
Figure 1
The multiple sclerosis plaque, classical portrayals.
A. Plaques embedded in brain white matter (Charcot 1880); B. perivascular and parenchymal inflammatory infiltrates (Babinski, 1885). (from H. Lassmann, Brain Pathol. 2005)
DOI: https://doi.org/10.4414/smw.2015.14189
The number of multiple sclerosis (MS) patients in Switzerland amounts to an estimated 10000. While this is much less than a classic “Volkskrankheit” like tumour or cardiovascular disease, MS is still considered one of the pressing problems of contemporary medicine. There are other factors that render the disease important. MS typically attacks young people (women more often than men), lasts throughout a regular lifetime and ultimately may lead to crippling disability, and until very recently there was no efficient remedy. All this creates immense individual suffering which, implicitly, adds up to a heavy socioeconomic burden.
MS develops spontaneously, seemingly out of the blue, without any specific prodromal signals preceding the first attack. The affected patient typically describes a mix of neurological signs, which include disturbances of sensibility, motor defects and sometimes also blurred vision. Typically also, these initial defects resolve after a while, but return later. In fact, a relapsing-remitting course is considered pathognomonic in most people with MS.
What is it that triggers “spontaneous” MS? Is MS a genetic disease, for example caused by a mutant gene (“nature”), like cystic fibrosis, or is it caused by an external trigger (“nurture”), such as an infectious agent? In the following, we shall discuss recent evidence pointing to combination of the two. We propose that MS is the result of factors from nature PLUS nurture.
There are now excellent arguments indicating that MS develops as a consequence of an autoimmune response, where effector cells from the immune system invade the patient’s own brain and destroy essential tissues. This evidence comes from the particular structure of the MS lesion, from experimental animal models, and is supported by genetics and therapy.
Active MS lesions present the key features of an acute inflammatory immune attack (fig. 1). These include actively ongoing destruction of myelin sheaths and, to varying degrees, degenerating axons. Most importantly in the context of autoimmunity, the affected areas also contain inflammatory infiltrates [1]. These are composed mostly of round leucocytes, macrophages and lymphocytes, which form dense cuff-like aggregations preferentially around small blood vessels (mostly postcapillary venules) and swarm out into the surrounding parenchyma. Especially in early phases of the response, most macrophages are of the proinflammatory (M1) type. Later, upon ingestion of myelin debris, they may assume an anti-inflammatory function, the M2 phenotype. This functional switch is an important phase in the pathogenesis, as it may serve as a target of therapies, such as glatiramer acetate (Copaxone®) [2]. The second major infiltrating cell type are lymphocytes. Their predominant components are T cells of the CD8+ subset, followed by CD4+ T cells, and by B cells [1]. Many of the infiltrating T cells produce the cytokines interleukin-17 (IL-17) and interferon-gamma, and exhibit the gene signatures of Th17 and Th1 cell lineages, respectively. In contrast, anti-inflammatory regulatory T cells (Tregs) seem to be relatively rare [3]. Immune cell aggregations are also commonly found in the leptomeninges. These typically contain B cells, which sometimes form clusters reminiscent of primary lymphoid follicles, and in some cases even form germinal centres characterised by inclusion of typical follicular dendritic cells [4].
Figure 1
The multiple sclerosis plaque, classical portrayals.
A. Plaques embedded in brain white matter (Charcot 1880); B. perivascular and parenchymal inflammatory infiltrates (Babinski, 1885). (from H. Lassmann, Brain Pathol. 2005)
The cellular composition and arrangement of the immune cell infiltrates in active MS plaques faithfully portrays an actively ongoing immune process. This impression is perfectly supported by molecular studies of antigen receptor repertoires used by the infiltrating T and B cells. These studies have been facilitated by new technologies allowing the cloning of paired T cell receptor (TCR) genes from single T cells out of histological material [5]. Analyses of the TCRs used by infiltrating CD8+ T cells (less so in CD4+ cells) provided compelling signs of clonal expansion. Accordingly, individual MS lesions contain clusters of T cells, all using identical TCR sequences, which indicates that they are the clonal offspring of the same ancestor T cell. Such “clonotypes” are also commonly found in other lesion sites of the same brain, and they may persist over extensive periods of time. Obviously all cells of the same clone share identical TCR sequences, and hence bind the same antigen. Indeed, recognition of this particular antigen (or autoantigen) within the brain tissue is the driving force of the clonal expansion [5].
