of the mouse as a model system for biomedical research and are certain to help bridge the gap between the bench and the bedside.
Strategies for genome manipulation
The first successful introduction of exogenous DNA into the mouse genome was accomplished by injection of viral DNA into mouse blastocyst stage embryos [15]. Several years later, the first examples of genetically engineered mice expressing a specific foreign gene [16] and transmitting it to their progeny [17] were reported. These types of mouse models are referred to as random integration transgenics (RITs), as foreign DNAs (transgenes) integrate into random genomic locations, typically in multiple copies [18]. To inactivate an endogenous gene required a combination of two techniques. First, the endogenous DNA recombination machinery was harnessed to insert a new gene into a specific genomic location of cultured cells, a process known as gene targeting [19, 20]. The next step was to convert gene-targeted mouse cells into fully developed, viable animals. This was accomplished by combining cultured embryonic stem cells (ESCs) with a blastocyst stage mouse embryo, leading to a hybrid (chimeric) mouse consisting of the original (host) blastocyst and the injected ESCs [21]. Importantly, the ESCs can contribute to all tissues of the chimeric embryo. Thus, with a little luck, a chimera is born in which the germ cells were derived from the ESCs and thus engineered genes carried by ESCs can be transmitted to offspring [21–25]. This technology was initially used to engineer interruptions in genes that prevented synthesis of a functional product and the resulting mice were thus called knockouts [26–28]. Soon after, it was possible to place mutant genes intended for expression into specific genomic loci, creating lines now known as knock-ins [29–32]. The practical and experimental differences between RIT and knock-in mice are critical to consider when designing and interpreting experiments (see fig. 1).
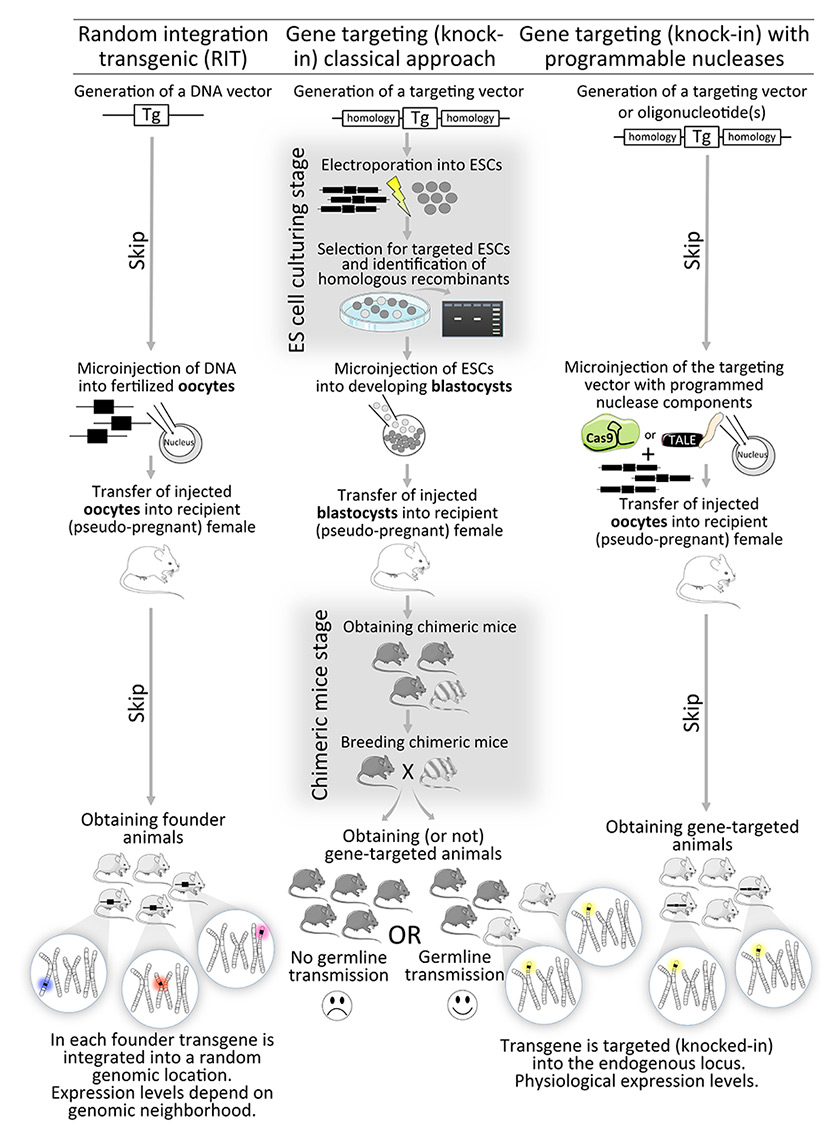
Figure 1
A comparison of strategies to genetically engineer mice. The random integration approach (left workflow) is useful for adding new genes, and is technically straightforward, but suffers from position effects (transgenes on different chromosomes on the bottom). The gene targeting approach (middle workflow) is more laborious, requiring embryonic stem cell (ESC) culturing, and breeding of chimeras, which often fail to transmit the transgene to their progeny. The breakthrough of the gene targeting approach employing guided nucleases (right workflow), is that the final result of gene-targeted mice is obtained without the ESC culturing and chimera breeding hassles.
Location matters – effects of genomic position
When RITs are developed, multiple ‘founder’ lines are typically created by independent integrations of the transgene into different genomic loci, typically resulting in different expression levels and patterns. Why does this happen? The answer is not completely understood but we can turn to other fields of genetics research for clues. Although mind bogglingly expansive, the mammalian genome is not a sea of randomly distributed elements that simply float around in the cell nucleus. Instead, the genome is contorted into very specific structures. Genes that are distantly localised in linear DNA space can be neighbours in three-dimensional space for coordinated regulation [33–35]. Indeed, neighbourhoods appear to give cells their identities [36, 37]. With the concept of genomic neighbourhoods in mind, it is conceivable that the same transgene behaves differently depending on the genomic context of the integration site. A transgene employing a promoter selective for a specific cell type would likely not be expressed if integrated into a genomic region that is silenced in that cell type. The endogenous genomic elements would likely dominate, and the transgene promoter would fail to recruit a sufficient compilation of activating transcription factors to outcompete the native elements suppressing transcription.
An important example of how this can affect mouse models of disease was published 20 years ago as a description of a new model of a neurodegenerative disease, spinocerebellar ataxia 1 [38]. The authors were as rigorous as possible. Not only did they generate and analyse multiple independent lines carrying the mutant transgene (table 1), but they did the same for a control transgene [38]. By intuition, we assume that the more of a toxic transgene is present, the more harmful its effects. However, the number of copies of the transgene did not reliably correlate with expression level and, even more surprisingly, the expression level was not predictive of the severity of the phenotype (table 1: BO4 vs BO1, BO2 and BO6). None of the lines expressing the control transgene showed any signs of the disease, indicating it was the mutation, and not simply overexpression of a human protein, that was causing the disease in these mice. This example highlights the importance of examining multiple independent lines expressing a mutant transgene, and a series expressing a control transgene, when working with RITs. The transgene vector employed in these lines is expressed in a very specific cell type, Purkinje cells of the cerebellum (directed by Pcp2 promoter elements). However, even more broadly expressed transgene vectors are vulnerable to position effects. For example, the thy1 vector, which is widely used for its tendency to express in neurons, is highly prone to position effects [39, 40], and sometimes even functions in glial cells [41]. A more broadly expressed transgene vector MoPrP.Xho1 [42], built from a modified mouse prion protein gene [43], is also prone to expression pattern differences. For example, sometimes it is expressed in the striatum (a brain region affected in Huntington’s disease), but sometimes not [42]. Even within brain regions there is variability, as sometimes it is active in Purkinje cells [44] but sometimes it is not [43, 45]. Since the thy1 and MoPrP.Xho1 vectors are widely employed to model brain diseases, recognising that this variability happens and controlling it is extremely important.
The fidelity of expression can be improved by employing larger transgene vectors, such as bacterial artificial chromosome (BAC) or yeast artificial chromosome constructs [46]. They can carry up to 300 kilobase-pairs of DNA, enough to include even distant enhancers or insulators, and significantly increase the probability that a transgene will be expressed in the correct pattern, though some variability still remains [46]. Finally, a tangentially related but nonetheless important issue is that RITs alter the integration site. Besides the presence of new DNA, which might interrupt a gene [47], very large deletions can be created, and the modified loci are often unstable and change through generations [18]. Moreover, the regulation of genes neighbouring the inserted transgene can also be affected [18, 48], certainly an undesirable and difficult-to-detect consequence. Thus, although relatively easy to create, RITs are prone to difficult-to-control confounders related to their genomic location of insertion.
|
Table 1: Variable expression and ataxia. |
|
Line
|
Copy no.
|
Expression (mRNA)*
|
Ataxia? (weeks)†
|
| BO3 |
50 |
0X |
No (20) |
| BO4 |
3-5 |
10X |
Yes (16) |
| BO1 |
30 |
50X |
No (36) |
| BO2 |
10 |
50X |
No (36) |
| BO6 |
10 |
50X |
Yes (26) |
| BO5 |
30 |
100X |
Yes (12) |
| * fold expression above endogenous levels
† hemizygous mice only
Data obtained from [38] |
Targeted genome modifications: the trouble with knock-ins
The knock-in approach has its own set of important disadvantages. The first is that mutations engineered into endogenous genes obtain only natural expression levels. In many cases, phenotypes can be accelerated or enhanced by increased expression, which can be an important strength of RITs despite the caveats raised in the previous section. Indeed, in our own work with knock-in mouse models of neurodegenerative diseases, the onset of phenotypes is typically after midlife, like in humans, which translates to 12 to 18 months in the mouse. Understandably, many researchers do not have the resources or patience to develop and study such slowly progressing models. Nonetheless, they can be viewed as models of early stage disease. These models often have neuropathological changes similar to findings in humans, such as disease-associated protein aggregates, neuronal degeneration, and reactive gliosis. Interestingly, the tissue tropism characteristic of the human disease is often recapitulated with knock-in mice [10, 49–53].
