Perspectives on cytokine-directed therapies in multiple sclerosis
DOI: https://doi.org/10.4414/smw.2015.14199
Bettina
Schreiner, Burkhard
Becher
Summary
Multiple sclerosis (MS) is the most common inflammatory demyelinating disorder of the central nervous system (CNS). Over the past 10 years there has been a heated debate as to whether MS pathogenesis commences in the CNS or whether it is actually primarily a disease of the immune system. The combined clinical data, therapy responses, pathology, animal models and genetic studies now provide overwhelming support for the concept that MS is a disease of the immune system and that the CNS is only the unfortunate target of a misguided immune attack.
Immune cells communicate through the use of cytokines and these proteins can orchestrate the most complex behaviour in immune cells. We propose that MS is a disease where immune communication is derailed, which makes MS very amenable to immunotherapy and in particular makes cytokines an attractive target to repair this miscommunication disorder.
Abbreviations
APRIL a proliferation-inducing ligand
BAFF B cell activation factor
BlyS B lymphocyte stimulator
CNS central nervous system
CSF cerebrospinal fluid
EAE experimental autoimmune encephalomyelitis
GM-CSF granulocyte-macrophage colony-stimulating factor
IFN interferon
Ig immunoglobulin
IL interleukin
mAbs monoclonal antibodies
MHC major histocompatibility complex
MS multiple sclerosis
NK cells natural killer cells
NMO neuromyelitis optica
TGF transforming growth factor
Th cell T helper cell
TNF tumour necrosis factor
Introduction
Cytokines are small proteins that permit immune cells to communicate with each other and their surrounding tissue. They represent the “words” in the complex language of the immune system and are therefore critical to coordination of immune functions. Different classes of cytokines have historically been categorised into interleukins (ILs), interferons (IFNs), growth factors, chemokines and tumour necrosis factors (TNFs). In all groups combined, more than 400 members of the greater cytokine family are independently recognised and studied. Multiple functions can be attributed to each of these mediators, a phenomenon broadly termed “pleiotropy”, which when extrapolated results in a vast range of responses controlled by cytokines, including cell proliferation, migration, fibrosis, repair, angiogenesis, immunity and inflammation [1, 2].
Deregulated cytokine responses are observed across all chronic inflammatory diseases and immunopathologies, and there are strong indications from genome-wide association studies that cytokine deregulation is a driver of the pathogenesis. Here, we will discuss current understanding of the role of cytokines in multiple sclerosis (MS), an autoimmune demyelination disease that can affect the brain and spinal cord (CNS). As there are far too many cytokines involved in the pathogenesis of MS to be covered in this review article, representative examples of the most prominent cytokines as therapeutic targets in MS are discussed.
Background
In the 1960s and 1970s, researchers became aware of multiple soluble factors in cell supernatants, distinguished from each other by bioactivity assays, for example “T cell growth factor” [3]. Another example is tumour necrosis factor (TNF), which was described as a mediator of lipopolysaccharide-induced necrosis of transplantable tumours [4]. In the late 1970s, the field of cytokine biology came of age with the introduction of molecular approaches that resulted in the first successful cloning of cytokines such as type I IFNs [5–7]. By the mid-1980s, there was a plethora of well-characterised cytokines and receptors that could be studied with use of molecular tools such as monoclonal antibodies that had revolutionised the ability to specifically recognise a given protein.
Today different therapeutic strategies are used to restore or reduce cytokine signalling pathways in vivo. Purified recombinant cytokines can be administered, for example haematopoietic growth factors (colony stimulating factors) and IFNs. Second, therapeutic agents that inhibit the harmful effects of deregulated proinflammatory cytokines have been developed. The most established methods to neutralise unwanted cytokine signalling are monoclonal antibodies (mAbs), soluble receptors or receptor-Fc fusion molecules and cytokine antagonists. To lower the immunogenicity of murine monoclonal antibodies (generated in rodent species), therapeutic antibodies used in clinical practice are “humanised” by replacing the rodent immunoglobulin structures with human counterparts [8]. Soluble receptor antagonists are usually truncated forms of the cell surface receptor that are devoid of the transmembrane and intracytoplasmic domains but still can bind the cytokine. Sometimes these receptors are fused to the Fc portion of human immunoglobulins (Igs) to further enhance the stability and half-life of the antibody.
First indications that cytokine pathways are implicated in MS pathogenesis were the results of genetic studies that reported an association of MS disease with certain cytokine and receptor genes (IL7Ra,IL2Ra and others) [9–12]. In patients with MS and in experimental mouse models of MS (in particular experimental autoimmune encephalomyelitis, EAE), altered patterns of cytokine expression by various immune cells are observed in the periphery and the CNS, which will be discussed in more detail below. Thus, it was assumed that the balance between pro- and anti-inflammatory cytokines regulates the development and progression of CNS inflammation and tissue damage. Clinical evidence that cytokines are functionally involved in MS in terms of clinical activity was initially based on a clinical trial in which administration of IFN-γ exacerbated the disease [13]. This established a presumptive pathogenic role for IFN-γ-producing, CD4+ T helper (Th) 1 cells, and these Th1 cells and their most abundant cytokines, such as IFN-γ, TNF-α and the Th1-polarising, antigen-presenting cell-derived molecule IL-12 were considered the primary disease mediators of neuroinflammation. Subsequent studies in MS and EAE indicated that this view is most likely overly simplistic, and that additional pathogenic T cell subsets with different cytokine profiles as well as “innate” cytokines (IL-1, IL-6, TNF-α, type I IFNs, etc.) secreted by innate immune cells and stroma might have important effects within inflamed CNS tissue.
In addition to the aforementioned studies, a great deal of research in preclinical disease models and patients has highlighted deregulated cytokine signals as a primary instigator of pathogenesis in EAE and MS. Therefore, multiple attempts have been made to treat MS patients with recombinant anti-inflammatory cytokines, or inhibitors of proinflammatory cytokines. It became clear very early that cytokine networks are more complex than anticipated and that individual cytokines may have diverse and even opposing functions in different clinical scenarios. With regard to MS, despite the best efforts there is only one approved cytokine therapy to date, which is IFN-β. Here we not only focus on drugs in development but also briefly discuss several treatment failures which one must better understand mechanistically before successfully and safely applying cytokine-directed therapies to MS patients in daily practice.