The question of the nature of the putative target antigen remains and, unfortunately, this still waits to be answered. So far, no specific brain structure has been identified as a possible autoantigen recognised by the expanded CD8+ T cells. In contrast, in the case of CD4+ one old report described a hypervariable (antigen binding) sequence motif of a TCR beta chain that was identical to a myelin basic protein (MBP) -specific T cell line isolated from another patient [6]. This was an important finding, but to date it has remained a singular one.
While T cells dominate the inflammatory infiltrates of active MS plaques, it is clear that, in addition, B cells afford an essential contribution to the immune pathogenesis of MS. Indeed, therapies targeting B cells by antibodies against the B cell marker CD20 (rituximab) have been remarkably successful [7]. B cells may contribute to MS on several levels. First of all, B cells could impact pathogenic processes through their immunoglobulin products, myelin-specific autoantibodies. In addition, however, certain B cells are important by presenting (auto-)antigen to T cells, thereby controlling T cell differentiation and activity. Finally B cells release soluble factors that modulate local immune milieus [8]. As mentioned, B cells are found to disperse within parenchymal infiltrates, and as aggregates in leptomeningeal and perivascular spaces. Local B cells secrete immunoglobulins, which form classical oligoclonal bands (OCBs) in the cerebrospinal fluid (CSF). The occurrence of OCBs is so typical for the disease that they are used as a major diagnostic biomarker. In contrast to the OCBs, whose target antigen(s) remain unknown, there are some candidate autoantigens for immunoglobulins circulating in the blood. A recent study identified potassium channel KIR4A in a sizeable proportion of adult patients with relapsing-remitting MS [9], whereas autoantibodies against myelin oligodendrocyte glycoprotein (MOG) were noted in a smaller subgroup of paediatric cases [10].
Thus, good evidence supports an autoimmune concept of the early MS pathogenesis, although the search for a putative target autoantigen for pathogenic T cells has so far remained futile. Our knowledge of brain autoantigens still comes exclusively from animal model studies, namely from experimental autoimmune encephalomyelitis (EAE). Animals with EAE develop clinical and histological signs mimicking the changes seen in human MS lesions. These are classically induced in rodents by autoimmunisation against known structures of the central nervous system (CNS), mostly myelin proteins. Immunisation can be active, by inoculation of the autoantigen in an immune adjuvant, or it can be passive, by transfer of autoantigen reactive T cells [11].
EAE studies have revealed the principles of brain autoimmunity [12]: they identified potentially brain-specific autoimmune T cells as normal components of the healthy immune repertoire. Throughout life, these cells are maintained in a dormant state, without any tendency to attack the brain. However, when activated, these T cells unfold their pathogenic potential. On their way to the brain parenchyma, the activated autoimmune T cells first take a complex journey through secondary immune organs, and thereupon they break through the tight vascular blood-brain barrier. Finally, within the brain tissue the activated T cells attack local brain cells that present the autoantigen, and with the help of additional recruited inflammatory cells (macrophages), in a second step they destroy myelin sheaths, and ultimately the denuded axons.
MS is often viewed as a disease of the temperate zones, a “White Man’s disease”. Indeed the disease is not evenly distributed over all continents and ethnicities. While in Europe and Northern America the frequency is 1/1000, MS is much rarer in Southern and Oriental zones. Also remarkable, the incidence in women is double that in men. Finally, there is a general, worldwide trend of disease increase [13].
Is it the environment (in a broad sense) of high-susceptibility places, or is it the particular genetic set-up of their residents that dictate disease risk? A simple consideration of twins tells us that both factors play a role. In genetically different, dizygotic twins the risk of both siblings developing the disease is low, around 5%. In stark contrast, in genetically identical, monozygotic twins the probability is much higher, around 30% [13]. This difference underlines the importance of genes in disease risk; however, since even in identical twin disease concordance is far from complete, there must be additional factors, “milieu factors” contributing to the development of MS.
Which are the genes that contribute to MS disposition? Clearly, there is no one “disease gene”, as is the case in monogenic diseases like cystic fibrosis. Instead, there are a great many gene variants that can increase (or decrease!) disease risk. Well established is a predisposition linked to the gene area encoding the major histocompatibility gene locus HLA that controls antigen recognition by T lymphocytes. Specifically, the HLA DR2 variant increases MSA risk significantly, while another variant, HLA A2, is protective. HLA genes are the strongest risk genes, but they are not the only ones. Recent genome-wide association studies (GWAS) identified more than 150 MS-associated gene variants outside the HLA gene complex [14]. Interestingly, most of these genes encode or regulate structure and functions of the immune system, lending further support to the immune concept of MS.