A second disadvantage of knock-in mouse models of disease is the inherent difficulty in creating them (fig. 1). The procedure includes the expensive and tedious process of genetically modifying ESCs, which are then injected into early stage mouse embryos. In comparison, the typical RIT approach requires only that a comparatively simple DNA construct be created and directly injected into mouse oocytes, providing greater flexibility in the DNA construction, and bypassing the ESC work.
Luckily, newly developed technologies make the generation of knock-in and knock-out lines technically quite similar to the random integration approach.
Combining the strengths of knock-ins and RITs
DNA double-strand breaks (DSBs) in the eukaryotic genome trigger two types of repair mechanisms, nonhomologous end joining (NHEJ) [54] and homologous recombination (HR) [55]. NHEJ simply fuses two free ends of DNA, sometimes with a random nucleotide insertion or deletion, resulting in a frameshift in the open reading-frame of a gene, potentially creating a gene knock-out. HR recombines the damaged genomic site with a separate piece of DNA (template) that is homologous to it, in effect switching the two [56]. This can be co-opted for genetic engineering by adding transgenic DNA to serve as the template, allowing one to introduce precise genome changes, such as gene knock-outs or knock-ins (see “strategies for genome manipulation” above). However, the efficiency of HR in a locus strongly depends on its chromatin state [57]. Genes in condensed chromatin are relatively protected from DSBs and thus render miserably low efficiency of targeted mutagenesis, potentially explaining why different loci differ in their susceptibility to gene targeting. For example, the first attempts to knock-out the prion protein gene (Prnp) required screening of approximately 10 000 ESC colonies for a single targeted clone [58]. This is likely because the Prnp locus remains rather silent in ESCs, and would rarely require and recruit the DSB-HR repair machinery [59]. On the other hand, in certain loci HR occurs with higher efficiency. Two such permissive loci are HPRT or ROSA26 [60, 61]. The ROSA26 locus is an especially popular choice for making expression reporter mouse lines [62], as it provides strong, uniform and ubiquitous transgene expression across multiple tissues [61, 63]. A recently identified locus that is also easy to manipulate (TIGRE) will complement, and may even be better for ubiquitous activity, than the ROSA26 locus [64, 65]. However, modifying or disabling most endogenous genes in mice remained inefficient and variable, making the stimulation of HR sorely needed. This motivated the development of programmable endonucleases to introduce double strand breaks into specific genomic locations and thereby triggering DNA repair mechanisms.
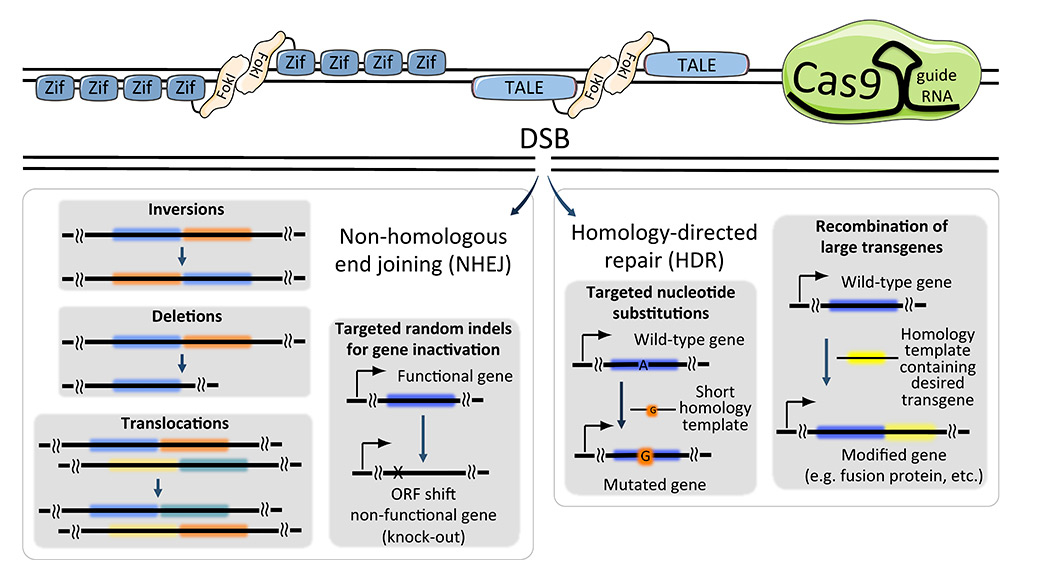
Figure 2
Comparisons of programmable nucleases and possible outcomes.Zinc finger nucleases (top left), transcription activator-like effector nucleases (TALENs; top middle) and CRIPSR/Cas9 (top right) systems introduce a site-directed double strand break (DSB) in DNA. Depending on the repair mechanism, nonhomologous end joining (NHEJ) or homology-directed repair (HDR), the site of interest can be left with random mutations, designed mutations, insertion of large transgenes, or (in case more than one DSB is generated), inversions, deletions and translocations.
Cas9 = CRISPR-associated system nuclease 9; CRISPR = clustered regularly interspaced short palindromic repeats; Fokl = Fokl nuclease; ORF = open reading frame; TALE = transcription activator-like effector; Zif = zinc finger nuclease
The first programmable nucleases routinely applied to mouse genome manipulation were created by fusing zinc finger DNA-binding domains to FokI endonuclease [66]. These chimeric restriction enzymes (termed zinc finger nucleases, ZFNs) were applied with amazing results in cells and in vivo [67–72] and even reached clinical trials [73]. However, this ground-breaking technology suffered from high cost of production and insufficient selectivity [74–76]. More recently, a related technique emerged, transcription activator-like effector nucleases (TALENs). TALENs proved to offer greater flexibility (they can target virtually any DNA sequence of interest, whereas ZFNs require a guanine-rich region, thereby limiting the density of targetable sites), and were easier to make thanks to their modular structure (see also fig. 2 and box 1). Most recently, RNA guided endonucleases (RGENs) burst on to the scene, with high efficiency and design flexibility for genetic modification of a multitude of species (fig. 2) [77–81]. The range of applications of TALENs and RGENs is expanding exponentially and a number of detailed reviews summarising their advantages and potential applications were published recently [72, 82–86].
The ground-breaking advantage of RGENs is the simplicity of construction, as the target specificity is directed by a 20-bp guide RNA molecule template which can be easily cloned into a vector encoding all the remaining components of the system: Cas9 nuclease and synthetic guide RNA (sgRNA) (fig. 2). Many dual expression vectors encoding Cas9 and sgRNA are freely available, and several online tools facilitate the identification of optimal guide sequences (see fig. 2 for details). Ease of design initially came at the expense of specificity, resulting in mutations in unintended locations [87, 88]. However, several recent advances diminish this problem. For example, a Cas9 mutant that cuts one DNA strand instead of two stimulates HR but insertions/deletions are not created in the case of NHEJ repair [89]. A second approach employs a catalytically inactive mutant version of Cas9 protein fused to FokI restriction endonuclease. Since this Cas9::FokI fusion protein requires dimerisation to function, two guide RNAs must bind in close proximity, greatly enhancing specificity [90]. A similar approach utilising TALENs successfully targeted a large construct injected into mouse oocytes [91]. Finally, when genetically engineering mice, most off-target mutations can be eliminated through backcrossing.
Compared with RGENs, ZFNs and TALENs require complex engineering of DNA recognition domains, as it is the protein, not RNA, which confers target specificity. The modular structure of TALENs greatly facilitates their design and construction, and many bioinformatics tools and assembly kits are currently available from open sources [92–95]. However, the design flexibility offered by RGENs is much greater. Consequently, they have rapidly become the method of choice for modifying multiple loci simultaneously. This was employed to generate multiple mouse lines simultaneously using “multiplexed” ESCs [96]. The ease of packaging of the small target specificity component into gene delivery systems should make it a useful tool for modifying genes in vivo, globally or in selected tissues. To simplify this process, a mouse line was genetically engineered to express Cas9 nuclease in specific cell types by use of Cre recombinase [97] (Cre is explained in fig. 3). In this inventive tool, a variety of established methods deliver guide RNAs, directing the modification of multiple loci simultaneously in specific cell-types of intact mice or cultured primary cells [97]. An interesting alternative is the recently reported delivery of both Cas9 (in this case a smaller version) and sgRNA packaged into adeno-associated virus particles [98]. The latter technique does not require transgenic mice and thus can be applied directly to mouse models of disease without the added expense of time and money to breed in additional lines. This amazing bid to specifically modify a single gene in a living organism once again highlights the therapeutic potential of RGENs. In human cells, CRISPR/Cas9 was already used with promising results to treat muscular dystrophy [99], human immunodeficiency virus infection [100], and Fanconi anaemia [101].