Approved cytokine therapies for MS: IFN-β
Type I IFNs were first characterised in 1957, and named for their first observed biological effect: prevention of, or “interference” during, viral infection [14]. Since then, it has been shown that the effects of type I IFNs are much more pleiotropic, and they are now appreciated for their roles not just as anti-viral agents, but also as immune modulators and cell growth regulators [15]. The connection between type I IFNs and MS, as well as other autoimmune disorders, is well known. Indeed, IFN-β has been extensively investigated in EAE and MS and is the focus of numerous studies found in the literature.
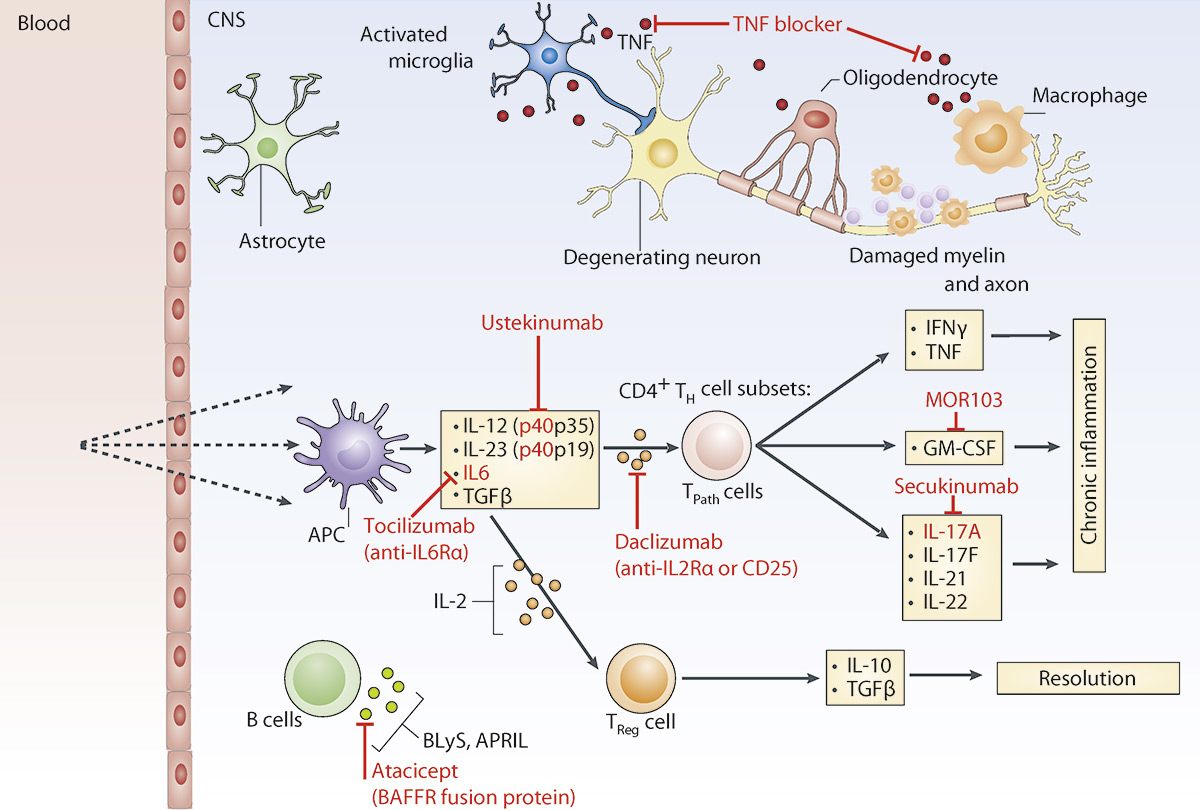
Figure 1
Key cytokines in the pathogenesis of multiple sclerosis.
In patients with MS and in its experimental models EAE, various immune cells have been shown to produce and react to pro- and anti-inflammatory cytokines in the inflamed CNS. The main cytokines secreted by antigen-presenting cells, pathogenic CD4+ Th cell subsets and Tregsare shown. Potential cytokine targets for therapy and drugs are indicated in red. TNFα is expressed by T cells but also by microglia and macrophages. Its blocking has failed in clinical MS trials possibly because it has also homeostatic effects in the CNS and is important for remyelination. Other cytokine inhibitors like ustekinumab, which targets the p40 subunit of IL‑12 and IL‑23, involved in the generation of pathogenic CD4+ Th cell subsets has recently been tested in clinical trials and was unexpectedly ineffective to reduce clinical relapses in MS patients. Studies with neutralizing anti-GM-CSF (MOR103), anti-IL17 (secukinumab) or anti-IL2Rα antibodies (daclizumab) are ongoing and show promising first result in the treatment of MS patients. Blocking of maturation factors of B cells (BLys, APRIL, or in the case of neuromyelitis optica IL-6Rα by tocilizumab) might be another therapeutic approach worth following up.
APC = antigen-presenting cell; BAFF = B cell activation factor; CNS = central nervous system; EAE = experimental autoimmune encephalomyelitis; GM-CSF = granulocyte macrophage colony stimulating factor; IFN = interferon; IL = interleukin; MS = multiple sclerosis; TPath = pathogenic CD4+ Th cell subsets; Th = T helper cell; TNF-α = tumour necrosis factor.α; Treg = regulatory T cell.