The genetic control of disease predisposition as found in human MS is largely recapitulated by its animal models, the variants of EAE [15]. Like in MS, the predominant susceptibility genes are located in the correlates of human HLA, the H-2 complex, and they are assisted by a host of minor gene variants, often analogous to the genes identified by human GWAS. The H-2 genes critically determine T cell reactivity against brain/myelin structures. Depending on their target specificity, these genes predispose the immune system to mount an autoimmune attack against the CNS. They act centrally by influencing the generation of the T cell repertoire, and possibly also by regulating the balance between effector and regulatory cells, and they peripherally control the presentation of autoantigens to either T cell class. Thus, dependent on the H-2 variant, myelin epitopes strongly autoantigenic in one mouse may be completely inert in another mouse with a distinct H-2 complex. However, like in MS, in addition genes outside the major histocompatibility complex (MHC) are crucial in determining the strength and character of brain autoimmunity [16].
The twin studies mentioned above, indicate that “disease-friendly” genetic profiles are supportive, but are by no means sufficient to trigger clinical MS. There is a need of an additional, nongenetic signal. This signal has been traditionally sought among classical infectious agents. Numerous searches have examined relationships between microbial infections and the onset of MS, but the results have been largely inconclusive. None of the suspected infectious agents could be lastingly associated with the initiation of MS. One possible exception is the Epstein-Barr virus [17], which seems to enhance MS risk, although it may not act as an acute disease trigger.
Recently, a new and unexpected disease trigger has emerged, namely the gut flora. Traditionally, the microbial gut flora has been undervalued as an innocuous, but dispensable fill of our intestines. This misconception could be corrected thanks to the new molecular technologies that have facilitated the detailed analysis and characterisation of the gut microbiota. Several unanticipated features came to light [18]. First the seemingly monotonous bacterial mass resolved into a true microcosmos of unprecedented diversity and colour. Healthy human gut microbiota are composed of thousands of different bacterial species (without counting archaebacteria, viruses and fungi). The profiles are highly variable and dynamic. Firstly, microbiota vary strongly in number and composition between intestinal segments, with a craniocaudal gradient of bacterial density (highest numbers of organisms in the colon). Secondly microbial profiles differ between individuals; they are influenced by age and genetics, and they respond promptly to numerous exogenous factors such as diet, stress and therapeutic treatments. Importantly, the complexity of the gut microbiota translates into an equivalent complexity of functions. The gut flora impacts on organismic functions as diverse as immune reactivity, metabolism, endocrine responsiveness and CNS function [19]. It influences disease states as divergent as obesity, type 2 diabetes mellitus, neurodegeneration and even autism. The gut bacteria act on the organism via different targets, all placed in the intestinal wall. These include the epithelia lining the lumen, the immune cells forming the gut associated lymphatic tissues (GALT), and the enteric nervous system. Importantly, the structure and function of the gut wall elements vary profoundly between different intestinal segments, especially between segments of the small and the large intestine. The signals traded include metabolic products processed by the bacteria as well as bacterial components. These act in particular on the innate immune system, which is strongly represented in the intestine [20].
Direct evidence linking genetic susceptibility to gut microbiota in brain autoimmunity came from EAE models. In an actively induced EAE variant, antibiotic ablation of a major part of gut microbiota led to significant resistance to disease induction, an effect attributed to intestinal regulatory T cells (fig. 2) [21].
The impact of gut microbiota on brain autoimmune disease was borne out in models of spontaneous EAE (spEAE). Typically, these paradigms involve transgenic mice that carry in their genome transgenes of myelin-protein specific TCRs, and consequently harbour an exaggerated proportion of myelin autoimmune T cells in their immune system. The first spEAE model was generated by Goverman et al., who constructed transgenic mice overexpressing a TCR recognising a dominant brain autoantigen, the myelin basic protein (MBP) [22]. Without any further experimental manipulation some of these animals developed spEAE presenting classical neurological and histological changes. The frequency of spEAE in this model was relatively low, and it depended on the hygienic condition of caging. Conventionally caged (“dirty”) mice developed spEAE, while mice of the same strain remained disease free, when maintained in “clean” environments. Studies of a similar MBP TCR transgenic mouse model showed that the frequency of spEAE abruptly rises after depletion of regulatory T cells.
