New and emerging roles of small RNAs in neurodegeneration, muscle, cardiovascular and inflammatory diseases
DOI: https://doi.org/10.4414/smw.2015.14192
Marian
Hruska-Plochan, Bei
Li, Diego
Kyburz, Jan
Krützfeldt, Ulf
Landmesser, Adriano
Aguzzi, Magdalini
Polymenidou
Summary
Small noncoding RNAs (snRNAs) were discovered more than two decades ago, yet it was not until relatively recently that their important role in genome regulation was recognised. With such a substantial role in genome regulation, it is not surprising that snRNAs are crucial contributors to an ever-increasing number of diseases, as evidenced by the long list of published studies. Currently, microRNAs (miRNAs) represent the most intensively studied snRNAs. Dysregulation of miRNAs has been confirmed in numerous diseases, and changes in their levels could play an essential role in disease onset and progression and could be used for prognosis and potential therapy. Indeed, disease-altered miRNAs may either signify a direct trigger or a consequence of the disease. Therefore, miRNAs represent unique targets for disease intervention through their down- or up-regulation. Importantly, miRNAs may facilitate disease monitoring by detection of disease-altered miRNAs in easily accessible bodily fluids, such as blood or cerebrospinal fluid. Therefore, study of these events is of utmost importance for understanding the molecular mechanisms that drive disease, as well as for diagnosis and therapy. Here we attempted to synthesise a large number of studies to highlight the crucial role of miRNAs in the pathogenesis of neurodegenerative, muscle, cardiovascular and inflammatory diseases.
Introduction
While the incredible race to sequence the human genome was in progress, it was predicted and anticipated that humans would have more protein-coding genes than, for instance, the mouse [1, 2]. The results were therefore quite surprising, since only about 1%–2% of the human DNA is ‘coding’ [3]. The rest was given names such as ‘noncoding’ or ‘junk’ DNA and it was not until relatively recently that the majority of this ‘junk DNA’ was acknowledged to be transcribed [4–6] and to have a function [7]: indeed, a function that has changed our view of gene expression, genome regulation and even evolution, development and the ever-fascinating complexity of the brain and its rapid expansion in primates [8, 9].
The first findings came about 20 years ago, when miRNA in Caenorhabditis
elegans was found to regulate gene expression or, more precisely, messenger RNA (mRNA) translation [10]. The miRNAs were the first genome regulatory small noncoding RNAs to be discovered and since then, various noncoding RNA species with multiple functions have been identified [7].
Small noncoding RNAs (snRNAs) are present during evolution, and the more complex the organism, the more species of snRNAs develop (such as small interfering RNAs [siRNAs], piwi-interacting RNA [piRNAs] and miRNAs). It is believed that they originally served as genome protectors against endogenous and exogenous threats and that they acquired their genome regulatory functions later in evolution [8]. Long noncoding RNAs (lncRNAs) arrived on the scene of gene regulation relatively recently but their function appears to be connected to increased evolutionary speed [8, 9]. The lncRNAs retain many of the protein-coding gene characteristics but are expressed with tighter tissue specificity and are mainly nuclear localised [3]. These differences indicate their function, as a new regulatory layer responsible for fine-tuning the snRNAs [8] as demonstrated by the numerous interactions between lncRNAs and miRNAs [11–13].
Box 1
Main enzymes of the miRNA pathway
Drosha – Enzyme that cleaves long primary pri-miRNA transcripts generating approx. 70bp pre-miRNAs
Dicer – Enzyme that cleaves double-stranded RNA and pre-miRNA into short double-stranded siRNAs and miRNAs
RISC – RNA-induced silencing complex, a multiprotein complex that incorporates one strand of either siRNA or miRNA which then serves as a template for perfect mRNA binding in case of siRNAs or for binding to imperfect complementary sites within the 3’UTRs of miRNA target mRNAs
Argonaute – Member of RISC, an enzyme that cleaves the target mRNA leading to RNA interference by mRNA degradation
Among the noncoding RNAs (ncRNAs), miRNAs are the most extensively studied and over 2 000 miRNAs have so far been identified in humans, each having several hundreds of potential target mRNAs [14]. In addition, thousands of single nucleotide polymorphisms (SNPs) were discovered within miRNA sites [15] and length and sequence heterogeneity (IsomiRs) for the vast majority of miRNAs has also been found [16]. Such polymorphisms might therefore contribute to phenotypic differences or even human diseases [15, 17, 18]. With such an extensive involvement in gene regulation, it is of no surprise that malfunctions of noncoding RNAs have been found to be directly responsible for or contribute to the pathogenesis of numerous disorders [8, 9, 17, 19]. These disease-inducing changes could be used for diagnosis, as alterations in miRNAs could be detected in cerebrospinal fluid or even blood plasma [17]. Moreover, they could be used as therapeutic targets [20, 21] with high potential, as a single miRNA could have hundreds of targets [14]. Furthermore, a major technological advancement originating from the noncoding RNA field is the broadly used downregulation of gene(s) of interest via small interfering RNA mechanism. In conclusion, noncoding RNA research is gaining momentum and we anticipate that while uncovering the ncRNA interactions with DNA, RNA and proteins, we will witness numerous, as yet unexplained, mechanisms becoming the solutions for many diseases.
Here, we review the latest discoveries on the role of miRNAs in human disease, focusing on neurodegenerative diseases, skeletal muscle and cardiovascular diseases, and the pathogenesis of inflammation.
Small noncoding RNAs in neurodegenerative diseases
Since the hallmark of most neurodegenerative diseases is protein aggregation [22], research has mainly been focused on the study of these misfolded and aggregated protein species. Hence, the term ‘proteinopathies’ has been established. Recent discoveries, however, show that major neurodegenerative diseases also involve miRNA dysfunction (table 1). Their importance for brain development and function was first noted in Dicer knockout experiments, resulting in absence of mature miRNAs and siRNAs, which led to widespread neuronal alterations during development. Dicer knockout is embryonically lethal in mice [23] and its conditional inactivation in defined neuronal populations or brain regions results in an abnormal phenotype and neuronal degeneration [24, 25]. Moreover, cell-specific Dicer ablation in oligodendrocytes [25] or astrocytes [26] severely affected miRNA biogenesis in these cells leading to their malfunction, which ultimately resulted in noncell autonomous neuronal dysfunction and degeneration, emphasising the role of both miRNAs and glial cells in neurodegeneration. Further work revealed the involvement of miRNA and siRNA in neuronal stem cell differentiation, cell-to-cell interaction, neural development, axonal growth and guidance, as well as synaptic transport [19, 26, 27]. Moreover, piwi-interacting RNAs (piRNAs) and small-nucleolar RNAs (snoRNAs) were shown to contribute in the regulation of complex neuronal functions, such as learning and memory formation [15, 28, 29].
Although the links between the dysregulated miRNAs in neurodegenerative diseases and their target mRNAs or their regulatory lncRNAs are still poorly understood, new exciting discoveries highlight the role of miRNAs in the pathogenesis of neurodegenerative diseases (fig. 1). Importantly, miRNAs reflecting the disease stage have been identified in plasma and whole blood samples, indicating their potential use as biomarkers that are desperately needed. In addition, given the large number and high stability of circulating miRNAs, it is possible that through use of unbiased high-throughput screenings, miRNAs could also be used to differentiate neurodegenerative diseases from other conditions, e.g., inflammatory neurological diseases.
In the following sections, we describe the role of the most important miRNAs involved in Alzheimer’s disease, Parkinson’s disease, prion disease, amyotrophic lateral sclerosis, frontotemporal dementia and Huntington’s disease.
Box 2
Staging of neurodegenerative diseases
Braak stages – AD staging that is based on the distribution of tau neurofibrillary tangles (Braak I-VI). Braak staging was also developed for PD where it is based on distribution of Lewy bodies
HD grades – HD or Vonsattel grades are used for HD staging and are based on the severity and pattern of striatal degeneration (HD 0-4)
|
Table 1: Reported miRNA alternations in neurodegenerative diseases. |
|
Disease
|
Dysregulation in patient samples
|
|
Up in CNS
|
Down in CNS
|
Up in periphery
|
Down in periphery
|
| Alzheimer’s disease |
miR-26b (temporal cortex)[56]
miR-9, miR-128 (CA1 hippocampus)[58] |
miR-124 (anterior temporal cortex)[42]
miR-107 (superior and middle temporal cortex)[37]
miR-29a, miR-29b-1 (high BACE1 brain)[50]
miR-29a (cerebral cortex)[53]
miR-132-3p (prefrontal cortex)[57]
miR-146b (hippocampus, frontal gyrus)[36] |
miR-9, miR-125b, miR-146a, miR-155 (CSF, neocortex ECF) [43]
miR-34a, miR-181b, let-7f, miR-200a (blood mononuclear cells)[54]
let-7d-5p, let-7g-5p, miR-15b-5p, miR-142-3p, miR-191-5p, miR-301a-3p and miR-545-3p (plasma)[59] |
miR-384 (blood, CSF)[38] |
| Parkinson’s disease |
|
miR-133b (midbrain)[69]
miR-205 (frontal cortex, striatum)[73] |
miR16-2*, miR26a2* (peripheral blood early onset patients)[78]
miR-331-5p (plasma)[79] |
miR1, miR-22*, miR-29a (peripheral blood non-treated patients)[78]
miR-126* (peripheral blood mononuclear cells)[237] |
| Amyotrophic lateral sclerosis |
miR-624, miR-520e, miR-524-5p, miR-548a-5p, miR-606, miR-612, miR-647 (sporadic ventral lumbar spinal cord)[136]
miR-b1123, miR-b2948, miR-b3265, miR-b5539, miR-sb1217*, miR-sb3998 (novel miRNA with MREs in NEFL mRNA 3′UTR, sporadic ventral lumbar spinal cord)[137]
miR-24-2*, miR-142-3p, miR-142-5p, miR-146a, miR-146b, miR-155 (spinal cord)[163] |
miR-9, miR-9-2 (human iPSC-derived neurons)[134]
239 different miRNAs (sporadic ventral lumbar spinal cord)[136]
miR-b1336, miR-b2403, miR-b4652, miR-sb659* (novel miRNA with MREs in NEFL mRNA 3′UTR, sporadic ventral lumbar spinal cord)[137] |
miR-23a, miR-29b, miR-206, miR-455 (skeletal muscle)[151]
miR106b, miR-206 (serum)[152]
miR-338-3p (sporadic peripheral blood leucocytes)[167]
miR-143-5p, miR-574-5p (sporadic CSF)[168] |
miR-451, miR-1275, miR-328-5P, miR-638, miR-149, miR-655 (sporadic peripheral blood leucocytes)[167]
miR-132-5p, miR-132-3p, miR-143-3p (sporadic CSF)[168]
miR-132-5p, miR-132-3p, miR-143-5p, miR-143-3p, let-7b (sporadic serum)[168]
miR-574-3p, miR-574-5p (TARDBP, FUS, C9ORF72 and sporadic mutant Epstein-Barr virus transformed lymphoblastoid cell lines)[168]
miR-663a, miR-9-5p (FUS mutant LCLs)[168]
Let-7b (FUS and C9ORF72 mutant LCLs)[168] |
| Huntington’s disease |
miR-29a, miR-330 (cortex, Brodman area 4)[181]
miR-132 (cortex, Brodman area 4)[135]
miR-196a[185], miR-486 (cortex, Brodman area 4, only HD1 grade)[135]
miR-100, miR-151-3p, miR-16, miR-219-2-3p, miR-219-5p, miR-27b, miR-451, miR-92a (frontal cortex, striatum)[16]
miR-10b-5p, miR-1247-5p, miR-196a-5p, miR-196b-5p, and miR-615-3p (prefrontal cortex Brodmann area 9)[189] |
miR-132 (cortex, Brodman area 4)[181]
miR-9, miR-9*, miR-29b, miR-124a (cortex, Brodman area 4)[135]
miR-128a (human pre- and post-symptomatic brain)[184]
miR-128, miR139-3p, miR-222, miR-382, miR-433, miR-485-3p (frontal cortex, striatum)[16] |
miR-34b (plasma, pre-manifest HD)[166] |
|
| 3’UTR = 3’untranslated region; BACE-1 = β-secretase; CNS = central nervous system; CSF = cerebrospinal fluid; ECF = extracellular fluid; HD = Huntington’s disease; iPSC = induced pluripotent stem cell; MREs = molecular recognition elements; mRNA = messenger RNA; NEFL = neurofilament light subunit; |
Alzheimer’s disease
Alzheimer’s disease (AD) is an age-related progressive dementia and is the most common neurodegenerative disease. With a worldwide prevalence estimated to be 24 million [31], AD is one of the most common diseases in the industrial world [30]. AD is clinically characterised by a slow but progressive loss of memory and cognitive functions that leads to dementia [32] and ultimately to death [33]. Pathological hallmarks of the disease are the extracellular senile plaques composed of aggregated and misfolded β-amyloid (Aβ) peptide and intracellular neurofibrillary tangles made of hyperphosphorylated tau protein, accompanied by microvascular damage and neuroinflammation [33]. Vast and broad loss of brain weight and volume is most likely caused by shrinkage and subsequent loss of neuronal processes, as well as specific loss of neurons within the entorhinal cortex and the hippocampus [34]. The majority of AD cases (>95%) do not show clear inheritance and they are called sporadic or late-onset AD. Nevertheless, several genetic factors were shown to contribute to the development of sporadic AD [33]. On the other hand, mutations in the amyloid precursor protein (APP), presenilin 1 (PSEN1) and presenilin 2 (PSEN2) all lead to aggregation of the Aβ peptide and cause the early-onset, autosomal dominant form of AD [30] with Mendelian inheritance.