IFN-β-deficient mice are more susceptible to EAE [16] and type I IFNs can attenuate EAE in mice [17], suggesting that IFN-β inhibits immune-mediated demyelination in this MS model. During EAE, elevated IFN-β concentrations can be measured in the CNS but not blood [104], and intrathecal application of the synthetic double-stranded RNA analogue poly I:C can stimulate cells in the CNS to produce IFN-β [105]. To trace the cell type that mediates the “protective” immune regulation of type I IFNs, mice lacking the type I receptor (IFNAR) only in distinct cell populations were used for EAE experiments [18]. This study revealed that negative regulation of EAE disease severity by type I IFNs relies on the presence of IFNAR on myeloid cells (including monocytes, macrophages and microglia). Prinz and colleagues concluded that locally produced IFN-β within the CNS acts on invading myeloid cells to attenuate autoimmune damage in the effector phase of EAE, and the IFNAR pathway seems to be tightly regulated in these cells [106]. The therapeutic potential of CNS-restricted type I IFN induction in MS is mainly unexplored. In fact, IFN-β drugs are currently administered subcutaneously or intramuscularly by MS patients and thereby probably act largely in the periphery.
By this route, human recombinant IFN-β is mildly effective in reducing the relapse rate in about half of patients with MS, reducing relapse rates by about one-third [19]. Multiple molecular mechanisms by which IFN-β exerts its immunomodulatory functions in MS patients have been proposed. They include inhibition of Th1 cell development [20], deviation to Th2 cell responses [21], induction of IL-10 [22], restoration of the disrupted blood-brain barrier [23] and suppression of IFN-γ-induced class II major histocompatibility complex (MHC) molecules on CNS cells [24]. It is still not entirely clear which of these effects really matter, but a combination of them likely contributes to the clinical success of IFN-β administration.
Treatment failures
Administration of inhibitory cytokines: TGF-β and IL-10
One method of cytokine-directed therapy is the application of recombinant anti-inflammatory cytokines. However, with the exception of IFN-β this approach was disappointing in the treatment of MS patients for different reasons up to now. For example, systemic administration of transforming growth factor (TGF)-β ameliorates EAE [25, 26] and this very potent immunosuppressive cytokine is elevated in the brains of individuals with MS [27]. However, renal toxic effects were encountered in a small pilot human trial to test the efficacy of administering TGF-β, and consequently it has never been approved as a treatment for MS [28]. Another phase II clinical trial testing IL-10, which is an important suppressive cytokine as well, as a treatment in MS patients was stopped due to lack of efficacy [29].
TNF-α blockers: what protects inflamed joints can be harmful for the brain
TNF-α is one of the most widespread clinically targeted cytokines. Its inhibition has also been considered as a therapy for MS. TNF-α is produced by several cell types including T cells, macrophages and microglia, and its receptors TNF receptor 1 and 2 are ubiquitously expressed. TNF-α is important in the control and containment of local infections, but deregulated TNF-α production can be harmful in the setting of sepsis and several other inflammatory and autoimmune diseases. In (chronic progressive) MS patients, intrathecal TNF-α synthesis has been demonstrated, and TNF-α levels in the CSF have been correlated with the severity and progression of disease [30, 31]. In the mouse model of MS, TNF-α blockade by antibodies or soluble TNF receptors prevented or reversed disease [32, 33].
Surprisingly, however, drugs that block TNF-α, which are beneficial in a number of diseases (including rheumatoid arthritis, ankylosing spondylitis, psoriasis, and inflammatory bowel disease) can cause the occurrence of prolonged and more relapses in patients with MS [34, 35]. The reasons for this worsening are still not fully understood. One explanation might be that TNF-α may have protective properties in the CNS and that TNF signalling is also important for remyelination [36, 37]. The failure of TNF-α inhibition to reduce clinical MS emphasises the fact that the mechanism of tissue damage in disease-affected joints in individuals with rheumatoid arthritis differs fundamentally from that in the CNS of MS patients. It also teaches us that cytokine functions can be complex and the sole presence of a proinflammatory cytokine in the CSF of MS patients does not necessarily mean that its inhibition results in a successful treatment strategy. Furthermore, it illustrates that studies using current EAE models have to be interpreted carefully and can have limitations with regard to predicting success in clinical trials [38].
It is of concern that new-onset MS-like demyelinating lesions as well as worsening of established MS following exposure to anti-TNF-α agents for rheumatoid arthritis or Crohn's disease have been reported [39, 40]. Moreover, TNF-α blockers have been claimed to give rise to various forms of demyelinating neuropathies [41]. Even though these observations might in part be explained by shared genetic susceptibility to autoimmune disorders including MS, anti-TNF therapy might trigger and bring out inflammatory demyelination disease in patients.
Atacicept: targeting B cell cytokines
Immunotherapies depleting B cell populations have been found to slow disease progression in MS (anti-CD20: rituximab and ocrelizumab) [42, 43]. However, targeting the cytokines BlyS (B-lymphocyte stimulator) and APRIL (a proliferation-inducing ligand), which are involved in B-cell maturation, function and survival, has failed in MS patients. This can be concluded from a phase II safety and efficacy trial of the BAFF (B cell activation factor) receptor fusion protein atacicept in patients with MS which had to be halted owing to an increase in annualised relapse rates [44]. The reasons for this disappointing result are unknown. It is clear that atacicept targets B cells in different developmental stages compared with other B cell-directed therapies like rituximab [45], but we have yet to learn why rituximab halts disease progression whereas atacicept drives relapses. As the role of B cells in MS pathogenesis remains largely enigmatic, we have a long way to go to make sense of these findings.
Interleukin-12/interleukin-23: hitting two birds with one stone
IL-12 and IL-23 are of particular interest in MS, because they induce the differentiation of naïve CD4+ T cells into proinflammatory and, presumably, pathogenic T helper cell populations. IL-12 and IL-23 are both produced mainly by activated myeloid cells and bind to receptors that are expressed on lymphocytes. IL-12 is crucial for the differentiation along a Th1 cell lineage, and IL-23 promotes and stabilises IL-17 production by CD4+ T cell (Th17 cells) [46]. IL-12 and IL-23 are heterodimers each composed of a common subunit (p40), with either p35 (IL-12) or p19 (IL-23) as a unique chain [47]. The common p40 subunit has been detected in MS lesions and levels in peripheral blood mononuclear cells in MS patients have been correlated with disease activity [48, 49]. The neutralisation of p40 inhibits EAE in rodents and nonhuman primates [50]. Therefore there is considerable evidence that blocking the p40 subunit would be beneficial to patients with MS.