A next generation of spEAE models uses transgenes specific for TCRs recognising myelin oligodendrocyte glycoprotein (MOG), an autoantigen that differs from MBP in several respects. MOG constitutes less than 1% of all myelin proteins, and, in contrast to the intracellular MBP, it is glycosylated and sits on the surface of myelin. One of these models is the RR mouse, a SJL/J mouse with a MOG-specific TCR transgene [23]. This model most faithfully simulates clinical and histological features of human MS. Disease onset is spontaneous, with multifarious defects including atactic gait, paralysis and, presumably, disturbed vision. The pathological CNS lesions also mimic active MS plaques, displaying demyelination, axonal degeneration and round-cell infiltration as key features. Then, also like MS, the course of the disease is initially in isolated bouts, rather than constant progression. Spontaneous disease development in RR mouse critically depends on genetic factors. Expression of the RR mouse TCR genes in mice with a genetic background differing from SJL/J, radically changes disease susceptibility: TCR transgenic C57BL.S mice remain completely disease free.
Over 90% of the RR mice develop relapsing-remitting spEAE when reared in clean (“specific pathogen-free”, SPF) laboratory cages. The animals are free of pathogenic infection, but have a perfectly normal commensal microflora, in particular in the gut. In sharp contrast, germfree RR mice (i.e. raised in the total absence of any bacterial organisms) are fully protected from spEAE. They promptly develop spEAE when exposed to bacteria present in SPF conditions [24]. These observations unequivocally expose commensal microbiota as triggers of the autoimmune disease, but the mechanisms of triggering, including the nature of the triggering organism(s), remain unknown. In particular, it is unknown whether autoimmune disease is prompted by single, specific bacteria or by particular groups of microbes.
Recently, gut bacteria with defined effects on immune functions have been identified. The segmented filamentous bacteria (SFB, they are found in the mouse, not in human gut) render the GALT milieu “Th17-friendly”, supporting the differentiation of IL-17 producing lymphocytes [25]. SFBs stand out by their topography. They populate the lower segments of the small intestine (ileum), and they count among the few bacteria that dive through the mucus layer and sit directly on the epithelial surface. Contact with SFBs results in the induction of the transcription factor RORγt in cells of the innate and adaptive immune system. These include IL-17‒producing CD4+ T cells as well as innate ILC-3 cells, which all produce a particular profile of proinflammatory cytokines. At the same time, it has become understood that IL-17‒-releasing Th17 cells have a key role not only in the protection against bacterial infection, but also as autoimmune effector cells. Consequently, in a transgenic model simulating certain aspects of rheumatoid arthritis, germfree mice were protected from disease development [26]. Importantly, however, SFBs did not trigger spEAE in the RR mouse model [24].
Other bacterial species have a protective potential. Bacteroides strains support the generation and function of Treg cells in the colon. They shed a polysaccharide (PSA) which turns on the appropriate differentiation pathway [27].
We propose a pathogenesis concept of MS which rests on three pillars: (a) genetic susceptibility as a permissive basis, (b) intestinal microbiota as a trigger, and (c) a resulting autoimmune attack as the initiating effector mechanism. In theory, all three features could present targets for specific therapies. In practice, however, practicability of such approaches varies greatly. Modification of the genome has become feasible in certain monogenic disorders involving defined stem cells. In contrast, manipulation of MS risk genes (“genomic editing”) still remains far out of reach [28]. Instead, the most successful treatments available to date concentrate on the depletion or attenuation of autoimmune cells, and blockade of their migration [29]. In addition, and quite unexpectedly, the gut and its flora have now emerged as another promising therapeutic target.