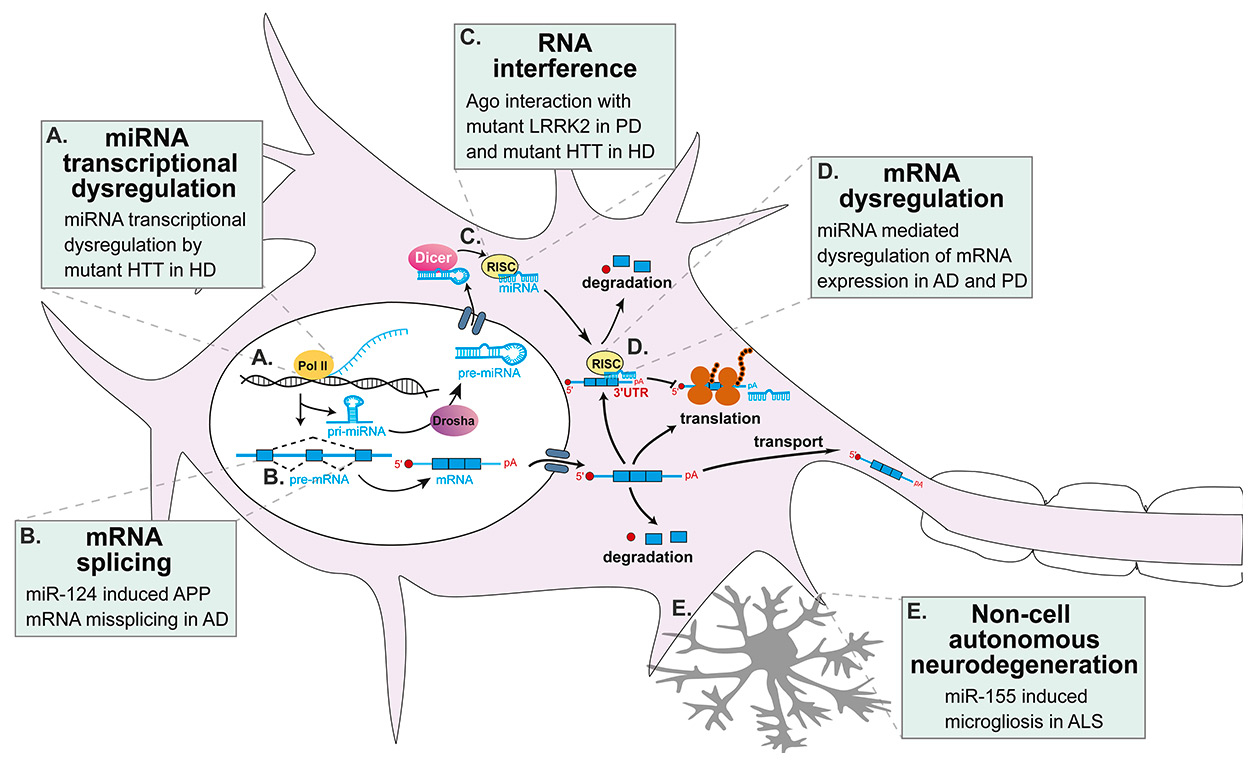
Figure 1
miRNA alternations in neurodegenerative diseases. Numerous alternations in miRNA biogenesis and function were found to contribute to the pathogenesis of several neurodegenerative diseases including: (A) In HD, miRNA transcription is disturbed as mutant huntingtin does not bind REST (which is normally bound to wild-type huntingtin), allowing the formation of NRSE thus repressing the transcription of neuronal miRNAs [135, 181, 183]. Mutant huntingtin also binds to p53 dysregulating p53-mediated transcription of miRNAs [16, 188]. (B) Downregulation of miR-124 has been found to cause missplicing of APP in AD [42]. (C) Ago has been found to be sequestered by mutant huntingtin and accumulated into stress granules in HD suggesting impaired miRNA-mediated mRNA regulation [189–191]. Interestingly, mutant LRRK2 also interacts with Ago, impairing mRNA regulation by miRNAs [72]. (D) Expression of APP and BACE1 in AD [37–41, 48–52] and SNCS and LRRK2 in PD [71-73] has been described to be specifically affected by their regulating miRNA(s) that are disturbed in these diseases. (E) Non-cell autonomous neurodegeneration is now well recognized in ALS and miR-155 seems to play a major role in the pathologic microgliosis [43, 163, 165].
3’UTR = 3’ untranslated region; AD = Alzheimer’s disease; ALS = amyotrophic lateral sclerosis; APP = amyloid precursor protein; BACE-1 = β-secretase 1; HD = Huntington’s disease; HTT = huntingtin gene; LRRK2= leucine-rich repeat kinase 2; miRNA = microRNA; mRNA = messenger RNA; NRSE = neuron restrictive silencer element; PD = Parkinson’s disease; pri-miRNA = primary miRNA; REST = RE1-silencing transcription factor; RISC = RNA-induced silencing complex
miRNA-mediated regulation of the amyloid precursor protein
Numerous studies of miRNAs in AD have been performed both in AD models and human AD patient samples. It is clear that miRNAs are deregulated in AD [35] and that miRNA levels change according to disease stage [36, 37] or AD type [38]. Direct APP mRNA regulation and subsequent decrease of Aβ accumulation has been shown for miR-101 in vitro [39, 40] and for miR-384 both in vitro and in cerebrospinal fluid (CSF) and serum of AD patients [38]. In addition, miR-384 also targets and regulates BACE1 (β-secretase 1; β-site APP cleaving enzyme 1), which in turn also indirectly represses the Aβ load, as a negative correlation between miR-384 and Aβ42 was observed in serum and CSF of AD patients [38]. Several other miRNAs (miR-20a, miR-17, miR-147, miR-655, miR-323-3p, miR-644, miR-153) were shown to regulate APP expression in vitro and in cells [41]. Moreover, as shown in cultured cells, some AD-linked mutations in APP interfere with miRNA binding, such as the T117C APP variant, which inhibited miR-147 binding, and the A454G APP variant, which increased miR-20a binding [41]. Alternative splicing of APP is also regulated by miR-124, which is reduced in AD brains leading to APP missplicing [42]. Upregulation of nuclear factor kappa-light-chain-enhancer (NF-κB)-sensitive inflammatory miRNAs (miR-9, miR-125b, miR-146a, miR-155) was reported in the CSF and extracellular fluid (ECF) from AD neocortex, where Aβ42 levels were slightly reduced, compared with other brain regions, suggesting a role for these miRNAs in the pathogenic signalling of AD [43], potentially disturbing the fine tuning of inflammatory responses as it has been shown that dysregulating only miR-155 or miR-146 is sufficient to trigger autoimmune disease [44–47].
miRNA-mediated regulation of β-secretase 1
Beta-secretase 1 (BACE1) is the proteolytic enzyme that cleaves APP at the β-site APP, resulting in the production of Aβ. BACE1-targeting miRNAs therefore regulate APP and Aβ indirectly. The miR-107 targets BACE1 mRNA and since miR-107 levels significantly decrease during AD progression, BACE1 mRNA levels subsequently increase [37]. Also, miR-195 was shown to regulate negatively BACE1 and Aβ in cell lines and in the SAMP8 (senescence-accelerated) mouse model, which developes AD signs [48]. Both miR-298 and miR-328 have been shown to regulate BACE1, as their overexpression in cell lines and primary neurons showed lower BACE1 levels [49]. BACE1 expression can be regulated by miR-29a, miR-29b-1, and miR-9 in cultured cells, and the miR-29a/b-1 cluster was significantly decreased in AD brain displaying abnormally high BACE1 protein [50]. Results from cultured cells also showed that miR-124 directly regulates BACE1 expression, with miR-124 downregulation leading to increased BACE1 levels and Aβ neurotoxicity [51]. Overexpression of miR-29c in cell lines and transgenic mice (APPswe/PSΔE9) leads to reduced levels of BACE1 [52]. Interestingly, miR-29a is significantly downregulated in human AD brains, leading to the upregulation of its target, neuron navigator 3 (NAV3), in degenerating pyramidal neurons in the cerebral cortex [53].
Misregulation of miRNAs in Alzheimer’s disease
In a miRNA microarray screen followed by quantitive polymerase chain reaction (qPCR) validation of hits, Schipper and colleagues demonstrated that miR-34a and miR-181b are significantly upregulated in blood mononuclear cells of patients with sporadic AD, while their expression differed between AD subjects bearing one or two APOE4 alleles [54]. Also, miR-34a was shown to harbour a 3’-untranslated region (3’UTR) binding site on tau mRNA, thereby directly repressing tau expression in cultured cells [55]. Misregulation of miRNAs occurs already in the early stages of AD (Braak III), such as elevation of miR-26b, which leads to apoptosis in primary neurons [56] and decrease of miR-132-3p [57] in postmortem AD brains. Importantly, the latter appears to occur mostly in neurons with tau hyperphosphorylation [57]. In addition to the above, numerous miRNAs were found to be deregulated across different brain areas and AD stages, both in brain and CSF. All members of the miR-30 family were upregulated in CSF while only miR-146b was consistently altered in both hippocampus and medial frontal gyrus in early and late stages of the disease [36]. Moreover, miR-9, miR-125b, miR-146a and miR-155 were upregulated in CSF and ECF of AD patients [43], while miR-384 was downregulated in both serum and CSF of AD patients [38]. Further, miR-9 was found to be upregulated in AD CSF and ECF [43] and hippocampus [58], but downregulated in neocortex [50], also suggesting differential spatial regulation of miRNAs in AD brain. The miRNA signature is also altered in the blood of AD patients. Indeed, blood mononuclear cells of AD subjects showed significantly increased levels of miR-34a and miR-181b [54]. Finally, using nCounter miRNA expression assay v1 (high-throughput expression profiling of miRNAs using digital detection and counting of miRNAs in a single reaction without amplification) followed by qPCR validation, let-7d-5p, let-7g-5p, miR-15b-5p, miR-142-3p, miR-191-5p, miR-301a-3p and miR-545-3p were all shown to be significantly upregulated in plasma of AD patients, providing differentiation of AD patients from normal controls with >95% accuracy [59].
Taken together, the above results clearly demonstrate the high potential of miRNAs as biomarkers for AD. Discrepancies between studies, however, indicate that additional multicentre in-depth studies are necessary to identify and/or confirm miRNAs that could represent reliable biomarkers for AD.
Parkinson’s disease
Parkinson’s disease (PD) is the second most common neurodegenerative disorder, which affects an estimated 1% of individuals over 60 years of age, a percentage that is expected to double by 2030 as the population ages [60]. PD is a progressive neurodegenerative disease presenting with resting tremor, bradykinesia, rigidity, postural instability and dementia [61]. These symptoms are attributed to loss of dopamine neurons in the substantia nigra pars compacta, as well as widespread neuropathological changes in multiple brain regions, including the amygdala, cingulate gyrus and cerebral cortex [61]. The pathological hallmark of PD are Lewy bodies, intraneuronal inclusions that mainly contain aggregated α-synuclein, often in association with neurofilaments and ubiquitin [62]. It has been shown that expression of mutant as well as overexpression of wild-type α-synuclein leads to development of PD [63]. PD is mainly sporadic and is believed to develop as a consequence of multiple genetic and possibly environmental factors in an aging brain. Mutations in approximately 15 genes were reported to cause rare cases of familial PD, including mutations in α-synuclein, LRRK2, parkin, PINK1, DJ-1 [64]. Interestingly, it is well-recognised that temporary Parkinsonian symptoms represent a common side effect of several drugs that usually vanishes after drug withdrawal [65]. However, specific cases of irreversible Parkinsonism were described in the late 70s and early 80s among drug abusers in the USA. The chemical compound responsible for the PD-like symptoms and severe neuronal loss in the substantia nigra was later identified to be MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), the side product in synthesis of the ‘new synthetic heroin’, MPPP (1-methyl-4-phenyl-4-propionoxypiperidine) [65–67]. Subsequently, MPTP became one of the most commonly used neurotoxins to model PD in experimental animals [68].
miRNAs were first suggested to be involved in PD pathology by deletion of Dicer specifically in mouse dopaminergic neurons, which resulted in their loss and consequently in reduced locomotion [69]. Levels of miR-133b are significantly decreased in the midbrain of PD patients compared with controls, leading to increased levels of the transcription factor Pitx3, which in turn specifically induces transcription of miR-133b in a negative feedback loop that is important for neurogenesis and function of dopaminergic neurons [69]. Later studies revealed that miRNAs are directly involved in PD pathogenesis, regulating pathways of PD-linked genes and their protein products. Through use of cultured cells, brain enriched miR-7 [70] and miR-153 [71] have been shown to inhibit SNCA mRNA and consequently α-synuclein protein targeting the 3’UTR of SNCA, suggesting that upregulation of miR-7 and miR-153 may represent a viable therapy for PD [71]. Mutant leucine-rich repeat kinase-2 (LRRK2) inhibits let-7 and miR-184* function leading to upregulation of their targets, E2F1 and DP, respectively, ultimately causing LRRK2-linked dopaminergic neuron pathogenesis in Drosophila [72]. Importantly, mutant LRRK2 has been shown to interact with and regulate argonaute protein in both Drosophila brains and cultured cells [72], suggesting global miRNA deregulation. Moreover, miR-205 has recently been shown to be downregulated in the brains of sporadic PD patients, with increased LRRK2 levels [73]. Since miR-205 directly regulates LRRK2 mRNA and protein, downregulation of miR-205 may contribute to pathogenic elevation of LRRK2 and, therefore, overexpression of miR-205 may represent potential therapy for PD [73]. SNPs in the 3’UTR of FGF20 were shown to be regulated by miR-433 in both cultured cells and in PD brains [74] and are associated with elevated levels of both FGF20 and α-synuclein, which has been linked to increased risk of developing PD [74, 75], but the association between FGF20 and PD remains controversial [76, 77].
Circulating miRNAs in peripheral blood were shown to differentiate control individuals from untreated PD patients (miR-1, miR-22, miR-29) and untreated from treated PD patients (miR-16-2, miR-26a2, miR30a) [78]. However, a later study with a substantially larger cohort of plasma samples did not confirm those results and instead showed only miR-331-5p to be significantly upregulated in PD patients [79]. Clearly, differences in sample types and patient cohorts are most likely the reason for these variations and more comprehensive studies are needed to identify the right miRNAs that are present across different PD types. Nevertheless, these results are encouraging and represent a huge step towards easily accessible and reliable biomarkers of PD.
Prion diseases
Prion diseases, also known as transmissible spongiform encephalopathies, are a group of rare fatal neurodegenerative disorders marked by neuronal loss, gliosis and spongiform changes in the brain. Prion diseases include Creutzfeldt-Jakob disease in humans, scrapie in sheeps and goats, chronic wasting disease in deer and elk, and the bovine spongiform encephalopathy in cattle [80]. The disease-causing agent consists of PrPSc, which is an isoform of the host-encoded cellular prion protein PrPC. PrPSc acts as a propagon that is capable of seeding a self-perpetuating reaction to convert PrPC to the pathological conformation of PrPSc within a bioassay system [80], a process termed prion replication.
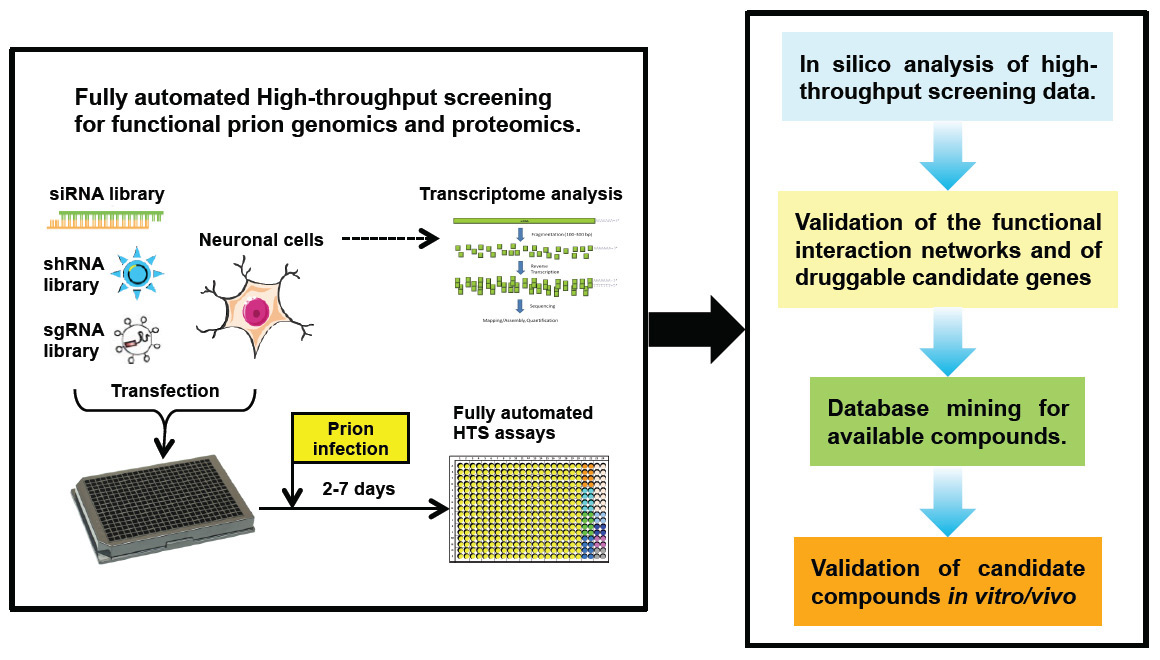
Figure 2
Proposed new strategies for high throughput small RNA screens to discover cell signalling mechanisms and novel therapeutic target for prion disease.