Contrary to this expectation, neutralisation of p40 did not inhibit disease activity in patients with MS. Two hundred and forty-nine patients with relapsing-remitting MS were treated with four different dosing regimens of ustekinumab (anti-p40) or placebo for 19 weeks. The primary endpoint, the cumulative number of new contrast-enhancing cranial MR lesions, was not different between placebo and the ustekinumab treatment arms [51]. It is not entirely clear mechanistically why the results of the ustekinumab trial in MS were negative. They are in conflict with the outcome of studies of p40 blockade in other autoimmune diseases. For example, ustekinumab effectively improved disease severity in patients with psoriasis and psoriatic arthritis [52, 53]. It also showed benefits in the treatment of moderate-to-severe Crohn’s disease that was resistant to TNF antagonists [54]. One explanation for its failure in MS might be that IL-23 is maybe only important in the generation of pathogenic T cells prior to disease onset or early during the disease course, and therefore ustekinumab was given too late. It is also unclear if ustekinumab needs to act locally at the site of CNS inflammation in MS patients and, if this is the case, whether the drug penetrates into the CNS sufficiently [55, 56].
In clinical development
Interleukin-17 inhibition
The rationale behind blockade of IL-23 stems from the assumption that IL-23 is an important differentiation and maintenance factor for Th17 cells and controls the production of presumably pathogenic IL-17. IL-17 is a pleiotropic cytokine that is the hallmark secreted protein of so-called Th17 cells. IL-17A and IL-17F are two IL-17 family members that are secreted by Th17 cells and share the same IL-17 receptors that are broadly expressed by stromal cells. In a deregulated autoimmune setting, IL-17 could recruit neutrophils and macrophages and cause release of mediators of local tissue destruction. Indeed, IL-17 has been linked to several autoimmune diseases including psoriasis, inflammatory bowel disease, rheumatoid arthritis and MS [57].
In EAE, the contribution of IL-17 itself as well as of IL-22, another cytokine produced by Th17 cells, to disease development is controversial, as T cell-specific IL-17A overexpression and IL-17A, IL-17F or IL-22 inhibition had only a minor impact [58, 59]. The results of the EAE studies raise at least some scepticism about the biological importance of IL-17 alone in autoimmune CNS inflammation. Nevertheless, in human MS, accumulation and up-regulation of Th17 signature cytokines such as IL-17 in the CNS have been reported [27, 60] and phase II clinical trials testing a humanised antibody that targets IL-17A (secukinumab or AIN457) are ongoing or have been completed [61] (clinicaltrials.gov identifier: NCT01433250 and NCT01874340). The results of the larger of these phase II trials including 380 patients and measuring the number of new gadolinium T1 lesions in magnetic resonance imaging (MRI) as a primary endpoint have not been published yet. Antibodies that specifically neutralise IL-17 showed extremely good therapeutic efficacy for the treatment of psoriasis [62], but have failed to ameliorate Crohn’s disease [63], maybe because basal levels of IL-17 might be gut protective. This again emphasises that the different disease setting matters, and it will be interesting to learn about the final outcome of secukinumab treatments in MS patients.
Humanised monoclonal antibodies against the IL-6 receptor and neuromyelitis optica
IL-6 is produced by various haematopoietic and nonhaematopoietic cells in response to tissue damage and infections. It is a central mediator of the immune system, inducing the acute phase response in the liver, fever and optimal T and B cell effector responses to fight off pathogens. In particular, it is an important factor that induces the final differentiation of B cells into antibody-secreting plasma cells [64]. Given the known functions of IL-6, targeting the bioactivity of this cytokine is a rational therapeutic approach for the treatment of autoimmune diseases. An important consideration for the design of IL-6 modulating therapies is that the IL-6Rα chain occurs in a membrane-bound and soluble form, and requires an accessory molecule glycoprotein 130 (gp130) for signal transduction. Tocilizumab is a humanised antibody that is directed against the IL-6Rα chains and allows for the targeting of both membrane-bound forms, expressed on immune cells and hepatocytes, and soluble forms of the receptor. Tocilizumab has efficacy in the therapy of moderate-to-severe rheumatoid arthritis [65, 66] and juvenile idiopathic arthritis [67], and is approved in Switzerland for these indications.
Neuromyelitis optica (NMO) is an inflammatory disease affecting the optic nerve and spinal cord, in which autoantibodies against aquaporin 4 water channel protein located on astrocyte processes probably are pathogenic [68]. As it is pathogenetically different from MS, some MS treatments, such as IFN-β, natalizumab and oral fingolimod, can even exacerbate NMO [69]. For example, IFN-β was tried as a therapeutic agent but it soon became apparent that IFN-β treatment can worsen NMO and induce severe relapses [70–73].
Recent studies have suggested that IL-6 could play a role in NMO pathogenesis by contributing to the persistence of anti-aquaporin-4 antibody-producing plasmablasts in patients with NMO [74]. Furthermore, increased IL-6 levels have been measured in serum and CSF of patients with NMO compared with those with MS [75, 76]. The relative infrequency of NMO, in contrast to MS, has impeded large clinical treatment trials for this disease. However, several cases and small patient series have been reported, which support the concept that IL-6-targeted therapy with tocilizumab could have a favourable effect in NMO patients who have failed to respond to other therapies [77–79]. For example, Araki and colleagues treated 7 patients with anti-aquaporin-4 positive NMO with tocilizumab for 1 year who had previously been treated with oral prednisolone and immunosuppressants such as azathioprine. The authors reported that the annualised relapse rate decreased in these patients from 2.9 to 0.4 [80].