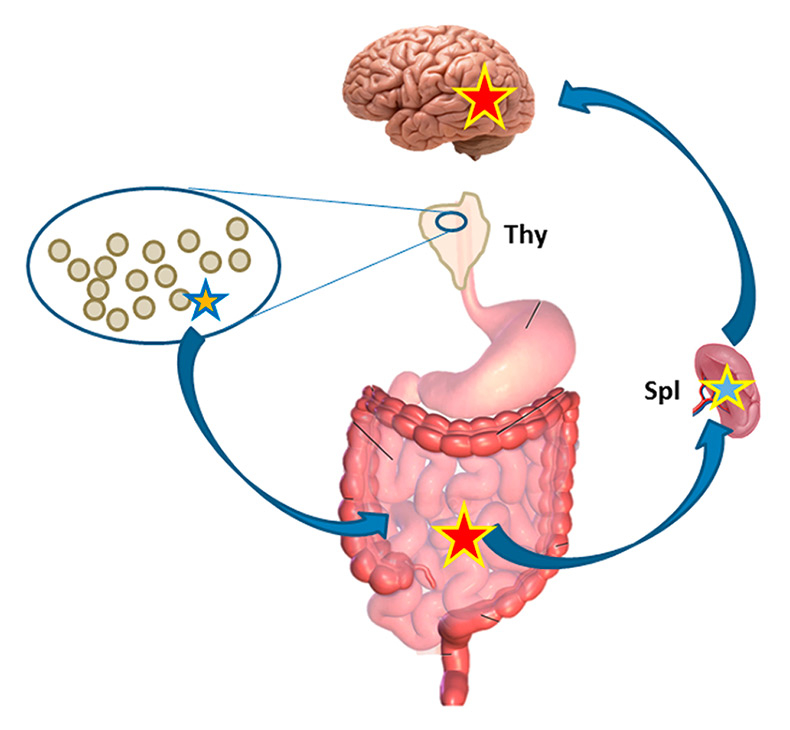
Figure 2
Autoimmune T cell migration. Dormant autoimmune T cell (blue/orange star) in natural thymus (Thy)-derived immune repertoire migrates to the intestinal tract, where it is activated (yellow/red). From there the cell travels through peripheral immune organs (lymph nodes, spleen, Spl), where it is tuned to migrate to the brain (yellow/blue) and ultimately penetrates into the brain parenchyma, where it is re-activated by confrontation with brain autoantigen (yellow/red).
An ideal therapy would manipulate the gut flora of an MS patient in a way to reduce pathogenic stimulation on autoimmune cells, and possibly even redirect it to turn on anti-inflammatory, regulatory cell populations. This strategy is not utopic, since, as discussed above, (a) the gut flora can be profoundly altered by exogenous factors, and (b) microbial components with proinflammatory potential have been identified, as have been other components with anti-inflammatory activity (vide supra).
An obvious, but blunt way to modulate the gut microbiota is by applying long-term antibiotics, regimens which may eliminate most members of the gut flora, among them pathogenic microbes, but this may be hampered by severe side effects including dysbiosis, and allergy [30]. Several antibiotics have now been tried; among these, minocycline takes a particular position. Treatment of small groups had remarkably beneficial effects, but it remained open as to whether these were a result of the drug’s antibacterial function, or of direct action on myelin-forming oligodendrocytes to support myelin regeneration [31]. A more selective approach would employ bacteriophages, viruses that specifically destroy defined bacterial target species with few side effects to be expected [32]. Application of phage therapy will be worth consideration, as soon as specific pathogenic microbes have been identified.
As an attractive alternative, disease-associated microbiota could be corrected by oral substitution regimens. In fact, as mentioned above, the composition of the healthy gut flora is highly dynamic, reflecting a permanent competition between individual microbes for their habitat [33]. One popular approach relies on introduction of beneficial microbes ‒ probiotics. Cultured microbes are ingested with the expectation that they settle within the recipients’ microbiota and there (re-)establish a balanced, healthy profile [34]. A similar, but more radical and less appetising approach is faecal transplantation, the replacement of a patient’s pathological gut flora by the microbiota of a healthy donor. This has been applied successfully in irritable bowel disease, and experimental treatment seems to be underway in MS as well [35].
The immune system responds promptly and profoundly to the dietary intake. Diets act directly on the GALT through components such as retinoic acid, adenosine triphosphate, short-chain fatty acids and vitamins. Alternatively there are indirect dietary effects, targeting microbiota profiles [36]. So it comes as no surprise that dietary regimens have been a traditional approach to treating MS. Indeed, there were numerous reports along this line, some with impressive successes, but most of these reports were anecdotal, rarely based on stringently controlled large scale trials [37].