HTS = high throughput screening; sgRNA = single guide RNA; shRNA = short hairpin RNA; siRNA = small interfering RNA
The expression of PrPC is indispensable for prion replication and prion-induced neurodegeneration. However, the intrinsic mechanisms underlying prion replication and subsequent neurodegeneration are largely unknown [81]. Several key questions, including the genes and proteins that regulate endogenous PrPC expression, the machinery of PrPC to PrPSc conversion, and the molecules affecting susceptibility to prion infection and cell-to-cell spread of prions, have not been answered yet. This is mainly because of the dearth of high-throughput technologies that can be used to identify these mechanisms, which would not only greatly improve our basic understanding of prion pathophysiology, but might also uncover novel therapeutic and diagnostic targets. Here we discuss the application of small RNA-based technologies in prion studies.
RNA profile change in neuronal cells upon prion infection
Upon prion infection of neuronal cell lines, the transcriptome profile changes reflect the direct response to prions and could indicate potential pathways leading to prion pathogenesis. Greenwood et al. performed the first study using a complementary DNA (cDNA) microarray and observed strikingly different expression profiles in both ScN2a and ScGT1 cells upon persistent prion infection [82]. Interestingly, many of the differentially regulated genes were also observed to change in other neurodegenerative disorders, suggesting some conserved responses to neurological damage in various neurodegenerative conditions. However, another transcriptional study conducted by Julius et al. analysed the transcriptome of several prion-infected cell lines (N2aPK1, CAD and GT1) under more stringently controlled conditions. By applying high-density oligonucleotide microarray analysis and exhaustive bioinfomatical interrogation of the data, Julius et al. identified very modest differential expression resulting from prion infection [83]. The discrepancy between the two studies may be due to differences in experimental design, cell culture conditions and/or prion infection processes. Among many possible factors, the clonal segregation strategy used in the Greenwood study might have resulted in differential transcriptional responses independent of prion infection.
The transcriptional profile of cell culture might change after prion infection; likewise, the transcriptome of cell lines could also affect the susceptibility to prion infection. Different cell lines have distinct susceptibilities to prion infection. These differences cannot be explained only by the PrPC expression levels, which are required but not sufficient for prion replication [84]. Recently, Marbiah et al. isolated prion-resistant revertants from susceptible cells. By comparing the transcriptome of prion-resistant revertants to that of susceptible cells, Marbiah et al. revealed a gene regulatory network for extracellular matrix remodelling that is associated with prion propagation.
The roles of miRNAs in prion diseases remain unclear. By using microarray and real-time PCR (RT-PCR), Saba et al. profiled miRNA expression changes in the brains of mice intracerebrally inoculated with mouse scrapie. Fifteen miRNAs were found to be deregulated during the disease processes and only one of them had previously been shown to be deregulated in neurodegenerative disease [85], suggesting a prion disease-specific pattern of differentially expressed miRNAs. In addition, a correlation between miRNA expression profile and expression change on putative gene targets was identified. Further studies are required to determine the cell types in which miRNAs are deregulated.
In conclusion, with proper experimental strategies and application of new RNA analysis technologies such as RNA sequencing, we expect that our understanding of the cellular transcriptional response to prion infection will be deepened. The identification of RNA profile changes upon prion infection may have potential use as biomarkers for prion diagnostics and therapeutic targets.
Small RNA-based target-identification and potential therapeutics for prion disease
PrPC is indispensable for prion replication and prion-induced pathogenesis. Mice devoid of PrPCshowed resistance to prion infection [86]. PrPC reduction of 50% (Prnp
+/–heterozygous mice) significantly prolongs the incubation time of prion disease in mouse, while overexpression of PrPC markedly shortens prion disease progression [87]. Therefore, identification of genes involved in PrPC biosynthesis by use of small RNA-based screening and RNA-mediated modulation of PrPC expression accordingly have great potential to mitigate the PrPC-PrPSc conversion, diminish prion-induced neurotoxicity and consequently slow disease progression.
Since Prnp is the main determinant for prion pathogenesis, small RNAs specifically and directly targeting Prnp have been extensively studied for prion therapeutics both in vitro and in vivo. Small interfereing RNA duplexes targeting the Prnp gene effectively suppressed expression of PrPC in prion-infected N2a cells and inhibited PrPSc accumulation [88]. A liposome-siRNA-peptide complex was later developed to suppress PrPCexpression and eliminate PrPScformation in prion-infected N2a cells [89]. Additionally, the vector-based short hairpin RNA (shRNA) was successfully applied to decrease PrPC expression in rabbit kidney epithelial cells [90] and mouse N2a cells [91]. Lentiviral vector delivered shRNA also decreased PrPC levels and extended the prion incubation time in mice [91, 92]. Similar effects were observed in transgenic livestock [93, 94], Therefore, RNA interference (RNAi) based Prnp knockdown represents a promising approach for prion therapeutics. However, there are concerns about RNAi-based therapeutics, such as immunogenicity and off-target induced cytotoxicity. Hence, modified versions of small RNAs or novel small RNAs targeting Prnp may circumvent those pitfalls. Recently, miRNA [95], DNA-based antisense oligonucleotides [96], and RNA aptamers [97, 98] targeting Prnp/PrPC have been shown to prevent prion disease progression.
HTS assays including PrP-FEHTA (PrP-FRET-enabled high throughput assay) [99] and ELISA (enzyme-linked immunosorbent assay) [100] have been applied to search for small compounds that may reduce PrPC expression for prion therapy, albeit no strong candidates have been discovered yet. An alternative strategy is RNAi screening, which is based on RNA-mediated gene silencing that can reduce levels of the mRNA target [101]. RNAi HTS has become a powerful tool for whole transcriptome manipulation and thus discovery of signalling mechanisms and therapeutic strategies in human diseases [101]. Transcriptome wide RNAi screenings (siRNA or shRNA libraries) are likely to uncover genes and proteins that are involved in PrPC biosynthesis. Moreover, the advent of CRISPR-Cas9 mediated single guide RNA (sgRNA) library HTS provides another alternative approach to interrogation of the molecular networks that regulate PrPC biosynthesis. Hits identified by those RNA-based HTS represent potential targets for modulating PrPC expression and therefore therapeutics for prion disease.
In addition, RNA-based HTS can be applied to identify genes and proteins that are involved in prion replication. However, this application is challenging because the conventional quantification of prion infectivity is performed by titration of serial dilutions using mouse bioassays [102] or standard scrapie cell endpoint assays [103]. Both approaches are either protracted or demanding substantial manual work, hence are not applicable for HTS. Therefore, new methodologies and technologies are required to meet the need of a reliable prion infectivity assay complementary to an automated high-throughput system, which will facilitate HTS of entire small RNA libraries including siRNA, shRNA or the newly developed sgRNA libraries, as well as compounds and peptides, with the aim of identifying disease relevant genes, early molecular markers and novel therapeutics (fig. 2). The progress of small RNA-based HTS technology is exciting and may contribute to the discovery of promising therapeutic targets for prion diseases and other neurodegenerative diseases.
Amyotrophic lateral sclerosis and frontotemporal dementia
Amyotrophic lateral sclerosis (ALS; also known as Lou Gehrig’s disease [USA] and motor neuron disease [UK]) is a late onset, progressive and fatal neurodegenerative disease characterised by loss of upper motor neurons in the motor cortex and lower motor neurons in the brain stem and spinal cord. Motor neuron neurodegeneration in ALS leads to muscle atrophy, weakness, fasciculation and spasticity, followed by paralysis and ultimately death, typically due to respiratory failure [104]. First described in 1869 by Jean-Martin Charcot (therefore initially known as a Charcot’s disease), ALS was named based on his observations of distinct loss of axons in the lateral aspect of the spinal cord [105]. Typical onset of the disease is usually in midlife (45-60 years) with an uncompromisingly progressive disease course of 3 to 5 years [106]. Worldwide incidence and prevalence of ALS are 1–2 and 4–6 per 100 000 each year, respectively, with a lifetime ALS risk of 1/350 to 1/1 000 [107, 108] and incidence peak (10–15/100 000/year) between ages 60 and 79 [109]. The vast majority of ALS cases (~90%) are sporadic with no family history. Sporadic ALS includes patients carrying de novo mutations or alleles with incomplete penetrance, or with an incomplete family history. The remaining 10% of ALS cases are caused by autosomal dominant mutations and therefore called familial ALS. Approximately 20% of familial ALS cases are associated with mutations in a gene coding for an antioxidative enzyme, the copper/zinc superoxide dismutase 1 (SOD1) [110]. Recent identification of causative mutations in the 43-kDa transactive response (TAR) DNA-binding protein (TDP-43) [111–5] and fused in sarcoma/translocated in sarcoma (FUS/TLS) [116, 117], which are both involved in RNA processing, in conjunction with the recognition that TDP-43 is the main protein component of the ubiquitinated inclusions in the vast majority of ALS patients [118, 119], led to the idea that aberrant RNA metabolism contributes to ALS pathogenesis [106, 120, 121]. More recently, hexanucleotide expansions in an intronic region of a poorly characterised gene called C9ORF72 (chromosome 9 open reading frame 72) were identified as the most common genetic cause of ALS [122–4]. This finding corroborated the link between ALS and frontotemporal dementia (FTD), which are now recognised as two clinical ends of one disease spectrum [106, 125, 126]. Indeed, it is estimated that about 20% of ALS patients develop signs of frontotemporal dementia [127] and that about 15% of FTD patients develop motor neuron degeneration [126, 127].
FTD is the clinical presentation of frontotemporal lobar degeneration (FTLD), which is a neurodegenerative disease characterised by progressive neuronal loss, predominantly involving the frontal and temporal lobes [128], that primarily affects behaviour, social awareness and language [129]. FTD has only recently been appreciated as one of the leading causes of dementia, particularly in patients younger than 65 years [130], making it the second most common cause of early onset dementia after Alzheimer's disease [131]. Data from the UK show a prevalence of around 15 cases per 100 000 population aged 45–64 years [131]. Approximately 50% of all FTD cases are familial with the microtubule-associated protein tau (MAPT) and progranulin (PGRN) accounting for 10%–20% of FTD cases [126]. Rare cases of FTD are caused by mutations in TDP-43 and FUS/TLS [120] but the strongest genetic link between ALS and FTD is the hexanucleotide expansion in C9ORF72 which is the most common genetic cause of FTD-ALS and accounts for a large number of FTD cases (10%–30%) [122–4, 126].
miRNA miR-9 in (motor) neurons
miRNA is essential for normal function of spinal motor neurons as ablation of Dicer1 in motor neurons induced denervation, muscle atrophy, astrogliosis and motor neuronal loss [132]. Deletion of Dicer in astrocytes led to significant downregulation of astrocyte-specific glutamate transporters GLT-1 and GLAST resulting in non-cell autonomous neuronal dysfunction and degeneration, demonstrating its crucial role in these glial cells [133]. Interestingly, in a mouse model of a loss of miRNA function in spinal motor neurons (MNDicerMUT mice), miRNA-deficient spinal motor neurons showed specific upregulation of the neurofilament heavy subunit (NEFH) that has been previously linked to motor neuron degeneration. Coordinated neurofilament gene expression is necessary for normal neuronal function and upregulation of only NEFH in the motor neurons of MNDicerMUT mice, suggests possible perturbation in the fine-tuning of related genes by miRNAs. Indeed, authors found a single miR-9–binding site on the neurofilament light subunit (NEFL) mRNA and nine different sites in the NEFH mRNA. Furthermore, motor neurons differentiated from mouse embryonic stem cells harbouring an SMN1 mutation revealed a 15-fold decrease in the expression of both miR-9 and miR-9* [132]. Importantly, human ALS neurons derived from induced pluripotent stem cells (iPSCs) (TARDBP A90V mutation) also displayed downregulation of miR-9 and miR-9-2 [134], which may be a common mechanism in several neurodegenerative diseases, including ALS, FTD, spinal muscular atrophy and Huntington’s disease. Indeed, miR-9 and miR-9* have been shown to regulate directly RE1-silencing transcription factor (REST) and CoREST, respectively, which could lead to increased or mislocalised pools of REST thereby dysregulating neuronal genes [135]. In addition, miR-9, among others, was also found to be significantly downregulated in the spinal cord of human sporadic ALS patients [136]. However, this study describes NEFL mRNA suppression by dysregulated miR-146a*, miR-524-5p and miR-582-3p, which are capable of direct binding to NEFL mRNA [136]. Moreover, the same group reported that additional miRNAs that normally stabilise NEFL mRNA, miR-b1336 and miR-b2403, were downregulated in ALS spinal cord, thereby contributing to the loss of NEFL steady-state levels [137]. In contrast to the findings in human ALS samples, miR-9 expression was upregulated in the spinal cord of SOD1G93A mice at early and late symptomatic stages [138], suggesting that this mechanism may vary among different types of ALS with distinct pathologies (primarily TDP-43 pathology in sporadic ALS or TARDBP A90V mutation carriers, versus misfolded SOD1 in the SOD1G93A mice).
TDP-43 and FUS miRNA regulation
The physiological role of the ALS/FTD-linked RNA-binding proteins TDP-43 and FUS has been extensively studied, which showed that they are involved in many aspects of RNA metabolism (reviewed in [120, 121, 126]), including miRNA regulation. Indeed, TDP-43 was shown to associate with Drosha [139] and Dicer complexes [140, 141], an interaction that may be disturbed by ALS-linked mutations that increase the protein half-life of TDP-43 [139], thereby interfering with miRNA biogenesis. In addition, FUS interaction is required for efficient recruitment of Drosha at specific pri-miRNA sites at early stages of transcription thereby stimulating miRNA biogenesis [149].