GM-CSF: detrimental in autoimmune tissue inflammation, needed to keep the lungs clean
GM-CSF (granulocyte-macrophage colony-stimulating factor) is another cytokine that has been associated with the progression of inflammatory diseases such as rheumatoid arthritis and MS. GM-CSF receptors are heterodimers consisting of a ligand-specific α-subunit (GM-CSFR-α) and a β-chain subunit that is shared with other cytokine receptors and is responsible for cell signalling. GM-CSF was first characterised by its ability to cause the differentiation of myeloid cells into granulocytic, macrophage and dendritic cell colonies in vitro. Clinicians take advantage of this effect and use it in patients to increase myeloid cell expansion and differentiation after chemotherapy-induced myelosuppression. Exacerbations of established rheumatoid arthritis have been reported in patients who received GM-CSF as supportive therapy [81, 82], which already points to an involvement of GM-CSF in autoimmune joint inflammation.
GM-CSF deficient mice are protected from myocarditis [83], experimental models of arthritis [84] and EAE [85, 86]. In line with these studies, experiments in mice have shown that neutralising antibodies against GM-CSF ameliorate EAE [87, 88]. Activated Th cells, the main source of GM-CSF during inflammation, do not themselves respond to GM-CSF. They therefore must communicate with GM-CSF-sensitive cell types to mediate pathology in EAE. Tissue-invading monocytes, which give rise to inflammatory antigen-presenting cells, are responsible for CNS tissue damage [89]. Additionally, these cells are highly GM-CSF responsive and represent a likely candidate for the GM-CSF-responder cell mediating demyelination during experimental autoimmunity [107].
In MS patients, elevated levels of GM-CSF in the CSF as compared to controls have been reported [90, 91], and Th cells from the CSF of MS patients have higher expression levels of GM-CSF ex vivo compared with control patients [92]. Furthermore, an association of GM-CSF expression by human Th cells with MS disease severity has been proposed [93], making this cytokine a promising therapeutic target in MS. A completed randomised, double blind, placebo-controlled phase Ib study including 31 patients with either relapsing-remitting or secondary progressive MS evaluated the safety of MOR103, a recombinant human IgG1 antibody against GM-CSF (clinicaltrials.gov identifier: NCT01517282). Data on the efficacy of MOR103 in MS are not available up to now. However, the same antibody was reported to be well tolerated and to show preliminary evidence of efficacy in patients with active rheumatoid arthritis (phase Ia/IIb) [94] and GM-CSF receptor-α blockade with mavrilimumab reduced disease activity in patients with rheumatoid arthritis (phase I and II) [95, 96]. Of note, neutralising anti-GM-CSF antibodies can impair alveolar macrophage function and are found in the majority of patients with acquired pulmonary alveolar proteinosis, a rare lung disease [97]. Burmester and colleagues did not observe changes in pulmonary parameters, but this needs to be closely monitored over longer time periods during clinical trials.
IL-2 receptor α-chain (CD25) blockade: daclizumab
IL-2 is a T cell growth factor that has long been known as an important survival and proliferation signal for T cells [3]. Unexpectedly, mice deficient in IL-2 or components of the IL-2 receptor succumb to an aggressive, lymphoproliferative autoimmune syndrome [98]. Later studies revealed that IL-2 is also a critical factor for CD4+ regulatory T cells (Foxp3+ Tregs) to be able to terminate overshooting autoreactive T cell responses [99, 100]. Daclizumab, a humanised monoclonal anti-IL-2Rα (also called CD25) antibody has been tested in several clinical trials in MS patients based on the rationale that blocking the high-affinity IL-2 receptor would prevent the expansion of autoreactive T lymphocytes [101]. Results of mechanistic studies showed that daclizumab acts at least in part by expanding a regulatory (CD56bright NK) immune cell population [102].
Previous Phase I/II clinical trials have demonstrated consistent and significant beneficial effects of daclizumab on MRI and clinical disease measures of MS. Daclizumab seems to be generally well tolerated. However, serious adverse events have been reported and include infections, skin reactions, abnormal liver function tests and autoimmune conditions (affecting liver and kidney). The DECIDE phase III trial (clinicaltrials.gov identifier: NCT01064401) compared daclizumab, which is injected subcutaneously once a month, and IFNβ-1a in patients with relapsing-remitting MS [103]. It was reported that daclizumab reduced the annualised relapse rate by 45% compared with IFNβ-1a. In addition, 54% fewer new or enlarging CNS lesions were detected by MRI after nearly two years of daclizumab treatment, compared with those patients injecting IFNβ-1a. Applications for the approval of daclizumab (high-yield process, DAC HYP) for the treatment of patients with MS are currently under review by agencies both in Europe and the USA.
Conclusions and future perspectives
Cytokines have a major role in driving CNS inflammation in MS patients. Therefore, blockade of proinflammatory cytokines remains an attractive avenue for MS therapy. In contrast to rheumatoid arthritis and inflammatory bowel disease, where TNF-α is currently one of the key targets for treatment, TNF-α inhibitors worsen MS. This may reflect that cytokine networks likely differ between autoimmune inflammatory diseases and tissue-location matters. The CNS appears to be unique in its vulnerability to an autoimmune milieu, where proinflammatory cytokines and immune cells are let loose. Furthermore, it has been proposed that cytokines are also involved in homeostatic brain functions like re-/myelination, which poses a challenge for cytokine-inhibiting therapies. Some blocking monoclonal antibodies that are directed against cytokine/receptors within the brain and spinal cord might not penetrate sufficiently, and approaches for the optimised delivery to the inflammatory CNS foci need to be further explored. It is still puzzling that one specific cytokine-directed therapy may be detrimental rather than protective even if the same target organ, the CNS, is affected. This is illustrated by the clinical exacerbations that have been observed after the use of IFN-β in NMO patients, probably because a different immune pathomechanism predominates compared with MS. Indeed, a characteristic feature of MS is its clinical and immunopathological heterogeneity and the existence of multiple phases of disease, during the course of which the cytokine production pattern and requirements for a sustained autoimmune tissue inflammation may change. Another important question that remains is the safety of long-term treatments, that could be complicated by re-activations of potentially harmful latent infections or impaired lung function for example in the case of GM-CSF inhibition. Lessons from clinical trials testing cytokine-directed therapies in various autoimmune diseases including MS teach us that all these factors need to be considered. Despite these limitations, the development of anti-cytokine therapies still holds promise also for CNS inflammatory diseases, and their use will certainly improve our understanding of their pathogenesis.
