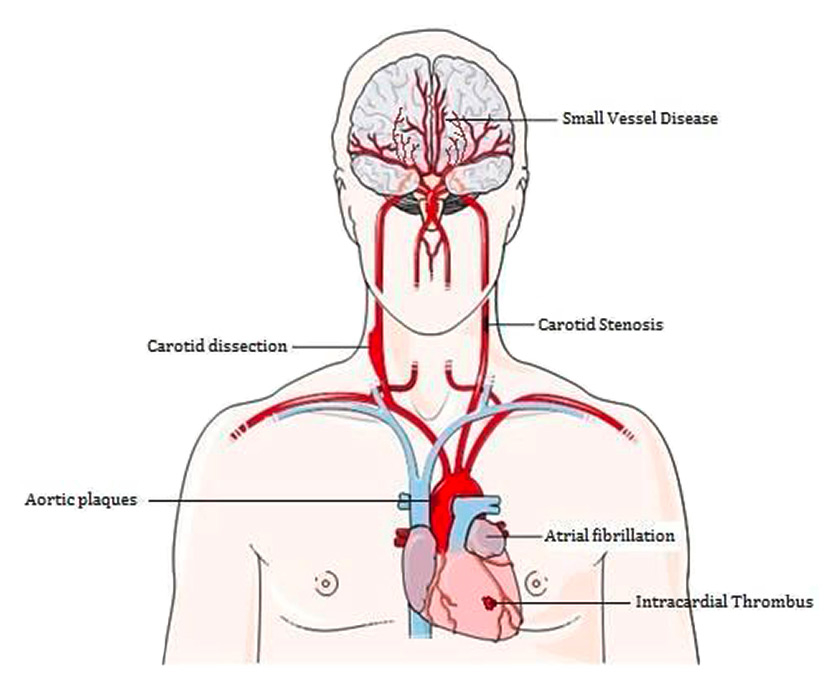
Figure 1
Examples of stroke aetiologies (imaging material adapted from Servier Medical Art).
DOI: https://doi.org/10.4414/smw.2015.14138
Stroke is a leading cause of death worldwide. In the United States of America every year about 795,000 people suffer from a new or recurrent stroke. In Switzerland, stroke incidence has been estimated at 150/100,000 inhabitants [4]. In 2010 direct and indirect costs were $315.4 billion in the United States of America for cardiovascular disease and stroke [1, 5].
Figure 1
Examples of stroke aetiologies (imaging material adapted from Servier Medical Art).
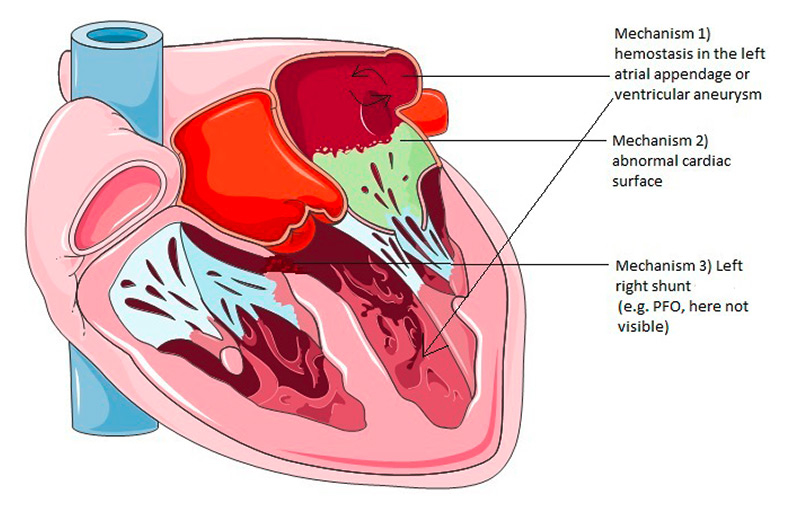
Figure 2
Most important mechanisms for the formation of cardiac embolism (imaging material adapted from Servier Medical Art).