This therapeutic potential can be inverted to model multigenic diseases, including cancer. In addition to generating multiple mutations simultaneously, guided nucleases can be employed to engineer tumour-specific chromosomal rearrangements [102–104] (fig. 2). These possibilities were recently described in detail in two excellent reviews [105, 106].
Another important feature is that programmable endonucleases can be employed to create gene-targeted transgenic mice by injection or electroporation into fertilised oocytes [81, 96, 107]. This approach saves precious time and resources that would otherwise be spent on culturing, targeting, screening, and karyotyping ESCs, and the subsequent generation and (often futile) breeding of chimeras (fig. 1). Moreover, the efficiency of in vivo gene targeting with RGENs can be enhanced through inhibition of the NHEJ pathway [108]. These improvements could make the generation of gene-targeted mice nearly as simple as the generation of RIT mice, but with the enormous advantage that the integration site of the transgene is precisely controlled.
Finally, modifications of TALEN and Cas9 systems are enabling the manipulation of gene expression [109, 110], sometimes controlled by light [111, 112]. Even the epigenome of specific genomic loci can be manipulated [113, 114]. TALEs- and CRISPR/Cas9-guided transcriptional activators were successfully used to remodel chromatin, enforcing gene activation in silenced regions [115]. Such specific control on gene-expression patterns in vivo will have a profound impact on disease research.
|
Box 1: A brief comparison of key features of ZFNs, TALENs and RGENs. |
|
ZFNs
Zinc finger nucleases
|
TALENs
Transcription activator-like effector nucleases
|
RGENs
RNA guided endonucleases
|
|
Origin
|
Zinc finger DNA binding protein domains |
TALEs from plant pathogenic bacteria xanthomonas. |
CRISPR-associated system (CRISPR-Cas) of found in many bacteria and archea species. |
|
Mechanism of DNA recognition
|
Arrays of 30 amino acid-long Cys2-His2 zinc finger domains separated by linker sequences [162] |
Arrays of 33‒35 amino-acid repeats (TALEs). Each repeat recognises a single DNA base pair in the major groove [163]. |
Cas9 nuclease recognises target DNA sequence through a short RNA molecule, which includes a 20-bp fragment complimentary to the target DNA. |
|
Off-target cleavage
|
High |
Low |
High for many applications, but being improved [89, 90]. |
|
Generation time
|
|
1–4 weeks |
±3 days |
|
Construction
|
DNA synthesis or several noncommercial DNA engineering methods [164] |
DNA synthesis or several noncommercial DNA engineering methods [92, 94, 95, 165–168]. |
Only one-step cloning of the guide sequence into gRNA/Cas9 expression vector. For increased efficiency, Cas9 mRNA or protein can be made in vitro[96]. |
|
Other important features
|
Lower target density as compared with RGENs and TALENs [74]
Higher risk poor DNA recognition or cytotoxicity when using newly designed ZFN [169, 170] |
Higher specificity than original CRISPR/Cas9 |
Fast and relatively inexpensive
Preferable for targeting multiple loci
Flexible design
Cre dependent Cas9-expressing mouse line available from the Jackson Lab: http://jaxmice.jax.org/strain/024857.html
Cas9 variant for AAV delivery [98] |
|
DNA recognition determinant
|
Protein (zinc finger domains) |
Protein (TALE arrays) |
Synthetic guide RNA (sgRNA) |
|
Nuclease component
|
FokI |
FokI |
Cas9 |
|
Online tools and other resources
|
http://www.zincfingers.org
http://www.zincfingertools.org |
http://www.e-talen.org
http://taleffector.genome-engineering.org/ |
http://www.e-crisp.org
http://crispr.genome-engineering.org/
http://www.rgenome.net/ |
| AAV = adeno-associated virus; bp = base-pair; Cas9 = CRISPR-associated system nuclease 9; CRISPR= clustered regularly interspaced short palindromic repeats; gRNA = guide RNA; sgRNA = synthetic guide RNA; TALE = transcription activator-like effector |
Context matters too – effects of genetic background
Engineered alleles (RITs, knock-outs or knock-ins) are often transferred from one genetic background to another by repeated backcrossing, occasionally with unwanted consequences. For example, mouse lines with different knock-out alleles of the prion protein gene were engineered in ESCs derived from 129S7, 129S4 or 129P2 mouse strains (hereafter collectively referred to as 129) and the resulting chimeras were typically bred to a very different genetic background, C57Bl/6 (B6 hereafter) [32, 116–119]. Even after backcrossing to B6 for 15 generations, a small amount of 129 genomic DNA flanking the Prnp gene remained [120]. This residual 129-derived sequence caused a phenotype, originally attributed to the mutant Prnp gene [121], that was later determined to be independent of the Prnp deletion [120]. Unfortunately, this was not an isolated event [122–124]. So how can it be avoided?
If using inbred mice is not possible one solution is to create a knock-in control allele that is built like the mutant allele but lacks the element expected to create the phenotype [50, 52, 122, 125–127]. This approach develops mutants and controls with approximately the same amount of ESC derived genome flanking the engineered locus [122]. However, creating the extra mouse line essentially doubles the work. An alternative solution is to utilise the plethora of available genome mapping and sequence data to identify natural genomic features in that locus that differ between the two strains [28]. This would enable one to backcross the wild-type allele from the donor background into the recipient background guided by a genotyping assay, thus bypassing the extra work required to engineer a knock-in control line, and providing a strategy for RITs if the integration site is known. Finally, with the rapid advances in the RGENs field, routine engineering of the same genetic element into mice of multiple inbred genetic backgrounds is foreseeable.
Cutting-edge tools to study gene regulation
Diseases typically cause changes to tissues that are marked by changes in gene expression, essentially serving as a quantitative phenotype. For example, in neurodegenerative diseases, astrocytes often convert into a reactive phenotype and therefore changes in related gene products could be detected by in situ hybridisation or northern blots [128]. In the 1990s the high-throughput method of DNA microarrays transformed this field, suddenly allowing for hundreds, and eventually thousands, of mRNAs to be tested systematically and simultaneously [129, 130]. Just as dramatically, the field took another exponential leap in the 2000s with the invention of next generation sequencing (NGS) technologies, which basically determine the sequence of tens of millions of small fragments of DNA (obtained by reverse transcription of RNAs) in parallel, which are used to calculate the relative fraction of transcripts of tens of thousands of genes in a sample [131]. These techniques generate very accurate quantitative data, with a large dynamic range, and can provide additional information (for example mRNA splice variants, posttranscriptional edits, etc.). Although contention exists [132, 133], many experiments reveal that gene expression changes observed in humans also occur in the corresponding mouse models [134–136], providing solid validation of the models. New techniques are constantly improving the sequencing side of the NGS approach, for example by reducing the amount of RNA needed while improving the quantitative nature of the technique [137–140]. Until recently a major limitation was rooted in the profound biological problem that tissues consist of an extraordinary mixture of multiple cell types, and cell types with opposing gene expression programmes will be masked if RNAs are purified from a homogenate containing both. Thus, analysis of RNAs from specific cell types would provide a huge leap forward.
It’s all about the lure – cell type-specific gene expression studies
One logical approach is to physically isolate specific cell types using a variety of sophisticated techniques. In one, tissues are gently dissociated into cell suspensions, labelled with antibodies targeting cell type-specific epitopes, and then separated by fluorescence activated cell sorting or immunopanning [141, 142]. This works extremely well for tissues that are easy to dissociate, such as blood, allowing a rapid separation procedure to be completed before too many changes to RNA levels occur. It is also perfectly suited for experiments where cells are sorted and subsequently cultured, allowing recovery from any gene expression changes induced by the dissociation and separation. This approach has yielded important insight into the transcriptomes of specific cell types in the mouse brain [141, 142]. Alternatively, cells in tissue slices can be hand-picked with laser capture microdissection (LCM) [143]. The LCM strategy limits unwanted changes in gene expression because the tissues are typically fixed or frozen, and can be combined with spatial information (e.g. proximity to a disease lesion). Though the cell bodies are typically the only parts captured and much mRNA is located far away from there, e.g. mRNAs translated at neuronal synapses [144] much important insight into disease has been acquired with LCM [135, 145].