Acknowledgments: We thank Andrew Croxford and Florian Mair for discussions and critical comments on this manuscript.
References
1 Vilcek J, Feldmann M. Historical review: Cytokines as therapeutics and targets of therapeutics. Trends Pharmacol Sci. 2004;25:201–9.
2 Oppenheim JJ. Cytokines: past, present, and future. Int J Hematol. 2001;74:3–8.
3 Gordon J, MacLean LD. A lymphocyte-stimulating factor produced in vitro. Nature. 1965;208:795–6.
4 Carswell EA, et al. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U S A. 1975;72:3666–70.
5 Taniguchi T, Fujii-Kuriyama Y, Muramatsu M. Molecular cloning of human interferon cDNA. Proc Natl Acad Sci U S A. 1980;77:4003–6.
6 Nagata S, et al. Synthesis in E. coli of a polypeptide with human leukocyte interferon activity. Nature. 1980;284:316–20.
7 Goeddel DV, et al. Synthesis of human fibroblast interferon by E. coli. Nucleic Acids Res. 1980;8:4057–74.
8 Lutterotti A, Martin R. Getting specific: monoclonal antibodies in multiple sclerosis. Lancet Neurol. 2008;7:538–47.
9 International Multiple Sclerosis Genetics, C., et al. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med. 2007;357:851–62.
10 Gregory SG, et al. Interleukin 7 receptor alpha chain (IL7R) shows allelic and functional association with multiple sclerosis. Nat Genet. 2007;39:1083–91.
11 Lundmark F, et al. Variation in interleukin 7 receptor alpha chain (IL7R) influences risk of multiple sclerosis. Nat Genet. 2007;39:1108–13.
12 International Multiple Sclerosis Genetics, C., et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–9.
13 Panitch HS, Hirsch RL, Schindler J, Johnson KP. Treatment of multiple sclerosis with gamma interferon: exacerbations associated with activation of the immune system. Neurology. 1987;37:1097–102.
14 Isaacs A, Lindenmann J, Valentine RC. Virus interference. II. Some properties of interferon. Proc R Soc Lond B Biol Sci. 1957;147:268–73.
15 Gonzalez-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol. 2012;12:125–35.
16 Teige I, et al. IFN-beta gene deletion leads to augmented and chronic demyelinating experimental autoimmune encephalomyelitis. J Immunol. 2003;170:4776–84.
17 Yu M., Nishiyama A, Trapp BD, Tuohy VK. Interferon-beta inhibits progression of relapsing-remitting experimental autoimmune encephalomyelitis. J Neuroimmunol. 1996;64:91–100.
18 Prinz M, et al. Distinct and nonredundant in vivo functions of IFNAR on myeloid cells limit autoimmunity in the central nervous system. Immunity. 2008;28:675–86.
19 Paty DW, Li DK. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. II. MRI analysis results of a multicenter, randomized, double-blind, placebo-controlled trial. UBC MS/MRI Study Group and the IFNB Multiple Sclerosis Study Group. Neurology. 1993;43:662–7.
20 McRae BL, Semnani RT, Hayes MP, van Seventer GA. Type I IFNs inhibit human dendritic cell IL-12 production and Th1 cell development. J Immunol. 1998;160:4298–304.
21 Kozovska ME, et al. Interferon beta induces T-helper 2 immune deviation in MS. Neurology. 1999;53:1692–7.
22 Rudick RA, et al. Interferon beta induces interleukin-10 expression: relevance to multiple sclerosis. Ann Neurol. 1996;40:618–27.
23 Stone LA, et al. The effect of interferon-beta on blood-brain barrier disruptions demonstrated by contrast-enhanced magnetic resonance imaging in relapsing-remitting multiple sclerosis. Ann Neurol. 1995:37:611–9.
24 Satoh J, Paty DW, Kim SU. Differential effects of beta and gamma interferons on expression of major histocompatibility complex antigens and intercellular adhesion molecule-1 in cultured fetal human astrocytes. Neurology. 1995;45:367–73.
25 Racke MK, et al. Prevention and treatment of chronic relapsing experimental allergic encephalomyelitis by transforming growth factor-beta 1. J Immunol. 1991;146:3012–7.
26 Racke MK, et al. Long-term treatment of chronic relapsing experimental allergic encephalomyelitis by transforming growth factor-beta 2. J Neuroimmunol. 1993;46:175–83.
27 Lock C, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002;8:500–8.
28 Calabresi PA, et al. Phase 1 trial of transforming growth factor beta 2 in chronic progressive MS. Neurology. 1998;51:289–92.
29 Wiendl H, Neuhaus O, Kappos L, Hohlfeld R. Multiple sclerosis. Current review of failed and discontinued clinical trials of drug treatment. Nervenarzt. 2000;71:597–610.
30 Sharief MK, Hentges R. Association between tumor necrosis factor-alpha and disease progression in patients with multiple sclerosis. N Engl J Med. 1991;325:467–72.
31 Maimone D, Gregory S, Arnason BG, Reder AT. Cytokine levels in the cerebrospinal fluid and serum of patients with multiple sclerosis. J Neuroimmunol. 1991;32:67–74.
32 Ruddle NH, et al. An antibody to lymphotoxin and tumor necrosis factor prevents transfer of experimental allergic encephalomyelitis. J Exp Med. 1990;172:1193–200.
33 Selmaj K, Raine CS, Cross AH. Anti-tumor necrosis factor therapy abrogates autoimmune demyelination. Ann Neurol. 1991;30:694–700.
34 van Oosten BW, et al. Increased MRI activity and immune activation in two multiple sclerosis patients treated with the monoclonal anti-tumor necrosis factor antibody cA2. Neurology. 1996;47:1531–4.
35 TNF neutralization in MS: results of a randomized, placebo-controlled multicenter study. The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. Neurology. 1999;53:457–65.