TDP-43 knockout in cultured cells led to downregulation of let-7b and upregulation of miR-663 expression [142]. Fan and colleagues recently applied transcriptome-wide analysis of TDP-43 binding small RNAs on previously published CLIP-seq data from human and mouse samples [144, 145]. Their analysis identified a novel NRXN1 intron-derived miRNA, called miR-NID1, found in both human and mouse [143]. This miRNA is located in the fifth intron of human neurexin 1 (NRXN1) and four pre-miR-NID1 candidates are located in the introns of protein coding genes in the mouse. Functional analysis by means of nucleocytoplasmic fractionation and RNA immunoprecipitation assays in SY-5YC cells revealed that endogenous TDP-43 is associated with miR-NID1 in both cytoplasm and nuclei and that miR-NID1 represses the expression of NRXN1 through TDP-43 [143].
Overexpression of human wild type FUS in transgenic mouse models has been shown to induce progressive motor neuron degeneration [146] and its deficiency resulted in chromosomal instability and perinatal death [147]. FUS carrying a 3’UTR G48A mutation, which affects the binding site of miR-141/200a, renders FUSmRNA insensitive to repression by these miRNAs [148]. Furthermore, as shown in SK-N-BE neuroblastoma cells, FUS controls miR-141/200a biogenesis suggesting a feed-forward regulatory loop in which FUS increases the expression levels of two miRNAs, which in turn regulate FUS accumulation and an increase in nuclear FUS protein [148]. Moreover, expression of a subset of miRNAs known to have a crucial role in neuronal function, differentiation and synaptogenesis (miR-9, miR-125b and miR-132), is altered upon FUS knockdown in cultured cells [149].
Espression of miR-206 in ALS
The importance of miR-206 expression in ALS was first highlighted by a study on neuromuscular junctions of mutant SOD1 mice [150]. The investigators found that skeletal muscle-specific miR-206 is dramatically increased in the SOD1G93A mouse model of ALS. In this study, they generated miR-206 knockout mice, which were also crossed with transgenic mice expressing low levels of SOD1G93A. The miR-206 knockout mice developed normally but the SOD1G93A mice lacking miR-206 displayed exacerbation of disease symptoms accompanied by accelerated atrophy of skeletal muscle, leading to kyphosis, paralysis and death [150]. It has been shown that axonal branches of surviving motor neurons are partially reinnervating muscles whose innervating motor neurons have already died during the course of ALS. Also, miR-206 slows ALS progression by sensing motor neuron injury and promoting the compensatory regeneration of neuromuscular synapses via induced secretion of fibroblast growth factor binding protein 1 (FGFBP1) from muscle by inhibiting HDAC4 translation [150]. This has been confirmed in patients as miR-23a, miR-29b, miR-206 and miR-455 were all increased in skeletal muscle of ALS patients compared with healthy controls [151]. It was found that miR-23a repressed PGC-1α translation in a 3′UTR dependent manner in vitro and that transgenic mice overexpressing miR-23a had a reduction in PGC-1α, cytochrome-b and COXIV protein levels leading to skeletal muscle mitochondrial dysfunction in ALS [151]. Recently, miR-206 was found to be consistently increased in fast-twitch muscles during the course of the disease in SOD1G93A mice, with highest expression levels towards the end stage [152]. Most importantly, miR-206 was also found to be significantly elevated in circulating blood of both SOD1G93A mice and human ALS patients [152].
Dysregulation of miRNAs in frontotemporal dementia
FTD and ALS are linked clinically, pathologically and molecularly by several lines of evidence and thus represent two ends of a disease spectrum [153]; therefore, a common miRNA biogenesis dysregulation by mutant TDP-43 and FUS can be expected. Nevertheless, progranulin (GRN) mutations are exclusively found to cause FTD and loss of progranulin function causes tau negative FTD with TDP-43 and ubiquitin pathology (FTD-TDP) [154, 155]. Moreover, a common genetic variant (rs5848), located in the 3’UTR of GRN within a binding-site for miR-659, is a major susceptibility factor for this type of FTD, as miR-659 binds the high-risk T allele of rs5848 more efficiently, resulting in enhanced translational inhibition of GRN [156]. Using miRNA array profiling and qPCR validation in both frontal cortex and cerebellum of FTD-TDP patients carrying GRN mutations, the same group found that miR-922, miR-516a-3p, miR-571, miR-548b-5p, and miR-548c-5p are exclusively dysregulated in these patients when compared with FTD-TDP patients without GRN mutations [157]. A genome-wide association study showed that chromosome 7p21 variants within the gene TMEM106B confer increased risk of developing FTD-TDP [158]. The same group later found that TMEM106Bis repressed by miR-132 and miR-212 and that both miRNAs are downregulated in FTD-TDP leading to aberrant overexpression of TMEM106B that affects the distribution and intracellular levels of progranulin, suggesting that the two proteins may act in the same pathogenic pathway in FTD-TDP [159]. Specific SNPs in the TMEM106B gene seem to regulate progranulin levels and thereby the penetrance of FTD in GRN mutation carriers [160]. Recently, TMEM106B has been identified as the first genetic factor modifying disease presentation in C9ORF72 expansion carriers, as homozygosity for the minor rs3173615 allele protects carriers from developing FTD, but not from developing ALS [161]. Furthermore, a SNP that confers increased risk for developing FTD-TDP (major, or T, allele of rs1990622) and which is associated with later age at onset and death in C9ORF72expansion carriers has been also identified [162]. Most recently, a genotype that confers decreased risk for developing FTD-TDP (minor, or C, allele of rs1990622) has been found to be associated with earlier age at onset and death in C9ORF72expansion carriers [134] completing the picture of TMEM106B as a genetic modifier in ALS and FTD.
miRNA overexpression or inhibition as a potential therapy in ALS/FTD
The levels of miR-206 are significantly elevated in muscles [150] and plasma [152] of SOD1G93A mice, and in the plasma of sporadic ALS patients [152]. Since miR-206 senses damage in motor neurons and promotes compensatory mechanisms that lead to regeneration of neuromuscular junctions, its increased expression acts to counteract disease progression in the SOD1G93A mouse model, but ultimately fails to completely reverse disease [150]. Nevertheless, it is possible that overexpression of miR-206 in skeletal muscles could further delay the muscle weakness observed in ALS patients, thereby providing a potential palliation approach.
Recent microarray screens revealed that miR-155 is significantly upregulated in SOD1G93A mouse and human ALS patient spinal cords [163], as well as in peripheral monocytes from ALS patients [164]. Furthermore, downregulation of miR-155 in the CNS of SOD1G93A mice using the antisense oligonucleotide anti-miR-155 resulted in significant extension of survival by 10 days and disease duration by 15 days (38%) in comparison with scrambled oligonucleotide control-treated mice [163]. Given the important proinflammatory function of miR-155 [44–7] and the fact that miR-155 has been found to be upregulated in AD CSF and neocortex ECF [43], it is possible that the elevated miR-155 levels in ALS reflect the ongoing neuroinflammation. As the administration of anti-miR-155 caused global derepression of targets in peritoneal macrophages after intraperitoneal treatment with lipopolysaccharide in mice, and since the anti-sense oligonucleotides were detected in CNS microglia upon intraventricular administration [163], the significant extension of survival of SOD1G93A mice may be associated with reduced non-cell autonomous neuronal toxicity of glial cells [165]. As miR-23a, which is overexpressed in the skeletal muscle of ALS patients, was found to repress PGC-1α translation in a 3′UTR dependent manner both in vitro and in mice, leading to skeletal muscle mitochondrial dysfunction in ALS [151], inhibition of miR-23a in skeletal muscle may represent potential therapy in ALS.
miRNAs as biomarkers in ALS/FTD
Microarray screening of 911 human miRNAs in leucocytes obtained from 8 sporadic ALS patients and 12 healthy controls revealed that miR-451, miR-1275, miR-328-5P, miR-638, miR-149 and miR-665 were downregulated across different gender groups, as well as in all examined samples. Importantly, qPCR analysis of miR-338-3p in 14 sporadic ALS patients and 14 controls confirmed the specific miR-338-3p upregulation in leucocytes of all ALS patients, suggesting that this miRNA could represent an invaluable marker for ALS that could offer early diagnosis through a blood test [167].
A qPCR screen of TDP-43 binding miRNAs in human serum and CSF found that out of the 9 recently discovered TDP-43 binding miRNAs, miR-132-5p, miR-132-3p and miR-143-3p were significantly downregulated, and miR-143-5p and miR-574-5p were significantly upregulated in serum and CSF of matched sporadic ALS patients suggesting a systemic epigenetic dysregulation in ALS [168].
Huntington’s disease
Huntington's disease (HD) is a fatal inherited autosomal dominant neurodegenerative disorder caused by an expansion of the CAG repeat localised in the first exon of the huntingtin gene (HTT or IT-15) [169]. Expression of the mutant huntingtin protein with an expanded poly-glutamine tract [169] induces progressive neurodegenerative changes in the whole brain, with the main pathology observed in the striatum, cortex [170] and white matter [171, 172], consequently leading to progressive motor dysfunction, cognitive decline and psychiatric disturbance [173].
Prevalence of the mutation/disease is about 4–10 per 100 000 in populations of Western European and North American descent, with many more at risk of the disease [170, 174]. CAG repeats longer than 40 are associated with practically full penetrance by the age of 65 years [173, 175]. Longer CAG repeats predict earlier onset of the disease [175], accounting for up to 50%–70% of variance in age of onset [173] with the remaining variance most likely to be attributed to modifying genes (40% of remainder) and the environment (60% of remainder) [176].
Most HD patients have expansions ranging from 40‒55 CAG repeats [170, 175], leading to the onset of the disease symptoms in middle age (40 years in average), whereas juvenile onset (under the age of 20) is associated with CAG repeats of 60 or longer [175]. The disease progresses over 15–20 years from onset and culminates in death [177].
REST-induced mRNA and miRNA transcriptome disruption in Huntington’s disease
It has long been known that the mutant huntingtin disrupts transcription as wild-type huntingtin sequesters the available REST/NRSF (RE1-silencing transcription factor; aka neuronal restrictive silencing factor) in the cytoplasm preventing it from forming the nuclear co-repressor complex at the RE1/NRSE (repressor element 1; aka neuron restrictive silencer element) nuclear site and allowing gene transcription of neuronal genes, while the mutant huntingtin does not [178]. It was therefore hypothesised that in the CNS, huntingtin may represent a general coordinator of neuronal gene transcription [178, 179]. Further research revealed that REST transcriptionally represses the miRNA family that includes miR-124a, one of the most common and highly expressed CNS miRNAs. [180]. During neuronal differentiation, similar to canonical REST-regulated genes, miR-124a expression is only allowed when REST dissociates from its chromatin binding sites. Diminishing miR-124a activity in mature neurons resulted in specific increase of non-neuronal genes, further demonstrating the neuronal specificity of miR-124a [180].
REST-regulated miRNAs in HD thus became widely researched. In the pivotal search for miRNA dysregulation in HD, REST binding sites [181] were compared with the positions of known miRNA genes [182], which identified 17 possible miRNA targets of REST, including 13 mouse orthologous miRNA-RE1 pairs [181]. Chromatin immunoprecipitation assay confirmed that the identified miRNA RE1s are indeed capable of interaction with REST both in vitro and in vivo. MiR-132 was found to be downregulated in brains of both human HD patients and R6/2 mice, the most commonly used model of HD expressing the human HTT exon 1 with 144 CAG repeats under the control of the human HTT promoter. Other miRNAs, however, showed divergent results, such as miR-29a, which is significantly upregulated in HD patients, but downregulated in R6/2 mice and miR-330, which was significantly upregulated in HD samples, but undetectable in R6/2 mice [181]. These data demonstrate that apparent differences could be expected across the samples of different origin, i.e. human HD and animal HD models that apparent differences could be expected across the samples of different origin (i.e., human HD patients and animal HD models).
Changes of miRNA expression levels correspond to target gene expression in Hungtington’s disease and correlate with disease progression
Nevertheless, further research brought significant functional discoveries of how miRNAs are involved in the pathogenesis of HD. REST-regulated miRNAs miR-9, miR-9*, miR-29b, miR-124a (all downregulated), and miR-132 (upregulated) differ significantly with increasing HD grade as detected with qPCR of human brain samples from Brodmann area 4 [135]. Both miR-196a (six-fold) and miR-486 (three-fold) were also found to be significantly upregulated in HD1 grade samples. Importantly, this study revealed that miR-9 targets REST and miR-9* targets CoREST, the components of the REST repressor complex [135]. The authors explained the conflict of their results with those of Johnson et al. [181] by the fact that while they analysed mature miRNAs, Johnson et al. used precursor miRNAs [135]. In an in-vitro study comparing the miRNAs expression in HdH109/109 and Hdh7/7cells with qPCR, Soldati et al. [183] confirmed that many HD-dysregulated miRNAs are directly repressed by elevated levels of REST, including miR-9, miR-9*, miR-29b, miR-124, miR-132, miR-135b which is consistent with previous findings [135, 181], but also previously not reported miR-137 and miR-153, as well as REST-indirectly controlled miR-222. Most importantly, they confirmed that impairment in the widespread regulation of miRNA expression by REST results in increased gene expression of miRNA targets in HD as they show that, for instance, Ak2, Elov1 and Ctdsp2 (miR-124 targets), are upregulated in Hdh109/109 cells [183].
Similarly, in one of the best large animal models for HD, the transgenic HD monkey (rhesus macaque), as well as in human HD brains, miR-128a was found to be significantly downregulated. It has been shown with a luciferase assay that miR-128a regulates the HD canonical signalling genes HIP-1, HTT, SP-1 and GRM5 [184]. Microarray analysis of 352 rhesus miRNAs led to detection of 11 dysregulated miRNAs in the frontal cortex. Four miRNAs with significant target association to the HD canonical pathway were further analysed with qPCR, which validated significant downregulation of miR-181c, miR-128a and miR-133c, and significant upregulation of miR-194 [184]. The same group has also previously discovered that miR-196a indirectly regulates the expression and aggregation of mutant huntingtin both in vitro and in vivo, involving miR-196a-induced alteration of ubiquitin-proteasome system, gliosis, and the cyclic AMP response element-binding protein (CREB) pathway [185].
Regulation of miRNA by p53 in Huntington’s disease
Using frontal cortex and striatal samples isolated from HD patient brains and appropriate controls, REST-regulated, as well as REST-independent miRNAs, were found to be dysregulated by means of next generation sequencing [16]. The latter approach revealed length and sequence heterogeneity for almost all miRNAs (isomiRs) and that the mechanisms governing the expression of identified miRNAs and their corresponding isomiRs are mostly parallel. Importantly, it was also discovered that the promoter region of the downregulated miRNAs in frontal cortex harbour significantly more p53 regulatory motifs [16] and p53 was previously shown to regulate multiple miRNAs [186]. Mutant huntingtin binds P53 and induces its overexpression [187]. In addition, p53 regulates most miRNAs among all transcription factors studied and this can be explained by its overexpression in HD [188]. Therefore, p53 may contribute to the widespread downregulation of miRNA in HD [16, 188].