An intriguing argument in favour of dietary treatment of MS comes from the epidemiology of MS in Japan. Traditionally, Japan has been considered as an area with a low incidence of MS, and the few cases diagnosed showed a clinical profile different from “Western type” MS. Over the past few decades, however, there has been a remarkable increase of disease incidence, mostly due to the appearance of Western type syndromes. This change has been related to a profound change of lifestyle, centrally involving a trend away from traditional Japanese cuisine toward Western food [38].
There are good reasons to reconsider dietary intervention on a scientific basis. Firstly, experimental work using EAE models unequivocally demonstrates nutritional effects on brain autoimmunity. It is now established that high fat diets have a detrimental effect on EAE, in contrast to beneficial high-fibre diets [39]. Also, salt affects disposition to brain autoimmunity [40, 41]. While the current studies agree that the diets ultimately act on the balance between pathogenic Th17 and protective Treg cells, they leave open how far these mechanisms act directly on the immune system, and how much they may involve the microbiota.
1 Lassmann H, Wekerle H. The pathology of multiple sclerosis. In: Compston A, Confavreux C, Lassmann H, McDonald I, Miller D, Noseworthy J, et al., editors. McAlpine's Multiple Sclerosis. 4 ed. Churchill Livingstone Elsevier. 2006;p.557–600.
2 Lalive PH, Neuhaus O, Benkhoucha M, Burger D, Hohlfeld R, Zamvil SS, et al. Glatiramer acetate in the treatment of multiple sclerosis emerging concepts regarding its mechanism of action. CNS Drugs. 2011;25(5):401–14.
3 Fritzsching B, Haas J, König F, Kunz P, Fritzsching E, Poschl J, et al. Intracerebral human regulatory T cells: Analysis of CD4+CD25+FOXP3+ T cells in brain lesions and cerebrospinal fluid of multiple sclerosis patients. Plos One. 2011;6(3).
4 Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004;14(2):164–44.
5 Dornmair K, Meinl E, Hohlfeld R. Novel approaches for identifying target antigens of autoreactive human B and T cells. Semin Immunopathol. 2009;31(4):467–77.
6 Oksenberg JR, Panzara MA, Begovich AB, Mitchell D, Erlich HA, Murray RS, et al. Selection for T-cell receptor Vβ-Dβ-Jβ gene rearrangements with specificity for a myelin basic protein peptide in brain lesions of multiple sclerosis. Nature. 1993;362:68–70.
7 Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358:676–88.
8 Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev Immunol. 2012;30:221–41.
9 Srivastava R, Aslam M, Kalluri SR, Schirmer L, Buck D, Tackenberg B, et al. Potassium channel KIR4.1 as an immune target in multiple sclerosis. N Engl J Med. 2012;367:115–23.
10 Reindl M, Di Pauli F, Berger T. The spectrum of MOG autoantibody-associated demyelinating diseases. Nat Rev Neurol. 2013;9(8):455–61.
11 Krishnamoorthy G, Wekerle H. EAE: An immunologist's magic eye. Eur J Immunol. 2009;39(8):2031–5.
12 Wekerle H, Lassmann H. The immunology of inflammatory demyelinating disease. In: Compston A, Confavreux C, Lassmann H, McDonald I, Miller D, Noseworthy J, et al., editors. McAlpine’s Multiple Sclerosis. 4 ed. Chhurchill Livingstone Elsevier; 2006;p.491–546.
13 Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502–17.
14 Beecham AH, Patsopoulos NA, Xifara DK, Davis MF, Kemppinen A, Cotsapas C, et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nature Genet. 2013;45(11):1353–60.
15 Olsson T, Jagodic M, Piehl F, Wallström E. Genetics of autoimmune neuroinflammation. Curr Opin Immunol. 2006;18(6):643–9.
16 Aitman TJ, Critser JK, Cuppen E, Dominiczak A, Fernandez-Suarez XM, Flint J, et al. Progress and prospects in rat genetics: a community view. Nature Genet. 2008;40(5):516–22.
17 Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis. Part I: The role of infection. Ann Neurol. 2007;61(4):288–99.
18 Walter J, Ley RE. The human gut microbiome: Ecology and recent evolutionary changes . Annu Rev Microbiol. 2011;65:411–29.
19 Dorrestein PC, Mazmanian SK, Knight R. Finding the missing links among metabolites, microbes, and the host. Immunity. 2014;40(6):824–52.
20 Caballero S, Pamer EG. Microbiota-mediated inflammation and antimicrobial defense in the intestine. Annu Rev Immunol. 2015.