36 Liu J, et al. TNF is a potent anti-inflammatory cytokine in autoimmune-mediated demyelination. Nat Med. 1998;4:78–83.
37 Arnett HA, et al. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci. 2001;4:1116–22.
38 Friese MA, et al. The value of animal models for drug development in multiple sclerosis. Brain. 2006;129:1940–52.
39 Hyrich KL, Silman AJ, Watson KD, Symmons DP. Anti-tumour necrosis factor alpha therapy in rheumatoid arthritis: an update on safety. Ann Rheum Dis. 2004;63:1538–43.
40 Kaltsonoudis E, et al. Neurological adverse events in patients receiving anti-TNF therapy: a prospective imaging and electrophysiological study. Arthritis Res Ther. 2014;16:R125.
41 Lozeron P, Denier C, Lacroix C, Adams D. Long-term course of demyelinating neuropathies occurring during tumor necrosis factor-alpha-blocker therapy. Arch Neurol. 2009;66:490–7.
42 Hauser SL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358:676–88.
43 Kappos L, et al. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet. 2011;378:1779–87.
44 Kappos L, et al. Atacicept in multiple sclerosis (ATAMS): a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Neurol. 2014;13:353–63.
45 Bible E. Multiple sclerosis: Atacicept increases relapse rates in multiple sclerosis. Nat Rev Neurol. 2014;10:182.
46 Hunter CA. New IL-12-family members: IL-23 and IL-27, cytokines with divergent functions. Nat Rev Immunol. 2005;5:521–31.
47 Kikly K, Liu L, Na S, Sedgwick JD. The IL-23/Th(17) axis: therapeutic targets for autoimmune inflammation. Curr Opin Immunol. 2006;18:670–5.
48 Windhagen A, et al. Expression of costimulatory molecules B7-1 (CD80), B7-2 (CD86), and interleukin 12 cytokine in multiple sclerosis lesions. J Exp Med. 1995;182:1985–96.
49 van Boxel-Dezaire AH, et al. Decreased interleukin-10 and increased interleukin-12p40 mRNA are associated with disease activity and characterize different disease stages in multiple sclerosis. Ann Neurol. 1999;45:695–703.
50 t Hart BA, et al. Suppression of ongoing disease in a nonhuman primate model of multiple sclerosis by a human-anti-human IL-12p40 antibody. J Immunol. 2005;175:4761–8.
51 Segal BM, et al. Repeated subcutaneous injections of IL12/23 p40 neutralising antibody, ustekinumab, in patients with relapsing-remitting multiple sclerosis: a phase II, double-blind, placebo-controlled, randomised, dose-ranging study. Lancet Neurol. 2008;7:796–804.
52 Leonardi CL, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet. 2008;371:1665–74.
53 McInnes IB, et al. Efficacy and safety of ustekinumab in patients with active psoriatic arthritis: 1 year results of the phase 3, multicentre, double-blind, placebo-controlled PSUMMIT 1 trial. Lancet. 2013;382:780–9.
54 Sandborn WJ, et al. Ustekinumab induction and maintenance therapy in refractory Crohn’s disease. N Engl J Med. 2012;367:1519–28.
55 Martin R. Neutralisation of IL12 p40 or IL23 p40 does not block inflammation in multiple sclerosis. Lancet Neurol. 2008;7:765–6.
56 Longbrake EE, Racke MK. Why did IL-12/IL-23 antibody therapy fail in multiple sclerosis? Expert Rev Neurother. 2009;9:319–21.
57 Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. 2014;14:585–600.
58 Haak S, et al. IL-17A and IL-17F do not contribute vitally to autoimmune neuro-inflammation in mice. J Clin Invest. 2009;119:61–9.
59 Kreymborg K, et al. IL-22 is expressed by Th17 cells in an IL-23-dependent fashion, but not required for the development of autoimmune encephalomyelitis. J Immunol. 2007;179:8098–104.
60 Tzartos JS, et al. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. 2008;172:146–55.
61 Havrdova E, et al. Sensitivity analysis of a phase IIa study of secukinumab in relapsing remitting multiple sclerosis. Mult Scler. 2013;19:210–1.
62 Chiricozzi A, Krueger JG. IL-17 targeted therapies for psoriasis. Expert Opin Investig Drugs. 2013;22:993–1005.
63 Hueber W, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. 2012;61:1693–700.
64 Kishimoto T. Interleukin-6: from basic science to medicine – 40 years in immunology. Annu Rev Immunol. 2005;23:1–21.
65 Smolen JS, et al. Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double-blind, placebo-controlled, randomised trial. Lancet. 2008;371:987–97.
66 Gabay C, et al. Tocilizumab monotherapy versus adalimumab monotherapy for treatment of rheumatoid arthritis (ADACTA): a randomised, double-blind, controlled phase 4 trial. Lancet. 2013;381:1541–50.
67 De Benedetti F, et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367:2385–95.
68 Lennon VA, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364:2106–12.
69 Papadopoulos MC, Bennett JL, Verkman AS. Treatment of neuromyelitis optica: state-of-the-art and emerging therapies. Nat Rev Neurol. 2014;10:493–506.
70 Warabi Y, Matsumoto Y, Hayashi H. Interferon beta-1b exacerbates multiple sclerosis with severe optic nerve and spinal cord demyelination. J Neurol Sci. 2007;252:57–61.
71 Palace J, Leite MI, Nairne A, Vincent A. Interferon Beta treatment in neuromyelitis optica: increase in relapses and aquaporin 4 antibody titers. Arch Neurol. 2010;67:1016–7.
72 Shimizu J, et al. IFNbeta-1b may severely exacerbate Japanese optic-spinal MS in neuromyelitis optica spectrum. Neurology. 2010;75:1423–7.
73 Uzawa A, Mori M, Hayakawa S, Masuda S, Kuwabara S. Different responses to interferon beta-1b treatment in patients with neuromyelitis optica and multiple sclerosis. Eur J Neurol. 2010;17:672–6.
74 Chihara N, et al. Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc Natl Acad Sci U S A. 2011;108:3701–6.
75 Uzawa A, et al. Markedly increased CSF interleukin-6 levels in neuromyelitis optica, but not in multiple sclerosis. J Neurol. 2009;256:2082–4.