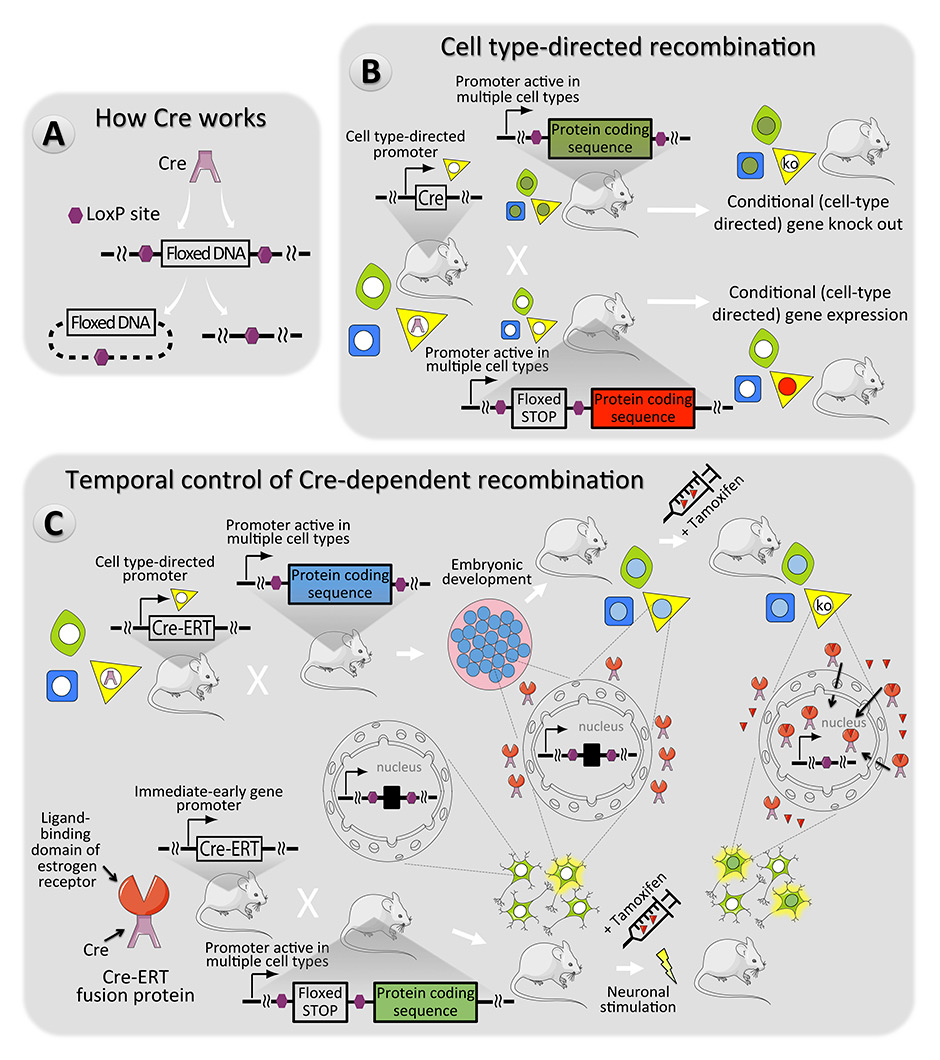
Figure 3
Cre recombinase – a versatile mouse genome manipulation tool. (A) In the systems described here, the activity of the Cre enzyme results in the removal of DNA sequences between two LoxP sites in the mouse genome [160]. (B) Spatially restricted expression of Cre can be employed to inactivate or activate genes in specific cell types [146] (cell type 3 – yellow). (C) Temporal control of Cre activity can be accomplished with fusion proteins consisting of Cre with an oestrogen receptor fragment (ERT) which prevents Cre-mediated recombination, until tamoxifen is present to direct the Cre-ERT fusion into the nucleus [161]. The top pathway features an example where the promoter driving Cre-ERT is cell type-specific. This is convenient in cases where cellular phenotypes change during development or through time (e.g. neural precursor cells can become neurons or glia). The bottom pathway features an example where Cre-ERT expression is induced by stimuli that happen often but when Cre activity is desired for a specific time. In this example an immediate early gene promoter drives expression, which would happen often, but Cre activity would occur only when tamoxifen is present [149].
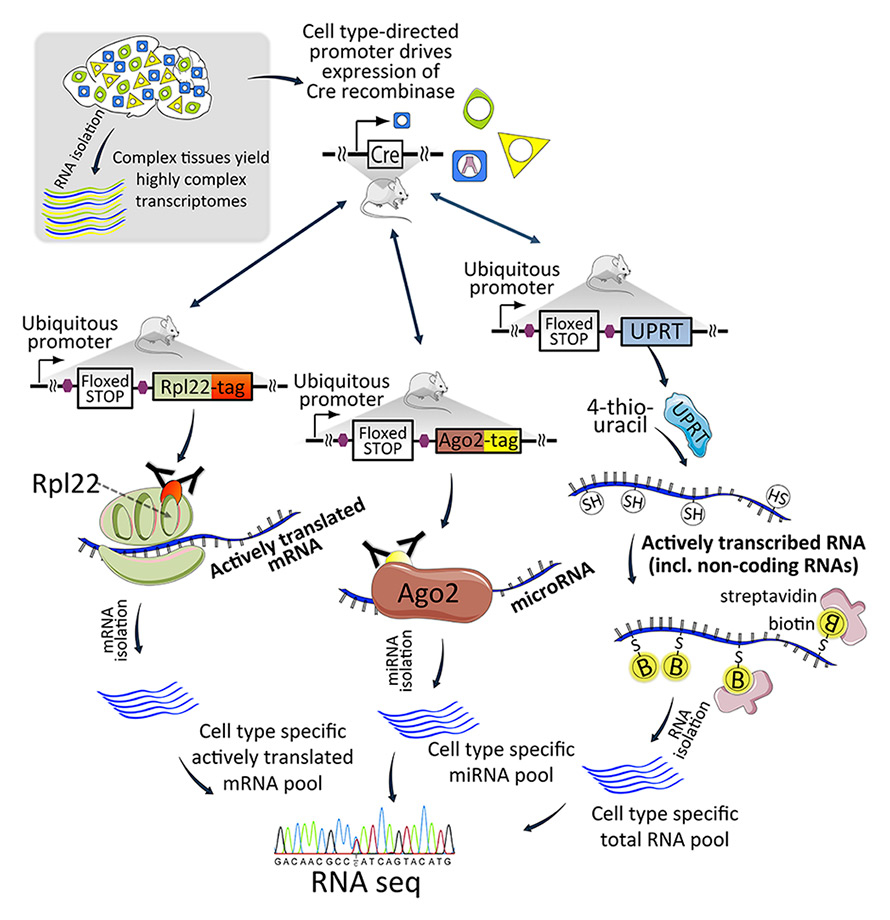
Figure 4
Transgenic techniques for cell type-specific RNA capture. A mouse expressing Cre recombinase is bred with a mouse encoding transgenes for RNA capture components. Each transgene is activated by Cre to drive cell type-specificity. The Rpl22 (ribosomal protein L22) protein captures ribosomes and ribosome-bound mRNAs during translation (the Ribotag method). The Ago2 protein captures the RISC complex and associated miRNAs. The UPRT enzyme converts 4-thiouracil into a nucleotide that is incorporated into newly transcribed RNA.
Ago2 = Argonaute2; miRNA = microRNA; RISC = RNA-induced silencing complex; UPRT = uracil phosphoribosyltransferase
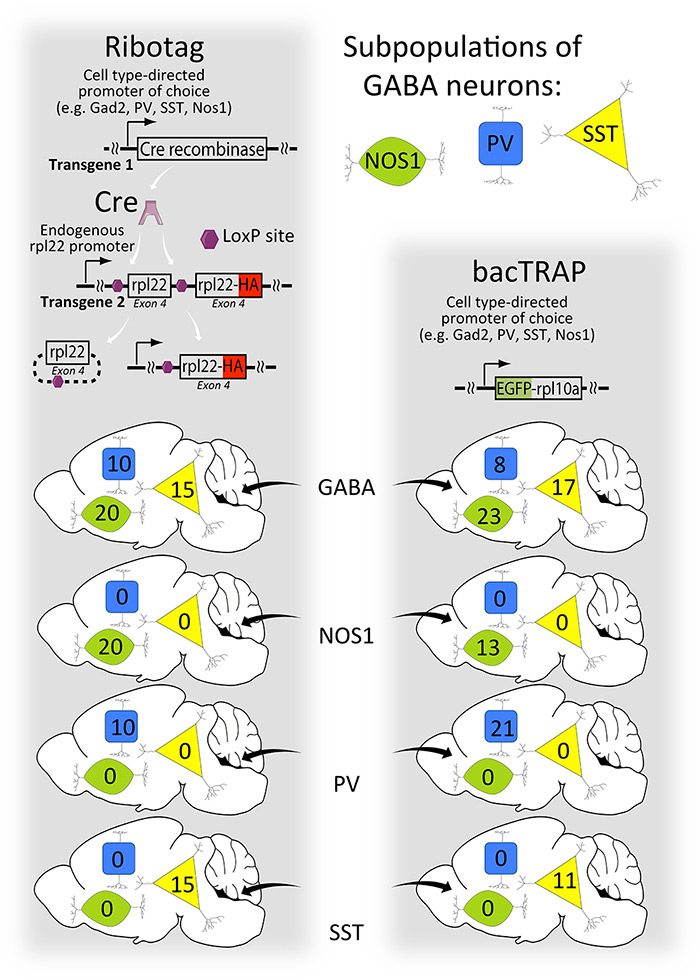
Figure 5
A comparison of Ribotag and bacTRAP methods. In the left pathway, a ubiquitous promoter drives expression of the Ribotag (rpl22-HA) protein only after Cre recombinase activity releases the inhibition of Ribotag expression. The endogenous rpl22 exon 4 functions as a ‘floxed STOP’ for the Ribotag method, as shown in fig. 4. The cell type specificity is determined by the pattern of Cre expression. Therefore, the same cell targeted by different Cre activators will have the same expression level of the Ribotag protein because the same ubiquitous promoter drives its expression from the same place in the genome. In contrast (right pathway), different promoters drive the expression of bacTRAP resulting in different expression levels in this hypothetical example. An important advantage of bacTRAP is that one mouse line is needed versus two for Ribotag.