Chromosomal clusters display similar regulation in Huntington’s disease
Significant upregulation of five miRNAs (miR-10b-5p, miR-196a-5p, miR-196b-5p, miR-615-3p and miR-1247-5p) has been identified with next generation sequencing of samples from HD and normal control brains (prefrontal cortex [Brodmann area 9]) and the differential expression of these miRNAs was further validated with qPCR. Interestingly, miR-10b-5p, miR-196a-5p, miR-196b-5p and miR-615- 3p are strongly related to Hox cluster genes [189]. Moreover, it was found that miRNA in chromosomal clusters exhibited similar expression patterns in HD; for example, let-7a, let-7c, let-7d and let-7e were all downregulated in HD frontal cortex and striatum and miR-30a, miR-30b, miR-30c and miR-30e were upregulated in the same tissues [16]. Additionally, miR-10b-5p, miR-196a-5p, miR-196b-5p and miR-615-3p, which are miRNAs strongly related to Hox cluster genes, were all found to be significantly upregulated in prefrontal cortex of HD patients [189]. Lastly, expression of IsomiRs and the corresponding miRNAs are in most cases parallel [16].
miRNA regulation by stress granules
The interesting hypothesis that mutant huntingtin impairs miRNA-mediated mRNA regulation and local translation, via its accumulation into stress granules together with argonaute 2, an RNA-induced silencing complex (RISC) component, which is required for RNA-mediated gene silencing, has been proposed [189–91]. TDP-43 and FUS accumulate into stress granules as a normal physiological response to stress [192, 193] and it has been hypothesised that the association of TDP-43 and FUS/TLS with stress granules may be an initiating event which, following chronic stress, eventually leads to irreversible pathological aggregation [126]. Taken together, the above highlight the possibility that pathological accumulation followed by coaggregation of these proteins with RISC and/or Drosha and Dicer complexes and multiple mRNAs could represent a common pathological mechanism in both ALS/FTD spectrum diseases and HD.
Dysregulation of miR-196a associates with clinical features of Huntington’s disease and represents potential therapeutic agent in Huntington’s disease
Next generation sequencing data of a relatively large number of patient samples allowed association studies of upregulated miRNAs with clinical features of HD. It was discovered that CAG repeat size correlates with miR-10b-5p and miR-196a-5p, age of motor dysfunction onset with miR-10b-5p, miR-196a-5p and miR-196b-5p, and age at death with miR-10b-5p and miR-615-3p [189]. Previoulsy, miR-196a had been found to be significantly overexpressed in HD1 brains [135] and in mutant huntingtin transfected NT2-derived neurons [166], and its overexpression resulted in downregulation of mutant huntingtin in human iPSC-derived HD neurons and in an HD mouse model [185]. It is, therefore, possible that increased miR-196a expression is an adaptive response promoting neuronal survival [189] but that its endogenous activation in HD is not sufficient to ameliorate the HD pathology [185]. However, overexpression of miR-196a may represent a potential therapeutic approach [185, 189].
miRNAs as biomarkers in HD
In order for miRNAs to become valuable biomarkers of HD onset and/or progression, they obviously need to be detectable and adequately stable. Importantly, human plasma miRNAs were reported to withstand repeated freeze-thaw cycles and, in only small volumes of plasma, p53-regulated miR-34b was found to be elevated in premanifest HD patients as detected with qPCR [166]. An earlier study of HD patient-derived whole blood identified multiple significantly upregulated mRNAs compared with controls [194], but this finding could not be replicated [195]. Next generation sequencing of plasma samples with further qPCR validation of miR-34b expression obtained from a large HD patient cohort is therefore absolutely necessary.
miRNAs in muscle dystrophy
The first evidence for an important role of miRNAs in mammalian myogenesis was shown for the paradigm of myoblast to myotube conversion [196–8]. Subsequently, there was an increasing interest for the regulation of miRNAs in muscle diseases. Muscular dystrophies are a heterogenous class of >30 different inherited myopathies [199] that were an obvious start for this search. They show a high cycling of muscle degeneration and regeneration, satellite cell proliferation, inflammation, adipocyte infiltration and fibrosis, giving many possibilities for miRNA regulation in diverse cell types. Duchenne muscular dystrophy (DMD) is the most prevalent and most severe of the muscular dystrophies, affecting about 1 in 3 500 live males. This X-linked recessive disease is caused by mutations in the dystrophin gene, which encodes for a large protein that connects the extracellular matrix with the cytoskeleton of muscle. This bridging function is thought to be important to stabilise the sarcolemma.
The mdx mouse – a mouse model for muscle dystrophy
The first investigations of altered miRNA expression in dystrophic skeletal muscle were performed in the most commonly applied mouse model for muscle dystrophy, the mdx mouse. These mice carry a stop codon in the dystrophin gene, mimicking the human situation, but the disease is rather mild in the mice possibly due to a higher regenerative capacity compared with humans. McCarthy et al. analysed with qPCR the expression of three muscle-specific miRNAs, miR-206, miR-1 and mi-133, and found a four- to five-fold increase of miR-206 in diaphragm with no major regulation in soleus or plantaris muscles [200]. Muscle-specific miR-1 and miR-133a showed no major regulation in any of the three muscle groups. The induction of miR-206 was most likely posttranscriptional since the primary transcript of this miRNA, pri-miR-206, was also upregulated. The isolated induction of miR-206 in the diaphragm was not completely surprising since the diaphragm represents the most severely affected muscle group in this mouse model. However, it was later shown that miR-206 is induced, while miR-1 is downregulated, also in other muscle groups of the mdx mice [203].
Regulation of miRNAs in patients with muscle dystrophy
Using miRNA microarrays, Eisenberg et al. identified no fewer than 151 miRNAs that are upregulated and 28 that are downregulated in 10 major muscle disorders [201]. Interestingly, with the exception of five miRNAs, the expression of these miRNAs was specific to individual disease groups. A significant correlation between miRNAs and their targets was observed in DMD patients and patients with Miyoshi myopathy, indicating that miRNAs could be also functionally relevant in muscle diseases. Messenger RNA ‒miRNA modules regulated in both muscle diseases were extracellular matrix processes and cytoskeletal organisation, indicating that these could be common functional clusters in dystrophic muscles regulated by miRNAs. Using qPCR, Greco et al. compared the levels of 250 adductor muscle miRNAs in mdx versus wild-type mice and identified 36 miRNAs that were regulated significantly [202]. Importantly, 11 miRNAs were identified as a common miRNA signature between mdx mice and DMD patients including miR-1, which was about three-fold downregulated, and miR-206, which was four- to nine-fold upregulated. Regulation of only three miRNAs was overlapping with the report from Eisenberg et al (miR-222, miR-335, miR-29c). Cacciarelli et al. ran low-density qPCR arrays on mdx and wild-type muscles, and detected as the most prominent changes the downregulation of miR-1, miR-133a, miR-29c and miR-30 and the induction of miR-206 [203]. Downregulation of miR-1 and miR-29c was attributed to transcriptional repression by histone deacetylase 2 (HDAC2). Importantly, this study also addressed the intramuscular localisation of miR-1 and miR-206. Localisation of miR-206 was restricted to newly-formed regenerating myofibres characterised by a centralised nucleus, while miR-1 showed intense signals in all mature fibres. These results indicated that increased expression of miR-206 is caused by the presence of newly formed fibres, while the downregulation of miR-1 is the consequence of a decrease in adult myofibres. The same group later discovered that the muscle-specific miRNAs are released into the blood of Duchenne patients and that their levels correlated with disease severity [204]. Cacchiarelli et al. also identified a nonmuscle specific miRNA, miR-31, that colocalised with miR-206 in regenerating fibres and was also strongly induced in mdx muscles and DMD patient samples [205].
The miR-206 knockout mouse
The first evidence for a functional role of a regulated miRNA in dystrophic muscles in vivo came from Liu et al., who deleted miR-206 in mice during embryonic development and crossed these mutants with the mdx mice [206]. The loss of miR-206 resulted in a more severe dystrophic phenotype in the mdx mice with creatine kinase activity, a marker of muscle damage, being eight-fold higher in the mutants than in the control mice. These results indicate that the upregulation of miR-206 in dystrophic muscle could serve as a protection mechanism to facilitate the formation of new fibres during muscle regeneration. Interestingly, miR-206 was also found to be significantly upregulated in skeletal muscles of the SOD1G93A mouse model of ALS [150, 152], as well as in skeletal muscles of ALS patients [151]. It has been shown that miR-206 promotes compensatory mechanisms that lead to regeneration of neuromuscular junctions, partially reinnervating muscles and resulting in delayed disease progression in the SOD1G93A mouse model [150], further supporting the idea that upregulation of miR-206 in regenerating muscle fibres is protective and its overexpression in skeletal muscles of ALS patients could delay muscle weakness and possibly slow down disease progression.
Potential for miRNAs as biomarkers in muscle dystrophy
Together these studies provide evidence that miRNAs are regulated in dystrophic muscles in animal models and patients. The overlap of regulated miRNAs between these studies is rather small, probably as a result of the different muscle groups that had been investigated or different techniques with which miRNAs were quantified (oligonucleotide arrays, qPCR, northern blotting). However, downregulation of miR-1 and upregulation of miR-206 have been repeatedly confirmed. These studies also demonstrate that miRNAs in muscle tissue or serum could be interesting markers for disease severity in muscle dystrophy. miRNAs in serum could be envisioned as biomarkers to monitor noninvasively clinical trials or to identify patients, who would benefit from interventions. The example of miR-206 highlights the idea that miRNAs could also serve as new therapeutic targets for muscle diseases.
Role of miRNA in cardiovascular disease
Several miRNAs have been identified as critical modulators of cardiovascular development and pathophysiology [207]. The important role of miRNAs in the cardiovascular system has provided novel perspectives on the pathophysiology of cardiovascular disease and has revealed interesting potential novel therapeutic targets [208, 209].
miRNA and cardiac function
Cardiac deletion of enzymes required in the biogenesis of miRNAs, such as Dicer, resulted in dilated cardiomyopathy and premature lethality, supporting a critical role of miRNA biogenesis for cardiac function and structure [210, 211]. By using a high-throughput functional screening approach, Wahlquist et al. have recently identified miR-25 as a potent regulator of calcium uptake kinetics in cardiomyocytes in vitro (out of 875 miRNAs tested) and observed that miR-25 was upregulated in heart failure, in both mice and humans [212]. Moreover, overexpression of miR-25 in vivo resulted in a reduced contractile cardiac function, whereas injection of an antisense oligonucleotide against miR-25 (anti-miR-25) markedly prevented progression of established heart failure in a mouse model and improved cardiac function and survival [212]. Furthermore, miR-34a was reported to be induced in ageing hearts and in-vivo silencing of miR-34a reduced age-associated cardiomyocyte cell death, cardiac fibrosis and promoted recovery of cardiac function after myocardial infarction [213]. Interestingly, miR-34a is upregulated in blood mononuclear cells of patients with sporadic AD [54], which may also represent a consequence of aging.
miRNA in cardiac development and stem cells
Dynamic regulation of miRNA is involved in developmental biology, for example, differentiation of embryonic stem cells towards a cardiomyocyte fate, and in-vitro studies have shown that miR-1, together with miR-499, are upregulated in the differentiation of human embryonic stem cells and cardiac progenitor cells toward cardiomyocytes. [214, 215] Overexpression of these miRNAs enhances differentiation toward a cardiomyocyte fate [214, 215].
miRNA and atherosclerotic vascular disease
Several recent reports have revealed that the human sterol-regulatory-element-binding-protein genes SREBF1 and SREBF2 harbour two intronic miRNAs, miR-33b and miR-33a, respectively, that modulate homeostasis of cholesterol and triglycerides, which are critical for the atherosclerotic disease process [209, 216]. Anti-miR33-treated mice showed reduced atherosclerotic plaque size and lipid content and decreased inflammatory gene expression [217].
Pharmacological inhibition of miR-33 using a subcutaneously injected, locked nucleic acid (LNA)-modified anti-miR in a nonhuman primate metabolic disease model lowered very low-density lipoprotein tryglycerides [218] and resulted in limited atherosclerosis progression in a mouse model of atherosclerosis [217].
We and others have observed a critical role of miR-126, which is particularly expressed in endothelial cells, for ischaemia-induced neovascularisation, for example, after myocardial infarction and in atherosclerotic vascular disease [219, 220]. In patients with chronic heart failure, miR-126 levels were markedly reduced in circulating endothelial repair-promoting cells. Moreover, administration of miR-126 promoted endothelial cell proliferation at predilection sites and reduced experimental atherosclerosis, suggesting that miR-126 may represent a potential novel therapeutic target to prevent progression of atherosclerotic vascular disease, the main cause of death in developed countries [220]. Notably, miR-126 limits leucocyte adherence to endothelial cells, at least in part by suppressing endothelial vascular cell adhesion molecule 1 (VCAM-1) expression, suggesting that miR-126 may limit vascular inflammation [221].
Role of miRNA in the pathogenesis of inflammation
MiRNA in autoimmune and inflammatory diseases
The immune system protects the organism from attack by microorganisms such as bacteria, viruses and parasites. Whereas the local inflammation at the site of infection and elimination of infected cells is necessary for defense of the individual, failure to control inflammation once microorganisms are cleared can have devastating consequences. It is, therefore, important to understand the molecular mechanisms regulating immune reactions and their defects in the various autoimmune diseases. The recognition of noncoding RNAs, in particular miRNAs, as a new principle of regulation of gene-expression, has been a major advance. Dysregulation of miRNA has been linked to cancer, cardiovascular, infectious and autoimmune diseases. Important pathways in the innate as well as the adaptive immune system are under control of miRNAs. As one miRNA has a multitude of targets and acts in a tissue specific way, the complexity of this regulatory system is considerable. Increasing evidence indicates that the expression patterns of miRNAs are dysregulated in a variety of autoimmune diseases. Whereas many different miRNAs have been reported to be either up- or downregulated in specific autoimmune diseases, an impact of these expression changes on the development of autoimmune diseases has been demonstrated only in a few instances so far.