21 Ochoa-Repáraz J, Mielcarz DW, Ditrio LE, Burroughs AR, Foureau DM, Haque-Begum S, et al. Role of gut commensal microflora in the development of experimental autoimmune encephalomyelitis. J Immunol. 2009;183(10):6041–50.
22 Goverman J, Woods A, Larson L, Weiner LP, Hood L, Zaller DM. Transgenic mice that express a myelin basic protein-specific T cell receptor develop spontaneous autoimmunity. Cell. 1993;72:551–60.
23 Pöllinger B, Krishnamoorthy G, Berer K, Lassmann H, Bösl M, Dunn R, et al. Spontaneous relapsing-remitting EAE in the SJL/J mouse: MOG-reactive transgenic T cells recruit endogenous MOG-specific B cells. J Exp Med. 2009;206(6):1303–16.
24 Berer K, Mues M, Koutroulos M, Al Rasbi Z, Boziki M, Johner C, et al. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature. 2011;479:538–41.
25 Cerf-Bensussan N, Gaboriau-Routhiau V. The immune system and the gut microbiota: friends or foes? Nature Rev Immunol. 2010;10(10):735–44.
26 Wu H-J, Ivanov II, Darce D, Hattori K, Shima T, Umesaki Y, et al. Gut residing filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity. 2010;32(6):815–23.
27 Wang Y, Telesford KM, Ochoa-Repáraz J, Haque-Begum S, Christy M, Kasper EJ, et al. An intestinal commensal symbiosis factor controls neuroinflammation via TLR2-mediated CD39 signalling. Nature Commun. 2014.
28 Cox DBT, Platt RJ, Zhang F. Therapeutic genome editing: prospects and challenges. Nature Med. 2015;21(2):121–31.
29 Kieseier BC, Stüve O. A critical appraisal of treatment decisions in multiple sclerosis – old versus new. Nat Rev Neurol. 2011;7(5):255–62.
30 Stecher B, Maier L, Hardt WD. “Blooming” in the gut: how dysbiosis might contribute to pathogen evolution. Nature Rev Microbiol. 2013;11(4):277–84.
31 Yong VW, Giuliani F, Xue M, Bar-Or A, Metz LM. Experimental models of neuroprotection relevant to multiple sclerosis. Neurol. 2007;68:S32–S37.
32 Reyes A, Semenkovich NP, Whiteson K, Rohwer F, Gordon JI. Going viral: next-generation sequencing applied to phage populations in the human gut. Nature Rev Microbiol. 2012;10(9):607–17.
33 Buffie CG, Pamer EG. Microbiota-mediated colonization resistance against intestinal pathogens. Nature Rev Immunol. 2013;13(11):790–801.
34 Bron PA, Van Baarlen P, Kleerebezem M. Emerging molecular insights into the interaction between probiotics and the host intestinal mucosa. Nature Rev Microbiol. 2012;10(1):66–78.
35 Vrieze A, de Groot PF, Kootte RS, Knaapen M, Van Nood E, Nieuwdorp M. Fecal transplant: A safe and sustainable clinical therapy for restoring intestinal microbial balance in human disease? Best Practice & Research in Clinical Gastroenterology. 2013;27(1):127–37.
36 Thorburn AN, Macia L, Mackay CR. Diet, metabolites, and “Western-lifestyle” inflammatory diseases. Immunity. 2014;40(6):833–42.
37 Schwarz S, Leweling H, Meinck HM. Alternative and complementary therapies in multiple sclerosis. Fortschritte der Neurologie Psychiatrie. 2005;73(8):451–62.
38 Yamamura T, Miyake S. Diet, gut flora, and multiple sclerosis: Current research and future perspectives. In: Yamamura T, Gran B, editors. Multiple Sclerosis Immunology.NewYork, Heidelberg, Dordrecht, London: Springer. 2014;p. 115–25.
39 Issazadeh-Navikas S, Teimer R, Bockermann R. Influence of dietary components on regulatory T cells. Mol Med. 2012;18(1):95–110.
40 Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, et al. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature. 2013;496:518–22.
41 Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, et al. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature. 2013;496:513–7.
Disclosure statement: The author is supported by the Hertie-Foundation, the Max-Planck-Society and Deutsche Forschungsgemeinschaft.
* Term coined by Francis Galton (1822‑1911) to describe the key factors determining the development of human personality