A new and radically different approach is to isolate RNAs from specific cell types directly from crude tissue homogenates. The various versions of this strategy share the common theme of labelling biomolecules in specific cells and then using the introduced labels to affinity purify (or co-purify) RNAs from homogenised tissue. The isolated tissues can be rapidly frozen, thereby maximally preserving the native (patho)physiological state of gene expression. Many of these tools employ Cre for activation (fig. 3). Cre can activate the expression of a RNA tagging tool encoding transgene in a subset of cells based on, for example, cell identity, developmental stage or activity state (fig. 3) [146–149].
One cell type-specific RNA labelling technique employs a uracil phosphoribosyltransferase (UPRT) enzyme from Toxoplasma gondii[150]. In contrast to the endogenous mouse UPRT, the exogenous enzyme efficiently activates a uracil analogue (4-thiouracil, 4TU), which is then incorporated into RNAs transcribed in the cell type of interest (fig. 4). Such 4TU-containing RNA can be easily biotinylated and efficiently separated from total RNA with routine biotin-streptavidin enrichment tools [151].
A second strategy aims to capture specifically miRNAs, small RNAs that modulate gene expression by guiding the RNA induced silencing complex (RISC) to specific mRNAs and driving their degradation or impairing their translation [152]. This was accomplished by cell type-specific expression of an epitope-tagged protein component of RISC called Argonaute2 (Ago2, fig. 4) [153, 154]. Tissue homogenates are then probed with antibodies against the engineered epitope tags, co-capturing the RISC and associated miRNAs [154].
Two slightly different methods using a concept similar to the Ago2 method were developed to capture, from specific cell types, functional ribosomes and their associated mRNAs, enabling the study of translatomes. In the first method, called RiboTag [155], a modified ribosomal protein is expressed from its endogenous genomic location, but only once activated by Cre (figs 4 and 5). In the second method, called bacTRAP, a modified ribosomal protein is expressed by a BAC transgene (fig. 5) [156, 157]. Besides being a tool to isolate mRNA from specific cell types, mRNAs associated with ribosomes more closely represent the proteome than total mRNAs [158]. Although both methods to study the translatome are broadly similar there are some important differences. One important difference is that for the RiboTag approach, not one but two mouse lines are required: the knocked-in ribosomal gene plus a Cre activator that determines the cell type-specificity. In an effort to study a genetic mouse model of a disease, the addition of two new loci to the mix requires complicated breeding schemes and lots of costly cage space. However, the RiboTag approach has an important advantage over the bacTRAP method. With the bacTRAP method a single promoter determines both cell type-specificity and expression levels. In addition to the issues of random integration (see above), the promoters will have variable strengths in different cells, which will make quantitative comparisons between lines complicated (fig. 5). Moreover, the effects of gene-expression changes on cell type markers might change during disease [36, 159], which would also alter the amount of the tagged ribosome in the bacTRAP method. Both approaches are very clever, but it is critical to identify the most important features and disadvantages of each when designing a project. In all, these various techniques for cell type-specific gene expression studies will vastly improve the information obtained from disease models.
Conclusion
In this short review we have presented a handful of examples of new genetic tools that are transforming biomedical research involving mouse models of disease. Some notable examples that we omitted include optogenetics and RNA interference, though they have existed for a while longer and are thus more widely known. Nonetheless, the examples we highlighted make for a very powerful compilation of tools that will harmonise. The gene-expression analysis tools will enhance the discovery of disease mechanisms and targets, and will also function as robust phenotyping tools to measure the efficacy of experimental therapeutic strategies. In turn, the new genome sequence and expression manipulation techniques, especially when applied to specific cell-types, will be a powerful approach to interrogate these pathways. In the 1980s, the development of gene-targeted mice made scientists absolutely giddy with excitement. That giddy feeling is back!
Acknowledgements:We are grateful to Sybille Krauss and Dan Ehninger for evaluating this manuscript and to two unknown journal appointed reviewers for very helpful suggestions. Some elements of the figures were provided by Servier Medical Art (www.servier.com) for which we are appreciative.
References
1 Fahlstrom A, Yu Q, Ulfhake B. Behavioral changes in aging female C57BL/6 mice. Neurobiol Aging. 2011;32(10):1868–80.
2 Aasum E, Hafstad AD, Severson DL, Larsen TS. Age-dependent changes in metabolism, contractile function, and ischemic sensitivity in hearts from db/db mice. Diabetes. 2003;52(2):434–41.
3 Houtkooper RH, Argmann C, Houten SM, Canto C, Jeninga EH, Andreux PA, et al. The metabolic footprint of aging in mice. Sci Rep. 2011;1:134.
4 Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341(6146):1237905. Epub 2013/07/06.
5 Al-Mahdawi S, Virmouni SA, Pook MA. The emerging role of 5-hydroxymethylcytosine in neurodegenerative diseases. Front Neurosci. 2014;8:397. Epub 2014/12/30.
6 Cheng Y, Ma Z, Kim BH, Wu W, Cayting P, Boyle AP, et al. Principles of regulatory information conservation between mouse and human. Nature. 2014;515(7527):371–5. Epub 2014/11/21.
7 Pope BD, Ryba T, Dileep V, Yue F, Wu W, Denas O, et al. Topologically associating domains are stable units of replication-timing regulation. Nature. 2014;515(7527):402–5. Epub 2014/11/21.
8 Stergachis AB, Neph S, Sandstrom R, Haugen E, Reynolds AP, Zhang M, et al. Conservation of trans-acting circuitry during mammalian regulatory evolution. Nature. 2014;515(7527):365–70. Epub 2014/11/21.
9 Yue F, Cheng Y, Breschi A, Vierstra J, Wu W, Ryba T, et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature. 2014;515(7527):355–64. Epub 2014/11/21.
10 Wu LC, Sun CW, Ryan TM, Pawlik KM, Ren J, Townes TM. Correction of sickle cell disease by homologous recombination in embryonic stem cells. Blood. 2006;108(4):1183–8. Epub 2006/04/28.
11 Hanna J, Wernig M, Markoulaki S, Sun CW, Meissner A, Cassady JP, et al. Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science. 2007;318(5858):1920–3. Epub 2007/12/08.
12 Heyer J, Kwong LN, Lowe SW, Chin L. Non-germline genetically engineered mouse models for translational cancer research. Nat Rev Cancer. 2010;10(7):470–80. Epub 2010/06/25.
13 Walrath JC, Hawes JJ, Van Dyke T, Reilly KM. Genetically engineered mouse models in cancer research. Adv Cancer Res. 2010;106:113–64. Epub 2010/04/20.
14 Philips T, Rothstein JD, Pouladi MA. Preclinical models: needed in translation? A Pro/Con debate. Mov Disord. 2014;29(11):1391–6. Epub 2014/09/13.
15 Jaenisch R, Mintz B. Simian virus 40 DNA sequences in DNA of healthy adult mice derived from preimplantation blastocysts injected with viral DNA. Proc Natl Acad Sci U S A. 1974;71(4):1250–4. Epub 1974/04/01.
16 Brinster RL, Chen HY, Trumbauer M, Senear AW, Warren R, Palmiter RD. Somatic expression of herpes thymidine kinase in mice following injection of a fusion gene into eggs. Cell. 1981;27(1 Pt 2):223–31. Epub 1981/11/01.
17 Costantini F, Lacy E. Introduction of a rabbit beta-globin gene into the mouse germ line. Nature. 1981;294(5836):92–4. Epub 1981/11/05.
18 Wurtele H, Little KC, Chartrand P. Illegitimate DNA integration in mammalian cells. Gene Ther. 2003;10(21):1791–9. Epub 2003/09/10.
19 Smithies O, Gregg RG, Boggs SS, Koralewski MA, Kucherlapati RS. Insertion of DNA sequences into the human chromosomal beta-globin locus by homologous recombination. Nature. 1985;317(6034):230–4.
20 Thomas KR, Capecchi MR. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell. 1987;51(3):503–12.
21 Robertson E, Bradley A, Kuehn M, Evans M. Germ-line transmission of genes introduced into cultured pluripotential cells by retroviral vector. Nature. 1986;323(6087):445–8. Epub 1986/10/02.
22 Hooper M, Hardy K, Handyside A, Hunter S, Monk M. HPRT-deficient (Lesch-Nyhan) mouse embryos derived from germline colonization by cultured cells. Nature. 1987;326(6110):292–5. Epub 1987/03/19.
23 Kuehn MR, Bradley A, Robertson EJ, Evans MJ. A potential animal model for Lesch-Nyhan syndrome through introduction of HPRT mutations into mice. Nature. 1987;326(6110):295–8. Epub 1987/03/19.
24 Koller BH, Hagemann LJ, Doetschman T, Hagaman JR, Huang S, Williams PJ, et al. Germ-line transmission of a planned alteration made in a hypoxanthine phosphoribosyltransferase gene by homologous recombination in embryonic stem cells. Proc Natl Acad Sci U S A. 1989;86(22):8927–31. Epub 1989/11/01.
25 Thompson S, Clarke AR, Pow AM, Hooper ML, Melton DW. Germ line transmission and expression of a corrected HPRT gene produced by gene targeting in embryonic stem cells. Cell. 1989;56(2):313–21. Epub 1989/01/27.
26 Zijlstra M, Li E, Sajjadi F, Subramani S, Jaenisch R. Germ-line transmission of a disrupted beta 2-microglobulin gene produced by homologous recombination in embryonic stem cells. Nature. 1989;342(6248):435–8. Epub 1989/11/23.