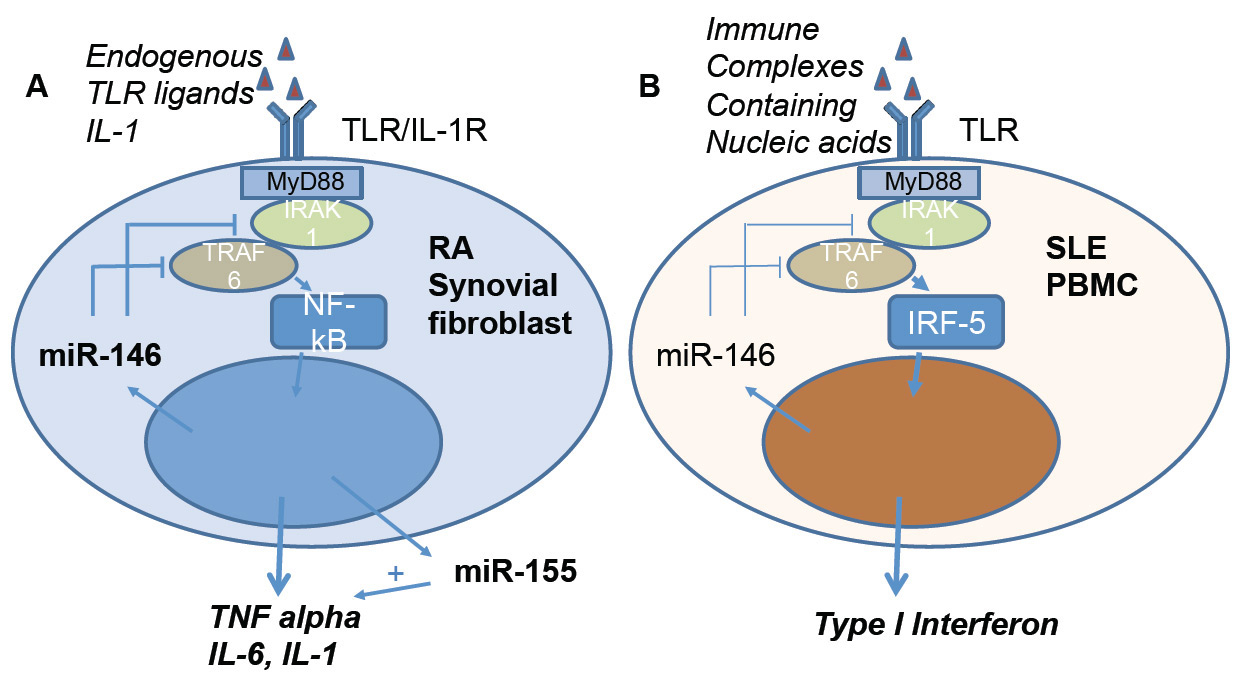
Figure 3
Regulatory role of miR-146 and miR-155 on inflammation in activated synovial fibroblasts of patients with rheumatoid arthritis (A) and on type I interferon production in PBMC of patients with SLE (B). (A) Activation of synovial fibroblasts by endogenous Toll-like receptor (TLR) ligands or interleukin 1 (IL-1) initiates a signalling cascade via nuclear factor kappa B (NF-κB) resulting in expression of proinflammatory cytokines, such as tumour necrosis factor alpha (TNF-α). Overexpression of TNF-α is a hallmark of rheumatoid arthritis. In activated synovial fibroblasts miR-155 expression is induced, further enhancing TNF-α production. In contrast, miR-146 has a modulatory role by inhibiting the signalling molecules interleukin 1 associated receptor kinase 1 (IRAK1) and TNF-receptor accessory factor 6 (TRAF 6). (B) Regulatory role of miR-146 in SLE PBMC. In contrast to the overexpression of miR-146 in RA, in SLE miR-146 levels are decreased, resulting in an increased activation of IRF-5 and expression of type 1 interferon.
IRF-5 = interferon regulatory factor 5; PBMC = peripheral blood mononuclear cells; RA = rheumatoid arthritis; SLE = systemic lupus erythemotosus
Rheumatoid arthritis
First reports on miRNA expression in rheumatoid arthritis appeared in 2008, describing upregulation of miR-155 and -146 in the rheumatoid synovium [222, 223]. Expression of miR-155 was shown to be induced by lipopolysaccharide and polyinosinic:polycytidylic acid (poly-I:C) in a MyD88-dependent fashion [224]. Macrophages overexpressing miR-155-produced significantly more tumour necrosis factor-α (TNF-α) and miR-155 transgenic mice showed increased sensitivity to lipopolysaccharide-induced septic shock, whereas miR-155 deficient mice failed to produce TNF-α [45, 46]. Also, miR-155 deficient mice were shown to be resistant to collagen-induced arthritis, whereby the generation of autoreactive B and T cells was prevented. The example of miR-155 demonstrates that a single miRNA can have a profound impact on the development of an autoimmune disease (fig. 3). Contrary to miR-155, miR-146 was shown to have an anti-inflammatory role. By targeting IRAK-1 and TRAF-6 miR-146 is a negative regulator of NF-κB activation. Accordingly, miR-146 deficient mice develop a fatal immune-mediated disease and tumours in lymphoid organs [47]. Administration of miR-146 in mice with established collagen-induced arthritis reduced bone destruction, but had no significant effect on inflammation [44]. Interestingly, a polymorphism in the 3’UTR of the IRAK-1 gene, a target of miR-146, is associated with the susceptibility to rheumatoid arthritis [225]. A number of additional miRNAs have been shown to be dysregulated in rheumatoid arthritis synovial cells. Most of these studies are however mainly descriptive and provide limited information on the functional impact of the miRNA as well as the regulation of their expression.
Systemic lupus erythematosus
Studies of miRNA in systemic lupus erythematosus (SLE) have focused primarily on miR-146a. In peripheral blood mononuclear cells of pateints with SLE, miR-146 was found to be downregulated, and expression of miR-146 was inversely correlated with the disease activity. Overexpression of miR-146 reduced type I interferon production by targeting IRF-5 and STAT-1 [226]. Therefore it was proposed that deficient miR-146 expression may be one of the causal factors of type I interferon pathway activation, characteristically seen in SLE. In support of this hypothesis, a functional promoter variant of miR-146a was found to confer risk for developing SLE [227]. Treatment of lupus-prone BXSB mice with virus-like particles containing miR-146 significantly reduced autoantibody production and could ameliorate SLE progression [228]. Analyses of expression profiles of miRNA in kidney tissue of patients with SLE revealed that miRNA expression is organ and cell-type specific, as expression differed from peripheral blood mononuclear cells of SLE patients. In contrast to the findings in peripheral blood mononuclear cells, miR-146a was found increased in glomerular tissue. In rodent models of SLE, various additional miRNAs have been found to contribute to disease. Both miR-21 and miR-214 were shown to be upregulated in renal tissue and upon stimulation of rat epithelial cells with transforming growth factor-β (TGF-β) in vitro. Inhibition of TGF-β signalling resulted in a downregulation of miR-21 and -214 and inhibition of epithelial-mesenchymal transition and collagen type I production. This suggests that TGF-β-induced miR-21 and miR-214 expression may contribute to extracellular matrix production and mesangial proliferative glomerulonephritis [229].
miR-21 was also found to be upregulated in systemic sclerosis skin fibroblasts and correlated with disease activity. A profibrotic effect of TGF-β-induced miR21 was postulated by negatively regulating its target Smad7 [230].
Interestingly, it has been shown that patients with connective tissue diseases may produce antibodies against proteins of the miRNA pathway, which localise in the cytoplasm in the so-called GW-bodies. Anti-GWB-antibodies have been identified in patients with SLE, Sjogren’s syndrome and rheumatoid arthritis and seem to be associated with neurological symptoms [231]. The functional relevance of these autoantibodies is still unclear.
Inflammatory bowel disease
In 2008 alterations in miRNA expression in inflammatory bowel disease were described for the first time [232]. Analyses of miRNA expression profiles of intestinal biopsy tissue as well as of peripheral blood mononuclear cells have revealed distinct profiles in ulcerative colitis and Crohn’s disease. However, information on the functional impact of these miRNAs is still lacking to a large extent. Decreased levels of miR-7 in inflamed colonic tissue in Crohn’s disease have been reported to correlate with upregulated expression of CD98, its target, which controls proliferation and differentiation of enterocytes [233]. A recent study has shown overexpression of miR-196 in intestinal epithelial cells in inflamed Crohn’s disease. Interestingly, miR-196 was found to bind to the protective variant of the immunity-related GTPase M (IRGM) but not a mutated IRGM which was shown to be highly associated with Crohn’s disease in European populations [234]. Similarly, it has been demonstrated that a variant of the IL-23R gene, highly associated with inflammatory bowel disease, results in the loss of a binding site for the miRNAs let-7e and let-7f, with increased expression of IL-23R in vitro. These results demonstrate possible links of miRNA dysregulation and pathogenesis of inflammatory bowel disease [235].
In summary, in autoimmune and autoinflammatory diseases a dysregulation of miRNA expression has been demonstrated which can have a profound impact on the pathogenesis of these diseases. Therefore, miRNAs are attractive targets for immunomodulatory therapeutic intervention. Moreover, miRNAs hold great promise as markers of disease activity and prognosis and to guide therapeutic intervention (for review [236]).
Outlook on translation of miRNA research into the clinics
Clearly, basic research provided us with breakthrough discoveries of miRNA biogenesis and the crucial function of miRNAs as genome regulators. Animal research using various mouse disease models was an important step that revealed that miRNAs indeed do change in disease. Later, it was recognised that changes in miRNA levels could be either a consequence or a direct trigger of the disease and it is now well established that changes of levels in numerous miRNAs reflect the disease onset and progress, and that could even be used for presymptomatic diagnosis and prognosis. Moreover, targeting the dysregulated miRNAs represents a new and very potent therapeutic intervention.
It is, however, important to point out that the results obtained from animal models are very often not directly replicated in the clinic. This may be owing to the inherent variability in human samples and it is obvious that all screening conditions (including sample source, size and preparation, screening techniques, data collection, analysis, validation and interpretation) need to be standardised across all patients and study sites in order for the miRNAs to become reliable disease biomarkers.
Unbiased screening using next generation RNA sequencing enables us to screen all known miRNAs, thus eliminating the possibility that the most affected miRNAs would be missed. Finally, given the large number of miRNAs, each regulating hundreds of mRNAs, it is possible that multiple miRNAs will become markers of certain disease processes rather than a single miRNA. Once such data are available, algorithms that would compare them across different patient groups and/or diseases may reveal common molecular mechanisms that would enable us to identify potential asymptomatic, yet already ongoing, pathological processes that may currently be missed during routine diagnostic procedures. This could lead to early therapeutic intervention that may involve direct targeting of the known, pathologically dysregulated miRNA(s), as well as other targets. We foresee that in the future, all this will be feasible from a single drop of body fluid, such as blood or CSF.
Authors’ contribution: MHP and BL contributed equally to this work
References
1 Lander ES, et al, Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):p.860–921.
2 Venter JC, et al. The sequence of the human genome. Science. 2001;291(5507):p.1304–51.
3 Derrien T, et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Research. 2012;22(9):p.1775–89.
4 Berretta JMA. Pervasive transcription constitutes a new level of eukaryotic genome regulation. EMBO reports. 2009;10(9):p.973–82.
5 Clark MB, et al. The Reality of Pervasive Transcription. PLoS Biol. 2011;9(7):p.e1000625.
6 Jacquier A. The complex eukaryotic transcriptome: unexpected pervasive transcription and novel small RNAs. Nat Rev Genet. 2009;10(12):p.833–44.
7 Carthew RW. Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell. 2009;136(4) p.642–55.
8 Barry, G., Integrating the roles of long and small non-coding RNA in brain function and disease. Mol Psychiatry, 2014;19(4):p.410–6.
9 Ng S-Y, et al. Long noncoding RNAs in development and disease of the central nervous system. Trends in Genetics, 2013;29(8):p.461–8.
10 Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75(5):p.843–54.
11 Geisler S. Coller J. RNA in unexpected places: long non-coding RNA functions in diverse cellular contexts. Nat Rev Mol Cell Biol. 2013;14(11):p.699–712.
12 Keniry A, et al. The H19 lincRNA is a developmental reservoir of miR-675 that suppresses growth and Igf1r. Nat Cell Biol. 2012;14(7):p.659–65.
13 Yoon J-H, Abdelmohsen K, Gorospe M. Functional interactions among microRNAs and long noncoding RNAs. Seminars in Cell & Developmental Biology. (0).
14 Friedlander M, et al. Evidence for the biogenesis of more than 1,000 novel human microRNAs. Genome Biology, 2014;15(4):p.R57.
15 Liu C, et al. Effects of genetic variations on microRNA:target interactions. Nucleic Acids Research. 2014.
16 Marti E, et al. A myriad of miRNA variants in control and Huntington's disease brain regions detected by massively parallel sequencing. Nucleic Acids Res. 2010;38(20): p. 7219–35.
17 Salta E. De Strooper B. Non-coding RNAs with essential roles in neurodegenerative disorders. Lancet Neurol. 2012;11(2):p.189–200.
18 Bhattacharya A, Ziebarth JD, Cui Y. PolymiRTS Database 3.0: linking polymorphisms in microRNAs and their target sites with human diseases and biological pathways. Nucleic Acids Research. 2013.
19 Qureshi IA, Mehler MF. Emerging roles of non-coding RNAs in brain evolution, development, plasticity and disease. Nat Rev Neurosci. 2012;13(8):p.528–41.
20 Bravo JA, Dinan TG. MicroRNAs: a novel therapeutic target for schizophrenia. Curr Pharm Des. 2011;17(2):p.176–88.
21 Stenvang J, et al. Inhibition of microRNA function by antimiR oligonucleotides. Silence. 2012;3(1):p.1.
22 Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10Suppl:p.S10–7.
23 Bernstein E, et al. Dicer is essential for mouse development. Nat Genet. 2003;35(3):p.215–7.
24 Davis TH, et al. Conditional loss of Dicer disrupts cellular and tissue morphogenesis in the cortex and hippocampus. J Neurosci. 2008;28(17):p.4322–30.
25 Shin D, et al. Dicer ablation in oligodendrocytes provokes neuronal impairment in mice. Annals of Neurology. 2009;66(6):p.843–57.
26 Christensen M, Schratt GM. microRNA involvement in developmental and functional aspects of the nervous system and in neurological diseases. Neuroscience Letters. 2009;466(2):p.55–62.
27 Makeyev EV, et al. The MicroRNA miR-124 Promotes Neuronal Differentiation by Triggering Brain-Specific Alternative Pre-mRNA Splicing. Molecular Cell. 2007;27(3):p.435–48.
28 Rajasethupathy P, et al. A Role for Neuronal piRNAs in the Epigenetic Control of Memory-Related Synaptic Plasticity. Cell. 2012;149(3):p.693–707.
29 Rogelj B, et al. Contextual fear conditioning regulates the expression of brain-specific small nucleolar RNAs in hippocampus. European Journal of Neuroscience. 2003;18(11):p. 3089–96.
30 Bertram L, Tanzi RE. Thirty years of Alzheimer's disease genetics: the implications of systematic meta-analyses. Nat Rev Neurosci. 2008;9(10):p.768–78.
31 Mayeux R, Stern Y. Epidemiology of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(8).
32 Bateman RJ, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. 2012;367(9):p.795–804.
33 Bertram L, Lill CM, Tanzi RE. The genetics of Alzheimer disease: back to the future. Neuron. 2010;68(2):p.270–81.
34 Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148(6):p. 04–22.
35 Nunez-Iglesias J, et al. Joint genome-wide profiling of miRNA and mRNA expression in Alzheimer's disease cortex reveals altered miRNA regulation. PLoS One. 2010;5(2):p.e8898.
36 Cogswell JP, et al. Identification of miRNA changes in Alzheimer's disease brain and CSF yields putative biomarkers and insights into disease pathways. J Alzheimers Dis. 2008;14(1):p.27–41.
37 Wang WX, et al. The expression of microRNA miR-107 decreases early in Alzheimer's disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. J Neurosci. 2008;28(5):p.1213–23.
38 Liu CG, et al. MicroRNA-384 regulates both amyloid precursor protein and beta-secretase expression and is a potential biomarker for Alzheimer's disease. Int J Mol Med. 2014;34(1):p.160–6.
39 Vilardo E, et al. MicroRNA-101 regulates amyloid precursor protein expression in hippocampal neurons. J Biol Chem. 2010;285(24):p.18344–51.