76 Icoz S, et al. Enhanced IL-6 production in aquaporin-4 antibody positive neuromyelitis optica patients. Int J Neurosci. 2010;120:71–5.
77 Araki M, et al. Clinical improvement in a patient with neuromyelitis optica following therapy with the anti-IL-6 receptor monoclonal antibody tocilizumab. Mod Rheumatol. 2013;23:827–31.
78 Kieseier BC, et al. Disease amelioration with tocilizumab in a treatment-resistant patient with neuromyelitis optica: implication for cellular immune responses. JAMA Neurol. 2013;70:390–3.
79 Ayzenberg I, et al. Interleukin 6 receptor blockade in patients with neuromyelitis optica nonresponsive to anti-CD20 therapy. JAMA Neurol. 2013;70:394–7.
80 Araki M, et al. Efficacy of the anti-IL-6 receptor antibody tocilizumab in neuromyelitis optica: a pilot study. Neurology. 2014;82:1302–6.
81 de Vries EG, et al. Flare-up of rheumatoid arthritis during GM-CSF treatment after chemotherapy. Lancet. 1991;338:517–8.
82 Hazenberg BP, Van Leeuwen MA, Van Rijswijk MH, Stern AC, Vellenga E. Correction of granulocytopenia in Felty’s syndrome by granulocyte-macrophage colony-stimulating factor. Simultaneous induction of interleukin-6 release and flare-up of the arthritis. Blood. 1989;74:2769–70.
83 Sonderegger I, et al. GM-CSF mediates autoimmunity by enhancing IL-6-dependent Th17 cell development and survival. J Exp Med. 2008;205,:2281–94.
84 Campbell IK, et al. Protection from collagen-induced arthritis in granulocyte-macrophage colony-stimulating factor-deficient mice. J Immunol. 1998;161:3639–44.
85 McQualter JL, et al. Granulocyte macrophage colony-stimulating factor: a new putative therapeutic target in multiple sclerosis. J Exp Med. 2001;194:873–82.
86 Ponomarev ED, et al. GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J Immunol. 2007;178:39–48.
87 Codarri L, et al. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12:560–7.
88 El-Behi M, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12:568–75.
89 Yamasaki R, et al. Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med. 2014;211:1533–49.
90 Perrella O, Carrieri PB, De Mercato R, Buscaino GA. Markers of activated T lymphocytes and T cell receptor gamma/delta+ in patients with multiple sclerosis. Eur Neurol. 1993;33:152–5.
91 Carrieri PB, et al. Profile of cerebrospinal fluid and serum cytokines in patients with relapsing-remitting multiple sclerosis: a correlation with clinical activity. Immunopharmacol Immunotoxicol. 1998;20:373–82.
92 Noster R, et al. IL-17 and GM-CSF expression are antagonistically regulated by human T helper cells. Sci Transl Med. 2014;6:241ra280.
93 Hartmann FJ, et al. Multiple sclerosis-associated IL2RA polymorphism controls GM-CSF production in human TH cells. Nat Commun. 2014;5:5056.
94 Behrens F, et al. MOR103, a human monoclonal antibody to granulocyte-macrophage colony-stimulating factor, in the treatment of patients with moderate rheumatoid arthritis: results of a phase Ib/IIa randomised, double-blind, placebo-controlled, dose-escalation trial. Ann Rheum Dis. 2015;74:1058–64.
95 Burmester GR, et al. Mavrilimumab, a human monoclonal antibody targeting GM-CSF receptor-alpha, in subjects with rheumatoid arthritis: a randomised, double-blind, placebo-controlled, phase I, first-in-human study. Ann Rheum Dis. 2011;70:1542–9.
96 Burmester GR, et al. Efficacy and safety of mavrilimumab in subjects with rheumatoid arthritis. Ann Rheum Dis. 2013;72:1445–52.
97 Miller AL, Schissel S, Levy BD, Loscalzo J. Clinical problem-solving. A crazy cause of dyspnea. N Engl J Med. 2011;364:72–7.
98 Suzuki H, et al. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor beta. Science. 1995;268:1472–6.
99 Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005;6:1142–51.
100 D’Cruz LM, Klein L. Development and function of agonist-induced CD25+Foxp3+ regulatory T cells in the absence of interleukin 2 signaling. Nat Immunol 2005;6:1152–9.
101 Pfender N, Martin R. Daclizumab (anti-CD25) in multiple sclerosis. Exp Neurol. 2014;262 Pt A:44–51.
102 Bielekova B, et al. Regulatory CD56(bright) natural killer cells mediate immunomodulatory effects of IL-2Ralpha-targeted therapy (daclizumab) in multiple sclerosis. Proc Natl Acad Sci U S A. 2006;103:5941–6.
103 Arnold DL, et al. Brain MRI results of DECIDE: a randomized, double-blind trial of DAC HYP vs. IFNβ-1a in RRMS patients. 2014 Joint ACTRIMS-ECTRIMS Meeting, Mult Scler. 2014;20:67–284.
104 Prinz M, Schmidt H, Mildner A, Knobeloch KP, Hanisch UK, Raasch J, et al. Distinct and nonredundant in vivo functions of IFNAR on myeloid cells limit autoimmunity in the central nervous system. Immunity. 2008 May;28(5):675‒86.
105 Khorooshi R, Mørch MT, Holm TH, Berg CT, Dieu RT, Dræby D, et al. Induction of endogenous Type I interferon within the central nervous system plays a protective role in experimental autoimmune encephalomyelitis. Acta Neuropathol. 2015 Jul;130(1):107‒18.
106 Goldmann T, Zeller N, Raasch J, Kierdorf K, Frenzel K, Ketscher L, et al. USP18 lack in microglia causes destructive interferonopathy of the mouse brain. EMBO J. 2015 Jun 12;34(12):1612‒29.
107 Croxford AL, Lanzinger M, Hartmann FJ, Schreiner B, Mair F, et al. The cytokine GM-CSF drives the inflammatory signature of CCR2(+) monocytes and licenses autoimmunity. Immunity. 2015;43(3):502-14.