27 Thomas KR, Capecchi MR. Targeted disruption of the murine int-1 proto-oncogene resulting in severe abnormalities in midbrain and cerebellar development. Nature. 1990;346(6287):847–50. Epub 1990/08/30.
28 Smithies O, Maeda N. Gene targeting approaches to complex genetic diseases: atherosclerosis and essential hypertension. Proc Natl Acad Sci U S A. 1995;92(12):5266–72. Epub 1995/06/06.
29 Melton DW. Gene targeting in the mouse. Bioessays. 1994;16(9):633–8.
30 Detloff PJ, Lewis J, John SW, Shehee WR, Langenbach R, Maeda N, et al. Deletion and replacement of the mouse adult beta-globin genes by a “plug and socket” repeated targeting strategy. Mol Cell Biol. 1994;14(10):6936–43. Epub 1994/10/01.
31 Stacey A, Schnieke A, McWhir J, Cooper J, Colman A, Melton DW. Use of double-replacement gene targeting to replace the murine alpha-lactalbumin gene with its human counterpart in embryonic stem cells and mice. Mol Cell Biol. 1994;14(2):1009–16. Epub 1994/02/01.
32 Moore RC, Redhead NJ, Selfridge J, Hope J, Manson JC, Melton DW. Double replacement gene targeting for the production of a series of mouse strains with different prion protein gene alterations. Biotechnology (N Y). 1995;13(9):999–1004.
33 Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326(5950):289–93. Epub 2009/10/10.
34 Gheldof N, Smith EM, Tabuchi TM, Koch CM, Dunham I, Stamatoyannopoulos JA, et al. Cell-type-specific long-range looping interactions identify distant regulatory elements of the CFTR gene. Nucleic Acids Res. 2010;38(13):4325–36. Epub 2010/04/03.
35 Gibcus JH, Dekker J. The hierarchy of the 3D genome. Mol Cell. 2013;49(5):773–82. Epub 2013/03/12.
36 Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155(4):934–47. Epub 2013/10/15.
37 Dowen JM, Fan ZP, Hnisz D, Ren G, Abraham BJ, Zhang LN, et al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell. 2014;159(2):374–87. Epub 2014/10/11.
38 Burright EN, Clark HB, Servadio A, Matilla T, Feddersen RM, Yunis WS, et al. SCA1 transgenic mice: a model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell. 1995;82(6):937–48.
39 Caroni P. Overexpression of growth-associated proteins in the neurons of adult transgenic mice. J Neurosci Methods. 1997;71(1):3–9. Epub 1997/01/01.
40 Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, et al. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28(1):41–51.
41 Livet J, Weissman TA, Kang H, Draft RW, Lu J, Bennis RA, et al. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature. 2007;450(7166):56–62. Epub 2007/11/02.
42 Borchelt DR, Davis J, Fischer M, Lee MK, Slunt HH, Ratovitsky T, et al. A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet Anal. 1996;13(6):159–63.
43 Fischer M, Rulicke T, Raeber A, Sailer A, Moser M, Oesch B, et al. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. Embo J. 1996;15(6):1255–64.
44 Faas H, Jackson WS, Borkowski AW, Wang X, Ma J, Lindquist S, et al. Context-dependent perturbation of neural systems in transgenic mice expressing a cytosolic prion protein. Neuroimage. 2010;49(3):2607–17. Epub 2009/10/20.
45 Karapetyan YE, Saa P, Mahal SP, Sferrazza GF, Sherman A, Sales N, et al. Prion strain discrimination based on rapid in vivo amplification and analysis by the cell panel assay. PLoS One. 2009;4(5):e5730. Epub 2009/05/30.
46 Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425(6961):917–25. Epub 2003/10/31.
47 Zabel M, Greenwood C, Thackray AM, Pulford B, Rens W, Bujdoso R. Perturbation of T-cell development by insertional mutation of a PrP transgene. Immunology. 2009;127(2):226–36. Epub 2009/01/16.
48 Akhtar W, de Jong J, Pindyurin AV, Pagie L, Meuleman W, de Ridder J, et al. Chromatin position effects assayed by thousands of reporters integrated in parallel. Cell. 2013;154(4):914–27. Epub 2013/08/21.
49 Lewis J, Yang B, Kim R, Sierakowska H, Kole R, Smithies O, et al. A common human beta globin splicing mutation modeled in mice. Blood. 1998;91(6):2152–6. Epub 1998/04/16.
50 Lin CH, Tallaksen-Greene S, Chien WM, Cearley JA, Jackson WS, Crouse AB, et al. Neurological abnormalities in a knock-in mouse model of Huntington’s disease. Hum Mol Genet. 2001;10(2):137–44. Epub 2001/01/12.
51 Watase K, Weeber EJ, Xu B, Antalffy B, Yuva-Paylor L, Hashimoto K, et al. A long CAG repeat in the mouse Sca1 locus replicates SCA1 features and reveals the impact of protein solubility on selective neurodegeneration. Neuron. 2002;34(6):905–19. Epub 2002/06/28.
52 Jackson WS, Borkowski AW, Faas H, Steele AD, King OD, Watson N, et al. Spontaneous generation of prion infectivity in fatal familial insomnia knockin mice. Neuron. 2009;63(4):438–50.
53 Jackson WS. Selective vulnerability to neurodegenerative disease: the curious case of Prion Protein. Dis Model Mech. 2014;7(1):21–9. Epub 2014/01/08.
54 Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. Epub 2010/03/03.
55 van den Bosch M, Lohman PH, Pastink A. DNA double-strand break repair by homologous recombination. Biol Chem. 2002;383(6):873–92. Epub 2002/09/12.
56 Szostak JW, Orr-Weaver TL, Rothstein RJ, Stahl FW. The double-strand-break repair model for recombination. Cell. 1983;33(1):25–35. Epub 1983/05/01.
57 Aymard F, Bugler B, Schmidt CK, Guillou E, Caron P, Briois S, et al. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat Struct Mol Biol. 2014;21(4):366–74. Epub 2014/03/25.
58 Weissmann C, Bueler H. A mouse to remember. Cell. 2004;116(2 Suppl):S111–3, 2 p following S3. Epub 2004/04/02.
59 Miranda A, Pericuesta E, Ramirez MA, Gutierrez-Adan A. Prion protein expression regulates embryonic stem cell pluripotency and differentiation. PLoS One. 2011;6(4):e18422. Epub 2011/04/13.
60 Palais G, Nguyen Dinh Cat A, Friedman H, Panek-Huet N, Millet A, Tronche F, et al. Targeted transgenesis at the HPRT locus: an efficient strategy to achieve tightly controlled in vivo conditional expression with the tet system. Physiol Genomics. 2009;37(2):140–6. Epub 2009/01/15.
61 Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21(1):70–1. Epub 1999/01/23.
62 Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13(1):133–40. Epub 2009/12/22.
63 Zambrowicz BP, Imamoto A, Fiering S, Herzenberg LA, Kerr WG, Soriano P. Disruption of overlapping transcripts in the ROSA beta geo 26 gene trap strain leads to widespread expression of beta-galactosidase in mouse embryos and hematopoietic cells. Proc Natl Acad Sci U S A. 1997;94(8):3789–94. Epub 1997/04/15.
64 Zeng H, Horie K, Madisen L, Pavlova MN, Gragerova G, Rohde AD, et al. An inducible and reversible mouse genetic rescue system. PLoS Genet. 2008;4(5):e1000069. Epub 2008/05/10.
65 Madisen L, Garner AR, Shimaoka D, Chuong AS, Klapoetke NC, Li L, et al. Transgenic mice for intersectional targeting of neural sensors and effectors with high specificity and performance. Neuron. 2015;85(5):942–58. Epub 2015/03/06.
66 Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci U S A. 1996;93(3):1156–60. Epub 1996/02/06.
67 Bibikova M, Beumer K, Trautman JK, Carroll D. Enhancing gene targeting with designed zinc finger nucleases. Science. 2003;300(5620):764. Epub 2003/05/06.
68 Bibikova M, Golic M, Golic KG, Carroll D. Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics. 2002;161(3):1169–75. Epub 2002/07/24.
69 Carbery ID, Ji D, Harrington A, Brown V, Weinstein EJ, Liaw L, et al. Targeted genome modification in mice using zinc-finger nucleases. Genetics. 2010;186(2):451–9. Epub 2010/07/16.
70 Kimberlin RH, Walker CA. Pathogenesis of mouse scrapie: effect of route of inoculation on infectivity titres and dose-response curves. J Comp Pathol. 1978;88(1):39–47. Epub 1978/01/01.
71 Porteus MH, Baltimore D. Chimeric nucleases stimulate gene targeting in human cells. Science. 2003;300(5620):763. Epub 2003/05/06.
72 Segal DJ, Meckler JF. Genome engineering at the dawn of the golden age. Annu Rev Genomics Hum Genet. 2013;14:135–58. Epub 2013/05/25.
73 Rahman SH, Maeder ML, Joung JK, Cathomen T. Zinc-finger nucleases for somatic gene therapy: the next frontier. Hum Gene Ther. 2011;22(8):925–33. Epub 2011/06/03.
74 Kim HJ, Lee HJ, Kim H, Cho SW, Kim JS. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Res. 2009;19(7):1279–88. Epub 2009/05/28.
75 Maeder ML, Thibodeau-Beganny S, Sander JD, Voytas DF, Joung JK. Oligomerized pool engineering (OPEN): an “open-source” protocol for making customized zinc-finger arrays. Nat Protoc. 2009;4(10):1471–501. Epub 2009/10/03.