40 Long JM, Lahiri DK. MicroRNA-101 downregulates Alzheimer's amyloid-beta precursor protein levels in human cell cultures and is differentially expressed. Biochem Biophys Res Commun. 2011;404(4):p.889–95.
41 Delay C, et al. Alzheimer-specific variants in the 3'UTR of Amyloid precursor protein affect microRNA function. Mol Neurodegener. 2011;6:p.70.
42 Smith P, et al. In vivo regulation of amyloid precursor protein neuronal splicing by microRNAs. J Neurochem. 2011;116(2): p. 240–7.
43 Alexandrov PN, et al. microRNA (miRNA) speciation in Alzheimer's disease (AD) cerebrospinal fluid (CSF) and extracellular fluid (ECF). Int J Biochem Mol Biol. 2012;3(4):p.365–73.
44 Nakasa T, et al. The inhibitory effect of microRNA-146a expression on bone destruction in collagen-induced arthritis. Arthritis Rheum. 2011;63(6):p.1582–90.
45 Thai TH, et al. Regulation of the germinal center response by microRNA-155. Science. 2007;316(5824):p.604–8.
46 Bala S, et al. Up-regulation of microRNA-155 in macrophages contributes to increased tumor necrosis factor {alpha} (TNF{alpha}) production via increased mRNA half-life in alcoholic liver disease. J Biol Chem. 2011;286(2):p.1436–44.
47 Boldin MP, et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J Exp Med. 2011;208(6):p.1189–201.
48 Zhu HC, et al. MicroRNA-195 downregulates Alzheimer's disease amyloid-beta production by targeting BACE1. Brain Res Bull. 2012;88(6):p.596–601.
49 Boissonneault V, et al. MicroRNA-298 and microRNA-328 regulate expression of mouse beta-amyloid precursor protein-converting enzyme 1. J Biol Chem. 2009;284(4):p.1971–81.
50 Hebert SS, et al. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer's disease correlates with increased BACE1/beta-secretase expression. Proc Natl Acad Sci U S A. 2008;105(17):p.6415–20.
51 Fang M, et al. The miR-124 regulates the expression of BACE1/beta-secretase correlated with cell death in Alzheimer's disease. Toxicol Lett. 2012;209(1):p.94–105.
52 Zong Y. et al. miR-29c regulates BACE1 protein expression. Brain Res. 2011;1395:p.108–15.
53 Shioya M, et al. Aberrant microRNA expression in the brains of neurodegenerative diseases: miR-29a decreased in Alzheimer disease brains targets neurone navigator 3. Neuropathol Appl Neurobiol. 2010;36(4):p.320–30.
54 Schipper HM, et al. MicroRNA expression in Alzheimer blood mononuclear cells. Gene Regul Syst Bio. 2007.1:p.263–74.
55 Dickson JR, et al. Alternative polyadenylation and miR-34 family members regulate tau expression. J Neurochem. 2013;127(6):p.739–49.
56 Absalon S, et al. MiR-26b, upregulated in Alzheimer's disease, activates cell cycle entry, tau-phosphorylation, and apoptosis in postmitotic neurons. J Neurosci. 2013;33(37):p.14645–59.
57 Lau P, et al. Alteration of the microRNA network during the progression of Alzheimer's disease. EMBO Mol Med. 2013.5(10):p.1613–34.
58 Lukiw WJ. Micro-RNA speciation in fetal, adult and Alzheimer's disease hippocampus. Neuroreport. 2007;18(3):p.297–300.
59 Kumar P, et al. Circulating miRNA biomarkers for Alzheimer's disease. PLoS One. 2013;8(7):p.e69807.
60 Shtilbans A, Henchcliffe C. Biomarkers in Parkinson's disease: an update. Curr Opin Neurol. 2012;25(4):p.460–5.
61 Dawson TM Dawson VL, Molecular pathways of neurodegeneration in Parkinson's disease. Science. 2003;302(5646):p.819–22.
62 Spillantini MG, et al. Alpha-synuclein in Lewy bodies. Nature. 1997;388(6645):p.839–40.
63 Saiki S, Sato S, Hattori N. Molecular pathogenesis of Parkinson's disease: update. J Neurol Neurosurg Psychiatry. 2012;83(4):p.430–6.
64 Gasser T. Molecular pathogenesis of Parkinson disease: insights from genetic studies. Expert Rev Mol Med. 2009;11:p.e22.
65 Davis GC, et al. Chronic Parkinsonism secondary to intravenous injection of meperidine analogues. Psychiatry Res. 1979;1(3):p.249–54.
66 Langston JW, et al. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219(4587):p.979–80.
67 Langston JW Ballard Jr. PA, Parkinson's disease in a chemist working with 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. N Engl J Med. 1983;309(5):p.310.
68 Dolezalova D, et al. Pig models of neurodegenerative disorders: Utilization in cell replacement-based preclinical safety and efficacy studies. J Comp Neurol. 2014;522(12):p.2784–801.
69 Kim J, et al. A MicroRNA feedback circuit in midbrain dopamine neurons. Science. 2007;317(5842):p.1220–4.
70 Junn E, et al. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc Natl Acad Sci U S A. 2009;106(31):p.13052–7.
71 Doxakis E. Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153. J Biol Chem. 2010;285(17):p.12726–34.
72 Gehrke S, et al. Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature. 2010;466(7306):p.637–41.
73 Cho HJ, et al. MicroRNA-205 regulates the expression of Parkinson's disease-related leucine-rich repeat kinase 2 protein. Hum Mol Genet. 201322(3):p.608–20.
74 Wang G, et al. Variation in the miRNA-433 binding site of FGF20 confers risk for Parkinson disease by overexpression of alpha-synuclein. Am J Hum Genet. 2008;82(2):p.283–9.
75 van der Walt JM, et al. Fibroblast growth factor 20 polymorphisms and haplotypes strongly influence risk of Parkinson disease. Am J Hum Genet. 2004;74(6):p.1121–7.
76 de Mena L, et al. FGF20 rs12720208 SNP and microRNA-433 variation: no association with Parkinson's disease in Spanish patients. Neurosci Lett. 2010;479(1):p.22–5.
77 Wider C, et al. FGF20 and Parkinson's disease: no evidence of association or pathogenicity via alpha-synuclein expression. Mov Disord. 2009;24(3):p.455–9.
78 Margis R, Margis R, Rieder CR. Identification of blood microRNAs associated to Parkinsonis disease. J Biotechnol. 2011;152(3):p.96–101.
79 Cardo LF, et al. Profile of microRNAs in the plasma of Parkinson's disease patients and healthy controls. J Neurol. 2013;260(5):p.1420–2.
80 Aguzzi A, Nuvolone M, Zhu C. The immunobiology of prion diseases. Nat Rev Immunol. 2013;13(12):p.888–902.
81 Weissmann C. Birth of a prion: Spontaneous generation revisited. Cell. 2005;122(2):p.165–8.
82 Greenwood AD, et al. Cell line dependent RNA expression profiles of prion-infected mouse neuronal cells. J Mol Biol. 2005;349(3):p.487–500.
83 Julius C, et al. Transcriptional stability of cultured cells upon prion infection. J Mol Biol. 2008;375(5):p.1222–33.
84 Mahal SP, et al. Prion strain discrimination in cell culture: The cell panel assay. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(52):p.20908–13.
85 Saba R, et al. A miRNA signature of prion induced neurodegeneration. PLoS One. 2008;3(11):p.e3652.
86 Bueler H, et al. Mice Devoid of Prp Are Resistant to Scrapie. Cell. 1993;73(7):p.1339–47.
87 Manson JC, et al. Prp Gene Dosage Determines the Timing but Not the Final Intensity or Distribution of Lesions in Scrapie Pathology. Neurodegeneration. 1994;3(4):p.331–40.
88 Daude N, Marella M, Chabry J. Specific inhibition of pathological prion protein accumulation by small interfering RNAs. J Cell Sci. 2003;116(Pt 13):p.2775–9.
89 Pulford B, et al. Liposome-siRNA-peptide complexes cross the blood-brain barrier and significantly decrease PrP on neuronal cells and PrP in infected cell cultures. PLoS One. 2010;5(6):p.e11085.
90 Tilly G, et al. Efficient and specific down-regulation of prion protein expression by RNAi. Biochem Biophys Res Commun. 2003;305(3):p.548–51.
91 Pfeifer A, et al. Lentivector-mediated RNAi efficiently suppresses prion protein and prolongs survival of scrapie-infected mice. J Clin Invest. 2006;116(12):p. 3204–10.
92 White MD, et al. Single treatment with RNAi against prion protein rescues early neuronal dysfunction and prolongs survival in mice with prion disease. Proc Natl Acad Sci U S A. 2008;105(29):p.10238–43.
93 Golding MC, et al. Suppression of prion protein in livestock by RNA interference. Proc Natl Acad Sci U S A. 2006;103(14):p.5285–90.
94 Wongsrikeao P. et al. Combination of the somatic cell nuclear transfer method and RNAi technology for the production of a prion gene-knockdown calf using plasmid vectors harboring the U6 or tRNA promoter. Prion. 2011;5(1):p.39–46.
95 Kang SG, et al. Establishment and characterization of Prnp knockdown neuroblastoma cells using dual microRNA-mediated RNA interference. Prion. 2011;5(2):p.93–102.
96 Nazor Friberg K, et al. Intracerebral Infusion of Antisense Oligonucleotides Into Prion-infected Mice. Mol Ther Nucleic Acids. 2012;1:p.e9.
97 Weiss S, et al. RNA aptamers specifically interact with the prion protein PrP. J Virol. 1997;71(11):p.8790–7.
98 Proske D, et al. Prion-protein-specific aptamer reduces PrPSc formation. Chembiochem. 2002;3(8):p.717–25.
99 Karapetyan YE, et al. Unique drug screening approach for prion diseases identifies tacrolimus and astemizole as antiprion agents. Proc Natl Acad Sci U S A. 2013;110(17):p.7044–9.
100 Silber BM, et al. Novel compounds lowering the cellular isoform of the human prion protein in cultured human cells. Bioorg Med Chem. 2014;22(6):p.1960–72.
101 Mohr S, Bakal C, Perrimon N. Genomic screening with RNAi: results and challenges. Annu Rev Biochem. 2010;79:p.37–64.
102 Prusiner SB, et al. Measurement of the Scrapie Agent Using an Incubation-Time Interval Assay. Annals of Neurology. 1982;11(4):p.3538.
103 Klohn PC, et al. A quantitative, highly sensitive cell-based infectivity assay for mouse scrapie prions. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(20):p.11666–71.
104 Rothstein JD. Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann Neurol. 2009;65:Suppl1p.S3–9.
105 Boillee S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52(1):p.39–59.
106 Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci. 2013;14(4):p.248–64.
107 Kiernan MC, et al. Amyotrophic lateral sclerosis. Lancet. 2011;377(9769):p.942–55.
108 Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006;7(9):p.710–23.
109 Logroscino G, et al. Incidence of amyotrophic lateral sclerosis in southern Italy: a population based study. J Neurol Neurosurg Psychiatry, 2005;76(8):p.1094–8.
110 Haidet-Phillips AM, et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol. 2011;29(9):p.824–8.
111 Gitcho MA, et al. TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol. 2008;63(4):p.535–8.
112 Sreedharan J, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319(5870):p.1668–72.
113 Kabashi E, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40(5):p.572–4.
114 Yokoseki A, et al. TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann Neurol. 2008;63(4):p.538–42.
115 Van Deerlin VM, et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. The Lancet Neurology. 2008;7(5):p.409–16.
116 Kwiatkowski Jr. TJ, et al, Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323(5918):p.1205–8.
117 Vance C, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323(5918):p.1208–11.
118 Arai T, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351(3):p.602–11.
119 Neumann M, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):p.130–3.
120 Lagier-Tourenne C, Polymenidou M, Cleveland DW. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet. 2010;19(R1):p.R46–64.
121 Polymenidou M, et al. Misregulated RNA processing in amyotrophic lateral sclerosis. Brain Res. 2012;1462:p.3–15.
122 DeJesus-Hernandez, M, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):p.245–56.
123 Gijselinck I, et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 2012;11(1):p.54–65.
124 Renton A.E, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):p.257–68.
125 Laferriere F, Polymenidou M. Advances and challenges in understanding the multifaceted pathogenesis of amyotrophic lateral sclerosis. Swiss Med Wkly. 2015;145:p.w14054.
126 Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79(3):p.416–38.
127 Cruts M, et al. Current insights into the C9orf72 repeat expansion diseases of the FTLD/ALS spectrum. Trends Neurosci. 2013;36(8):p.450–9.
128 Snowden JS, Neary D, Mann DM. Frontotemporal dementia. Br J Psychiatry. 2002;180:p.140–3.
129 Sleegers K, Cruts M, Van Broeckhoven C. Molecular pathways of frontotemporal lobar degeneration. Annu Rev Neurosci. 2010;33:p.71–88.
130 Rabinovici GD, Miller BL. Frontotemporal lobar degeneration: epidemiology, pathophysiology, diagnosis and management. CNS Drugs. 2010;24(5):p.375–98.
131 Piguet O, et al. Behavioural-variant frontotemporal dementia: diagnosis, clinical staging, and management. Lancet Neurol. 2011;10(2):p.162–72.
132 Haramati S. et al., miRNA malfunction causes spinal motor neuron disease. Proc Natl Acad Sci U S A. 2010;107(29):p.13111–6.
133 Tao J, et al. Deletion of astroglial Dicer causes non-cell-autonomous neuronal dysfunction and degeneration. J Neurosci. 2011;31(22):p.8306–19.
134 Zhang Z, et al. Downregulation of microRNA-9 in iPSC-derived neurons of FTD/ALS patients with TDP-43 mutations. PLoS One. 2013;8(10):p.e76055.
135 Packer AN, et al. The bifunctional microRNA miR-9/miR-9* regulates REST and CoREST and is downregulated in Huntington's disease. J Neurosci. 2008;28(53):p.14341–6.
136 Campos-Melo D, et al. Altered microRNA expression profile in Amyotrophic Lateral Sclerosis: a role in the regulation of NFL mRNA levels. Mol Brain. 2013;6:p.26.
137 Ishtiaq M, et al. Analysis of novel NEFL mRNA targeting microRNAs in amyotrophic lateral sclerosis. PLoS One. 2014;9(1):p.e85653.
138 Zhou FH, et al. miRNA-9 expression is upregulated in the spinal cord of G93A-SOD1 transgenic mice. International Journal of Clinical and Experimental Pathology. 2013;6(9):p.1826–38.
139 Ling SC, et al. ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proc Natl Acad Sci U S A. 2010;107(30):p.13318–23.
140 Freibaum BD, et al. Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J Proteome Res. 2010;9(2):p.1104–20.
141 Kawahara Y, Mieda-Sato A. TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc Natl Acad Sci U S A. 2012;109(9):p.3347–52.
142 Buratti E, et al. Nuclear factor TDP-43 can affect selected microRNA levels. FEBS J. 2010;277(10):p.2268–81.
143 Fan Z, Chen X, Chen R. Transcriptome-wide analysis of TDP-43 binding small RNAs identifies miR-NID1 (miR-8485), a novel miRNA that represses NRXN1 expression. Genomics. 2014;103(1):p.76–82.