76 Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. 2010;11(9):636–46. Epub 2010/08/19.
77 Ferguson C, McKay M, Harris RA, Homanics GE. Toll-like receptor 4 (Tlr4) knockout rats produced by transcriptional activator-like effector nuclease (TALEN)-mediated gene inactivation. Alcohol. 2013;47(8):595–9. Epub 2013/11/10.
78 Sung YH, Baek IJ, Kim DH, Jeon J, Lee J, Lee K, et al. Knockout mice created by TALEN-mediated gene targeting. Nat Biotechnol. 2013;31(1):23–4. Epub 2013/01/11.
79 Sung YH, Kim JM, Kim HT, Lee J, Jeon J, Jin Y, et al. Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome Res. 2014;24(1):125–31. Epub 2013/11/21.
80 Tesson L, Usal C, Menoret S, Leung E, Niles BJ, Remy S, et al. Knockout rats generated by embryo microinjection of TALENs. Nat Biotechnol. 2011;29(8):695–6. Epub 2011/08/09.
81 Wefers B, Meyer M, Ortiz O, Hrabe de Angelis M, Hansen J, Wurst W, et al. Direct production of mouse disease models by embryo microinjection of TALENs and oligodeoxynucleotides. Proc Natl Acad Sci U S A. 2013;110(10):3782–7. Epub 2013/02/22.
82 Gilles AF, Averof M. Functional genetics for all: engineered nucleases, CRISPR and the gene editing revolution. Evodevo. 2014;5:43. Epub 2015/02/24.
83 Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346(6213):1258096. Epub 2014/11/29.
84 Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157(6):1262–78. Epub 2014/06/07.
85 Gaj T, Gersbach CA, Barbas CF, 3rd. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31(7):397–405. Epub 2013/05/15.
86 Pu J, Frescas D, Zhang B, Feng J. Utilization of TALEN and CRISPR/Cas9 technologies for gene targeting and modification. Exp Biol Med (Maywood). 2015. Epub 2015/05/10.
87 Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae S, et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24(1):132–41. Epub 2013/11/21.
88 Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31(9):822–6. Epub 2013/06/25.
89 Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154(6):1380–9. Epub 2013/09/03.
90 Guilinger JP, Thompson DB, Liu DR. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat Biotechnol. 2014;32(6):577–82. Epub 2014/04/29.
91 Sommer D, Peters A, Wirtz T, Mai M, Ackermann J, Thabet Y, et al. Efficient genome engineering by targeted homologous recombination in mouse embryos using transcription activator-like effector nucleases. Nat Commun. 2014;5:3045. Epub 2014/01/15.
92 Briggs AW, Rios X, Chari R, Yang L, Zhang F, Mali P, et al. Iterative capped assembly: rapid and scalable synthesis of repeat-module DNA such as TAL effectors from individual monomers. Nucleic Acids Res. 2012;40(15):e117. Epub 2012/06/29.
93 Reyon D, Tsai SQ, Khayter C, Foden JA, Sander JD, Joung JK. FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol. 2012;30(5):460–5. Epub 2012/04/10.
94 Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, et al. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011;39(12):e82. Epub 2011/04/16.
95 Schmid-Burgk JL, Schmidt T, Kaiser V, Honing K, Hornung V. A ligation-independent cloning technique for high-throughput assembly of transcription activator-like effector genes. Nat Biotechnol. 2013;31(1):76–81. Epub 2012/12/18.
96 Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153(4):910–8. Epub 2013/05/07.
97 Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014;159(2):440–55. Epub 2014/09/30.
98 Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520(7546):186–91. Epub 2015/04/02.
99 Ousterout DG, Kabadi AM, Thakore PI, Majoros WH, Reddy TE, Gersbach CA. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat Commun. 2015;6:6244. Epub 2015/02/19.
100 Liao HK, Gu Y, Diaz A, Marlett J, Takahashi Y, Li M, et al. Use of the CRISPR/Cas9 system as an intracellular defense against HIV-1 infection in human cells. Nat Commun. 2015;6:6413. Epub 2015/03/11.
101 Osborn MJ, Gabriel R, Webber BR, DeFeo AP, McElroy AN, Jarjour J, et al. Fanconi anemia gene editing by the CRISPR/Cas9 system. Hum Gene Ther. 2015;26(2):114–26. Epub 2014/12/30.
102 Heckl D, Kowalczyk MS, Yudovich D, Belizaire R, Puram RV, McConkey ME, et al. Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat Biotechnol. 2014;32(9):941–6. Epub 2014/06/24.
103 Lee HJ, Kweon J, Kim E, Kim S, Kim JS. Targeted chromosomal duplications and inversions in the human genome using zinc finger nucleases. Genome Res. 2012;22(3):539–48. Epub 2011/12/21.
104 Renouf B, Piganeau M, Ghezraoui H, Jasin M, Brunet E. Creating cancer translocations in human cells using Cas9 DSBs and nCas9 paired nicks. Methods Enzymol. 2014;546:251–71. Epub 2014/11/16.
105 Maddalo D, Manchado E, Concepcion CP, Bonetti C, Vidigal JA, Han YC, et al. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature. 2014;516(7531):423–7. Epub 2014/10/23.
106 Mou H, Kennedy Z, Anderson DG, Yin H, Xue W. Precision cancer mouse models through genome editing with CRISPR-Cas9. Genome Med. 2015;7(1):53. Epub 2015/06/11.
107 Qin W, Dion SL, Kutny PM, Zhang Y, Cheng A, Jillette NL, et al. Efficient CRISPR/Cas9-Mediated Genome Editing in Mice by Zygote Electroporation of Nuclease. Genetics. 2015. Epub 2015/03/31.
108 Maruyama T, Dougan SK, Truttmann MC, Bilate AM, Ingram JR, Ploegh HL. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat Biotechnol. 2015;33(5):538–42. Epub 2015/03/24.
109 Cheng AW, Wang H, Yang H, Shi L, Katz Y, Theunissen TW, et al. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013;23(10):1163–71. Epub 2013/08/28.
110 Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517(7536):583–8. Epub 2014/12/11.
111 Konermann S, Brigham MD, Trevino AE, Hsu PD, Heidenreich M, Cong L, et al. Optical control of mammalian endogenous transcription and epigenetic states. Nature. 2013;500(7463):472–6. Epub 2013/07/24.
112 Polstein LR, Gersbach CA. A light-inducible CRISPR-Cas9 system for control of endogenous gene activation. Nat Chem Biol. 2015;11(3):198–200. Epub 2015/02/11.
113 Heller EA, Cates HM, Pena CJ, Sun H, Shao N, Feng J, et al. Locus-specific epigenetic remodeling controls addiction- and depression-related behaviors. Nat Neurosci. 2014;17(12):1720–7. Epub 2014/10/28.
114 Hilton IB, D’Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol. 2015. Epub 2015/04/08.
115 Polstein L, Perez-Pinera P, Kocak D, Vockley C, Bledsoe P, Song L, et al. Genome-wide specificity of DNA-binding, gene regulation, and chromatin remodeling by TALE- and CRISPR/Cas9-based transcriptional activators. Genome Res. 2015. Epub 2015/05/31.
116 Bueler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, et al. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature. 1992;356(6370):577–82.
117 Manson JC, Clarke AR, Hooper ML, Aitchison L, McConnell I, Hope J. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol Neurobiol. 1994;8(2-3):121–7.
118 Sakaguchi S, Katamine S, Shigematsu K, Nakatani A, Moriuchi R, Nishida N, et al. Accumulation of proteinase K-resistant prion protein (PrP) is restricted by the expression level of normal PrP in mice inoculated with a mouse-adapted strain of the Creutzfeldt-Jakob disease agent. J Virol. 1995;69(12):7586–92. Epub 1995/12/01.
119 Jackson WS, Krost C, Borkowski AW, Kaczmarczyk L. Translation of the Prion Protein mRNA Is Robust in Astrocytes but Does Not Amplify during Reactive Astrocytosis in the Mouse Brain. PLoS One. 2014;9(4):e95958. Epub 2014/04/23.
120 Nuvolone M, Kana V, Hutter G, Sakata D, Mortin-Toth SM, Russo G, et al. SIRPalpha polymorphisms, but not the prion protein, control phagocytosis of apoptotic cells. J Exp Med. 2013;210(12):2539–52. Epub 2013/10/23.
121 de Almeida CJ, Chiarini LB, da Silva JP, PM ES, Martins MA, Linden R. The cellular prion protein modulates phagocytosis and inflammatory response. J Leukoc Biol. 2005;77(2):238–46. Epub 2004/11/13.
122 Gerlai R. Gene-targeting studies of mammalian behavior: is it the mutation or the background genotype? Trends Neurosci. 1996;19(5):177–81. Epub 1996/05/01.
123 Crusio WE. Flanking gene and genetic background problems in genetically manipulated mice. Biol Psychiatry. 2004;56(6):381–5. Epub 2004/09/15.
124 Striebel JF, Race B, Pathmajeyan M, Rangel A, Chesebro B. Lack of influence of prion protein gene expression on kainate-induced seizures in mice: studies using congenic, coisogenic and transgenic strains. Neuroscience. 2013;238:11–8. Epub 2013/02/19.
125 Ordway JM, Tallaksen-Greene S, Gutekunst CA, Bernstein EM, Cearley JA, Wiener HW, et al. Ectopically expressed CAG repeats cause intranuclear inclusions and a progressive late onset neurological phenotype in the mouse. Cell. 1997;91(6):753–63. Epub 1997/12/31.
126 Jackson WS, Tallaksen-Greene SJ, Albin RL, Detloff PJ. Nucleocytoplasmic transport signals affect the age at onset of abnormalities in knock-in mice expressing polyglutamine within an ectopic protein context. Hum Mol Genet. 2003;12(13):1621–9.
127 Jackson WS, Borkowski AW, Watson NE, King OD, Faas H, Jasanoff A, et al. Profoundly different prion diseases in knock-in mice carrying single PrP codon substitutions associated with human diseases. Proc Natl Acad Sci U S A. 2013;110(36):14759–64. Epub 2013/08/21.
128 Diedrich J, Wietgrefe S, Zupancic M, Staskus K, Retzel E, Haase AT, et al. The molecular pathogenesis of astrogliosis in scrapie and Alzheimer's disease. Microb Pathog. 1987;2(6):435–42. Epub 1987/06/01.
129 Lashkari DA, DeRisi JL, McCusker JH, Namath AF, Gentile C, Hwang SY, et al. Yeast microarrays for genome wide parallel genetic and gene expression analysis. Proc Natl Acad Sci U S A. 1997;94(24):13057–62. Epub 1997/12/16.
130 Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270(5235):467–70. Epub 1995/10/20.
131 Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57–63. Epub 2008/11/19.
132 Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2013;110(9):3507–12. Epub 2013/02/13.
133 Takao K, Miyakawa T. Genomic responses in mouse models greatly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2015;112(4):1167–72. Epub 2014/08/06.
134 Kuhn A, Goldstein DR, Hodges A, Strand AD, Sengstag T, Kooperberg C, et al. Mutant huntingtin’s effects on striatal gene expression in mice recapitulate changes observed in human Huntington’s disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum Mol Genet. 2007;16(15):1845–61. Epub 2007/05/24.
135 Pereson S, Wils H, Kleinberger G, McGowan E, Vandewoestyne M, Van Broeck B, et al. Progranulin expression correlates with dense-core amyloid plaque burden in Alzheimer disease mouse models. J Pathol. 2009;219(2):173–81. Epub 2009/06/27.
136 Neueder A, Bates GP. A common gene expression signature in Huntington inverted question marks disease patient brain regions. BMC Med Genomics. 2014;7(1):60. Epub 2014/11/02.
137 Adiconis X, Borges-Rivera D, Satija R, DeLuca DS, Busby MA, Berlin AM, et al. Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nat Methods. 2013;10(7):623–9. Epub 2013/05/21.
138 Islam S, Zeisel A, Joost S, La Manno G, Zajac P, Kasper M, et al. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat Methods. 2014;11(2):163–6. Epub 2013/12/24.
139 Bhargava V, Head SR, Ordoukhanian P, Mercola M, Subramaniam S. Technical variations in low-input RNA-seq methodologies. Scientific reports. 2014;4:3678. Epub 2014/01/15.
140 Shanker S, Paulson A, Edenberg HJ, Peak A, Perera A, Alekseyev YO, et al. Evaluation of commercially available RNA amplification kits for RNA sequencing using very low input amounts of total RNA. J Biomol Tech. 2015;26(1):4–18. Epub 2015/02/05.
141 Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28(1):264–78. Epub 2008/01/04.
142 Foo LC, Allen NJ, Bushong EA, Ventura PB, Chung WS, Zhou L, et al. Development of a method for the purification and culture of rodent astrocytes. Neuron. 2011;71(5):799–811. Epub 2011/09/10.
143 Emmert-Buck MR, Bonner RF, Smith PD, Chuaqui RF, Zhuang Z, Goldstein SR, et al. Laser capture microdissection. Science. 1996;274(5289):998–1001. Epub 1996/11/08.
144 Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445(7124):168–76.
145 Majer A, Medina SJ, Niu Y, Abrenica B, Manguiat KJ, Frost KL, et al. Early mechanisms of pathobiology are revealed by transcriptional temporal dynamics in hippocampal CA1 neurons of prion infected mice. PLoS Pathog. 2012;8(11):e1003002. Epub 2012/11/13.
146 Gu H, Marth JD, Orban PC, Mossmann H, Rajewsky K. Deletion of a DNA polymerase beta gene segment in T cells using cell type-specific gene targeting. Science. 1994;265(5168):103–6. Epub 1994/07/01.
147 Taniguchi H, He M, Wu P, Kim S, Paik R, Sugino K, et al. A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron. 2011;71(6):995–1013. Epub 2011/09/29.
148 Vong L, Ye C, Yang Z, Choi B, Chua S, Jr., Lowell BB. Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron. 2011;71(1):142–54. Epub 2011/07/13.
149 Guenthner CJ, Miyamichi K, Yang HH, Heller HC, Luo L. Permanent genetic access to transiently active neurons via TRAP: targeted recombination in active populations. Neuron. 2013;78(5):773–84. Epub 2013/06/15.
150 Gay L, Miller MR, Ventura PB, Devasthali V, Vue Z, Thompson HL, et al. Mouse TU tagging: a chemical/genetic intersectional method for purifying cell type-specific nascent RNA. Genes Dev. 2013;27(1):98–115. Epub 2013/01/12.
151 Gay L, Karfilis KV, Miller MR, Doe CQ, Stankunas K. Applying thiouracil tagging to mouse transcriptome analysis. Nat Protoc. 2014;9(2):410–20. Epub 2014/01/25.
152 Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466(7308):835–40. Epub 2010/08/13.
153 Hammond SM, Boettcher S, Caudy AA, Kobayashi R, Hannon GJ. Argonaute2, a link between genetic and biochemical analyses of RNAi. Science. 2001;293(5532):1146–50.
154 He M, Liu Y, Wang X, Zhang MQ, Hannon GJ, Huang ZJ. Cell-type-based analysis of microRNA profiles in the mouse brain. Neuron. 2012;73(1):35–48. Epub 2012/01/17.
155 Sanz E, Yang L, Su T, Morris DR, McKnight GS, Amieux PS. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc Natl Acad Sci U S A. 2009;106(33):13939–44. Epub 2009/08/12.
156 Doyle JP, Dougherty JD, Heiman M, Schmidt EF, Stevens TR, Ma G, et al. Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell. 2008;135(4):749–62. Epub 2008/11/18.
157 Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, et al. A translational profiling approach for the molecular characterization of CNS cell types. Cell. 2008;135(4):738–48. Epub 2008/11/18.
158 Battle A, Khan Z, Wang SH, Mitrano A, Ford MJ, Pritchard JK, et al. Genomic variation. Impact of regulatory variation from RNA to protein. Science. 2015;347(6222):664–7. Epub 2015/02/07.
159 Achour M, Le Gras S, Keime C, Parmentier F, Lejeune FX, Boutillier AL, et al. Neuronal Identity Genes Regulated by Super-Enhancers Are Preferentially Down-Regulated in the Striatum of Huntington’s Disease Mice. Hum Mol Genet. 2015. Epub 2015/03/19.
160 Gu H, Zou YR, Rajewsky K. Independent control of immunoglobulin switch recombination at individual switch regions evidenced through Cre-loxP-mediated gene targeting. Cell. 1993;73(6):1155–64. Epub 1993/06/18.
161 Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr Biol. 1998;8(24):1323–6. Epub 1998/12/09.
162 Pabo CO, Peisach E, Grant RA. Design and selection of novel Cys2His2 zinc finger proteins. Annu Rev Biochem. 2001;70:313–40. Epub 2001/06/08.
163 Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326(5959):1509–12. Epub 2009/11/26.
164 Kim JS, Lee HJ, Carroll D. Genome editing with modularly assembled zinc-finger nucleases. Nat Methods. 2010;7(2):91; author reply -2. Epub 2010/01/30.
165 Morbitzer R, Elsaesser J, Hausner J, Lahaye T. Assembly of custom TALE-type DNA binding domains by modular cloning. Nucleic Acids Res. 2011;39(13):5790–9. Epub 2011/03/23.
166 Zhang F, Cong L, Lodato S, Kosuri S, Church GM, Arlotta P. Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nat Biotechnol. 2011;29(2):149–53. Epub 2011/01/21.
167 Sanjana NE, Cong L, Zhou Y, Cunniff MM, Feng G, Zhang F. A transcription activator-like effector toolbox for genome engineering. Nat Protoc. 2012;7(1):171–92. Epub 2012/01/10.
168 Reyon D, Maeder ML, Khayter C, Tsai SQ, Foley JE, Sander JD, et al. Engineering customized TALE nucleases (TALENs) and TALE transcription factors by fast ligation-based automatable solid-phase high-throughput (FLASH) assembly. Curr Protoc Mol Biol. 2013;Chapter 12:Unit 12 6. Epub 2013/07/04.
169 Cornu TI, Thibodeau-Beganny S, Guhl E, Alwin S, Eichtinger M, Joung JK, et al. DNA-binding specificity is a major determinant of the activity and toxicity of zinc-finger nucleases. Mol Ther. 2008;16(2):352–8. Epub 2007/11/21.
170 Ramirez CL, Foley JE, Wright DA, Muller-Lerch F, Rahman SH, Cornu TI, et al. Unexpected failure rates for modular assembly of engineered zinc fingers. Nat Methods. 2008;5(5):374–5. Epub 2008/05/01.