144 Tollervey JR, et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci. 2011;14(4): p. 452–8.
145 Polymenidou M, et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci. 2011;14(4):p.459–68.
146 Mitchell JC, et al. Overexpression of human wild-type FUS causes progressive motor neuron degeneration in an age- and dose-dependent fashion. Acta Neuropathol. 2013;125(2):p.273–88.
147 Hicks GG, et al. Fus deficiency in mice results in defective B-lymphocyte development and activation, high levels of chromosomal instability and perinatal death. Nat Genet. 2000;24(2):p.175–9.
148 Dini Modigliani S, et al. An ALS-associated mutation in the FUS 3'-UTR disrupts a microRNA-FUS regulatory circuitry. Nat Commun. 2014;5 p 4335.
149 Morlando M, et al. FUS stimulates microRNA biogenesis by facilitating co-transcriptional Drosha recruitment. EMBO J. 2012;31(24):p.4502–10.
150 Williams AH, et al. MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science. 2009;326(5959):p.1549–54.
151 Russell AP, et al. Disruption of skeletal muscle mitochondrial network genes and miRNAs in amyotrophic lateral sclerosis. Neurobiology of Disease. 2013;49:p.107–17.
152 Toivonen JM, et al. MicroRNA-206: a potential circulating biomarker candidate for amyotrophic lateral sclerosis. PLoS One. 2014;9(2):p.e89065.
153 Gascon E, Gao FB. The emerging roles of microRNAs in the pathogenesis of frontotemporal dementia-amyotrophic lateral sclerosis (FTD-ALS) spectrum disorders. J Neurogenet. 2014;28(1–2):p.30–40.
154 Baker M, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442(7105):p.916–9.
155 Cruts M, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442(7105):p.920-4.
156 Rademakers R, et al. Common variation in the miR-659 binding-site of GRN is a major risk factor for TDP43-positive frontotemporal dementia. Hum Mol Genet. 2008;17(23):p.3631–42.
157 Kocerha J, et al. Altered microRNA expression in frontotemporal lobar degeneration with TDP-43 pathology caused by progranulin mutations. BMC Genomics. 2011;12:p.527.
158 Van Deerlin VM, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet. 2010;42(3):p.234–9.
159 Chen-Plotkin AS, et al. TMEM106B, the risk gene for frontotemporal dementia, is regulated by the microRNA-132/212 cluster and affects progranulin pathways. J Neurosci. 2012;32(33):p.11213–27.
160 Finch, N, et al. TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology. 2011;76(5):p.467–74.
161 van Blitterswijk M, et al. TMEM106B protects C9ORF72 expansion carriers against frontotemporal dementia. Acta Neuropathol. 2014;127(3):p.397–406.
162 Gallagher MD, et al. TMEM106B is a genetic modifier of frontotemporal lobar degeneration with C9orf72 hexanucleotide repeat expansions. Acta Neuropathol. 2014;127(3):p.407;18.
163 Koval ED, et al. Method for widespread microRNA-155 inhibition prolongs survival in ALS-model mice. Hum Mol Genet. 2013;22(20):p.4127–35.
164 Butovsky O, et al. Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J Clin Invest. 2012;122(9):p.3063–87.
165 Cardoso AL, et al. miR-155 modulates microglia-mediated immune response by down-regulating SOCS-1 and promoting cytokine and nitric oxide production. Immunology. 2012;135(1):p.73–88.
166 Gaughwin PM, et al. Hsa-miR-34b is a plasma-stable microRNA that is elevated in pre-manifest Huntington's disease. Hum Mol Genet. 2011;20(11):p.2225–37.
167 De Felice B, et al. A miRNA signature in leukocytes from sporadic amyotrophic lateral sclerosis. Gene. 2012;508(1):p.35–40.
168 Freischmidt A, et al. Systemic dysregulation of TDP-43 binding microRNAs in amyotrophic lateral sclerosis. Acta Neuropathol Commun. 2013;1(1):p.42.
169 A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell. 1993;72(6):p.971–83.
170 Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57(5):p.369–84.
171 Dumas EM, et al. Early changes in white matter pathways of the sensorimotor cortex in premanifest Huntington's disease. Hum Brain Mapp. 2012;33(1):p.203–12.
172 Tabrizi SJ, et al. Potential endpoints for clinical trials in premanifest and early Huntington's disease in the TRACK-HD study: analysis of 24 month observational data. Lancet Neurol. 2012;11(1):p.42–53.
173 Ross CA, Tabrizi SJ. Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10(1):p.83–98.
174 Tabrizi SJ, et al. Biological and clinical changes in premanifest and early stage Huntington's disease in the TRACK-HD study: the 12-month longitudinal analysis. Lancet Neurol. 2011;10(1):p.31–42.
175 Langbehn DR, et al. A new model for prediction of the age of onset and penetrance for Huntington's disease based on CAG length. Clin Genet. 2004;65(4):p.267–77.
176 Wexler NS, Res UVC. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington's disease age of onset. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(10):p.3498–503.
177 Novak MJ, Tabrizi SJ. Huntington's disease. BMJ. 2010;340:p.c3109.
178 Zuccato C, et al. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet. 2003;35(1):p.76–83.
179 Cattaneo E, Zuccato C, Tartari M. Normal huntingtin function: an alternative approach to Huntington's disease. Nat Rev Neurosci. 2005;6(12):p.919–30.
180 Conaco C, et al. Reciprocal actions of REST and a microRNA promote neuronal identity. Proc Natl Acad Sci U S A. 2006;103(7):p.2422–7.
181 Johnson R, et al. A microRNA-based gene dysregulation pathway in Huntington's disease. Neurobiol Dis. 2008;29(3):p.438–45.
182 Griffiths-Jones S. The microRNA Registry. Nucleic Acids Res. 2004;32(Database issue):p.D109–11.
183 Soldati C, et al. Dysregulation of REST-regulated coding and non-coding RNAs in a cellular model of Huntington's disease. J Neurochem. 2013;124(3):p.418–30.
184 Kocerha J, et al. microRNA-128a dysregulation in transgenic Huntington's disease monkeys. Mol Brain. 2014;7:p.46.
185 Cheng PH, et al. miR-196a ameliorates phenotypes of Huntington disease in cell, transgenic mouse, and induced pluripotent stem cell models. Am J Hum Genet. 2013;93(2):p.306–12.
186 Tarasov V, et al. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle. 2007;6(13):p.1586–93.
187 Bae BI, et al. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington's disease. Neuron. 2005;47(1):p.29–41.
188 Sinha M, Mukhopadhyay S, Bhattacharyya NP. Mechanism(s) of alteration of micro RNA expressions in Huntington's disease and their possible contributions to the observed cellular and molecular dysfunctions in the disease. Neuromolecular Med. 2012;4(4):p.221–43.
189 Hoss AG, et al. MicroRNAs located in the Hox gene clusters are implicated in huntington's disease pathogenesis. PLoS Genet. 2014;10(2):p.e1004188.
190 Savas JN, et al. A role for huntington disease protein in dendritic RNA granules. J Biol Chem. 2010;285(17):p.13142–53.
191 Savas JN, et al. Huntington's disease protein contributes to RNA-mediated gene silencing through association with Argonaute and P bodies. Proc Natl Acad Sci U S A. 2008;105(31):p.10820–5.
192 Andersson MK, et al. The multifunctional FUS, EWS and TAF15 proto-oncoproteins show cell type-specific expression patterns and involvement in cell spreading and stress response. BMC Cell Biol. 2008;9:p.37.
193 Liu-Yesucevitz L, et al. Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One. 2010;5(10):p.e13250.
194 Borovecki F, et al, Genome-wide expression profiling of human blood reveals biomarkers for Huntington's disease. Proc Natl Acad Sci U S A. 2005;102(31):p.11023–8.
195 Runne, H, et al. Analysis of potential transcriptomic biomarkers for Huntington's disease in peripheral blood. Proc Natl Acad Sci U S A. 2007;104(36):p.14424–9.
196 Chen JF, et al. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet. 2006;38(2):p.228–33.
197 Kim HK, et al. Muscle-specific microRNA miR-206 promotes muscle differentiation. J Cell Biol. 2006;174(5):p.677–87.
198 Naguibneva I, et al. The microRNA miR-181 targets the homeobox protein Hox-A11 during mammalian myoblast differentiation. Nat Cell Biol. 2006;8(3):p.278–84.
199 Davies KE, Nowak KJ. Molecular mechanisms of muscular dystrophies: old and new players. Nat Rev Mol Cell Biol. 2006;7(10):p.762–73.
200 McCarthy JJ, Esser KA, Andrade FH. MicroRNA-206 is overexpressed in the diaphragm but not the hindlimb muscle of mdx mouse. Am J Physiol Cell Physiol. 2007;293(1):p.C451–7.
201 Eisenberg I, et al. Distinctive patterns of microRNA expression in primary muscular disorders. Proc Natl Acad Sci U S A. 2007;104(43):p.17016–21.
202 Greco S, et al. Common micro-RNA signature in skeletal muscle damage and regeneration induced by Duchenne muscular dystrophy and acute ischemia. FASEB J. 2009;23(10):p3335–46.
203 Cacchiarelli D, et al. MicroRNAs involved in molecular circuitries relevant for the Duchenne muscular dystrophy pathogenesis are controlled by the dystrophin/nNOS pathway. Cell Metab. 2010;12(4):p.341–51.
204 Cacchiarelli D, et al. miRNAs as serum biomarkers for Duchenne muscular dystrophy. EMBO Mol Med. 2011;3(5):p.258–65.
205 Cacchiarelli D, et al. miR-31 modulates dystrophin expression: new implications for Duchenne muscular dystrophy therapy. EMBO Rep. 2011;12(2):p.136–41.
206 Liu N, et al. microRNA-206 promotes skeletal muscle regeneration and delays progression of Duchenne muscular dystrophy in mice. J Clin Invest. 2012;122(6):p.2054–65.
207 Small EM, Olson EN. Pervasive roles of microRNAs in cardiovascular biology. Nature. 2011;469(7330):p.336–42.
208 Jakob P, Landmesser U. Role of microRNAs in stem/progenitor cells and cardiovascular repair. Cardiovasc Res. 2012;93(4):p.614–22.
209 van Rooij E, Kauppinen S. Development of microRNA therapeutics is coming of age. EMBO Mol Med. 2014;6(7):p.851–64.
210 Chen JF, et al. Targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(6):p.2111–6.
211 Rao PK, et al. Loss of cardiac microRNA-mediated regulation leads to dilated cardiomyopathy and heart failure. Circulation research. 2009;105(6):p.585–94.
212 Wahlquist C, et al. Inhibition of miR-25 improves cardiac contractility in the failing heart. Nature. 2014;508(7497):p.531–5.
213 Boon RA, et al. MicroRNA-34a regulates cardiac ageing and function. Nature. 2013;495(7439):p.107–10.
214 Wilson KD, et al. Dynamic microRNA expression programs during cardiac differentiation of human embryonic stem cells: role for miR-499. Circulation. Cardiovascular genetics. 2010;3(5):p.426–35.
215 Sluijter JP, et al. MicroRNA-1 and -499 regulate differentiation and proliferation in human-derived cardiomyocyte progenitor cells. Arteriosclerosis, thrombosis, and vascular biology. 2010;30(4):p.859–68.
216 Rayner KJ, et al. MiR-33 contributes to the regulation of cholesterol homeostasis. Science. 2010;328(5985):p.1570–3.
217 Rayner KJ, et al. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J Clin Invest. 2011;121(7):p.2921–31.
218 Rayner KJ, et al. Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL triglycerides. Nature. 2011;478(7369):p.404–7.
219 Jakob P, et al. Loss of angiomiR-126 and 130a in angiogenic early outgrowth cells from patients with chronic heart failure: role for impaired in vivo neovascularization and cardiac repair capacity. Circulation. 2012;126(25):p.2962–75.
220 Schober A, et al. MicroRNA-126-5p promotes endothelial proliferation and limits atherosclerosis by suppressing Dlk1. Nat Med. 2014;20(4):p.368–76.
221 Harris TA, et al. MicroRNA-126 regulates endothelial expression of vascular cell adhesion molecule 1. Proc Natl Acad Sci U S A. 2008;105(5):p.1516–21.
222 Nakasa T, et al. Expression of microRNA-146 in rheumatoid arthritis synovial tissue. Arthritis Rheum. 2008;58(5):p.1284–92.
223 Stanczyk J, et al. Altered expression of MicroRNA in synovial fibroblasts and synovial tissue in rheumatoid arthritis. Arthritis Rheum. 2008;58(4):p.1001–9.
224 O'Connell RM, et al. Sustained expression of microRNA-155 in hematopoietic stem cells causes a myeloproliferative disorder. J Exp Med. 2008;205(3):p.585–94.
225 Chatzikyriakidou A, et al. A polymorphism in the 3'-UTR of interleukin-1 receptor-associated kinase (IRAK1), a target gene of miR-146a, is associated with rheumatoid arthritis susceptibility. Joint Bone Spine. 2010;77(5):p.411–3.
226 Tang Y, et al. MicroRNA-146A contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum. 2009;60(4):p.1065–75.
227 Luo X, et al. A functional variant in microRNA-146a promoter modulates its expression and confers disease risk for systemic lupus erythematosus. PLoS Genet. 2011;7(6):p.e1002128.
228 Pan Y, et al. MS2 VLP-based delivery of microRNA-146a inhibits autoantibody production in lupus-prone mice. Int J Nanomedicine. 2012;7:p.5957–67.
229 Denby L, et al. miR-21 and miR-214 are consistently modulated during renal injury in rodent models. Am J Pathol. 2011;179(2):p.661–72.
230 Altorok N, et al. Epigenetics, the holy grail in the pathogenesis of systemic sclerosis. Rheumatology (Oxford). 2014.
231 Bhanji RA, et al. Clinical and serological features of patients with autoantibodies to GW/P bodies. Clinical Immunology. 2007;125(3):p.247–56.
232 Wu F, et al. MicroRNAs are differentially expressed in ulcerative colitis and alter expression of macrophage inflammatory peptide-2 alpha. Gastroenterology. 2008;135(5):p.1624–35e24.
233 Nguyen HT, et al. MicroRNA-7 modulates CD98 expression during intestinal epithelial cell differentiation. J Biol Chem. 2010;285(2):p.1479–89.
234 Brest P, et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn's disease. Nat Genet. 2011;43(3):p.242–5.
235 Zwiers A, et al. Cutting edge: a variant of the IL-23R gene associated with inflammatory bowel disease induces loss of microRNA regulation and enhanced protein production. J Immunol. 2012;188(4):p.1573–7.
236 Duroux-Richard I, Jorgensen C, and F. Apparailly, miRNAs and rheumatoid arthritis - promising novel biomarkers. Swiss Med Wkly. 2011;141:p.w13175.
237 Martins M, et al. Convergence of miRNA expression profiling, alpha-synuclein interacton and GWAS in Parkinson's disease. PLoS One. 2011;6(10):p.e25443.