PFO = patent foramen ovale
Stroke can be categorised into ischaemic and haemorrhagic stroke. Ischaemic stroke accounts for approximately 87% whereas haemorrhagic stroke accounts for 13% of all strokes [6].
Ischaemic stroke itself is a heterogeneous disease and different pathological events can lead to a common final ischaemic cascade within the brain (see fig. 1). In a European population of incidence stroke, 30.2% were due to cardioembolism, 25.8% due to small artery/vessel occlusion (SVO) and 15.3% due to large artery atherosclerosis (LAA). The cause of ischaemic strokes remained undetermined in 25% to 39% of patients even after thorough examination [7]. Moreover the risk of recurrence differs between the different etiological subtypes. The highest recurrence rate was found in cardioembolic strokes (22%), in the same study the recurrence rate for LAA was 10% [7].
In any case of ischaemic stroke it is important to reduce the overall risk factors that led to the disease with, for example, blood-pressure treatment, antidiabetic agents, antiplatelet therapy and lipid-lowering agents in the case of atherosclerosis, heart disease or metabolic disorders. There are some aetiologies where there is, however, a specific treatment indicated.
If the stroke is due to a cardioembolic event in patients with atrial fibrillation, it has been shown in several studies that these patients clearly benefit from oral anticoagulation. In a first randomised controlled trial – the WARS trial ‒ warfarin was superior to placebo or aspirin in reducing stroke rates in patients suffering from atrial fibrillation [8, 9]. Over the years, meta-analysis data confirmed that warfarin reduces the occurrence of stroke in patients with atrial fibrillation by 64%. After that, several recent studies followed, proving a relevant risk reduction of stroke recurrence also for novel oral anticoagulants (direct thrombin inhibitors or factor Xa inhibitors) such as abixaban, dabigatran, rivoroxaban or edoxaban [10–13].
In the case of large vessel disease, if a patient suffers from a 70% to 99% stenosis without near-occlusion, carotid surgery (carotid endartectomy) showed an absolute risk reduction of 16% (p <0.001) [14]. Among patients with symptomatic carotid stenosis, the 4-year rate of stroke or death was clearly reduced to 8.0% with stenting and to 6.4% with endarterectomy [15]. Overall, the recurrence rate was reduced compared with “natural history” rates for both intervention types; however, endarterectomy performed slightly better than stenting with the exception of some subgroups.
Because of the aetiological heterogeneity and the fact that prognosis, risk of recurrence and management options differ greatly between subtypes, it is important to identify rapidly and accurately the underlying pathological mechanism leading to ischaemic stroke.
Various classifications of ischaemic strokes have been developed. The TOAST (Trial of Org 10172 in Acute Stroke Treatment) classification [16] is the most commonly used classification system. Based on the patient’s neurological symptoms, imaging of the brain using computed tomography or magnet resonance imaging (CT/MRI) and other diagnostic tests, including standard 24-hour electrocardiography (ECG), echocardiography and ultrasound of extra- and intracranial arteries, the most likely aetiology is determined.
The TOAST classification divides ischaemic strokes into the following subtypes:
Ischaemic stroke due to large artery atherosclerosis, cardioembolism, small vessel occlusion, ischaemic stroke of undetermined aetiology and ischaemic stroke of other determined aetiology [16].
Currently, at least two newer sub-classification systems, the SSS-TOAST [17] and the A-S-C-O classification [18], are available. These newly developed classification systems are advancing the accuracy of ischaemic stroke subtype diagnosis by taking into account the level of diagnostic evidence in order to devise the “most likely mechanism” in the presence of multiple potential overlapping causes (i.e. SSS-TOAST classification) or the “phenotype”, which categorises stroke patients according to a combination of their aetiological characteristics (ASCO). These advanced systems, however, are not yet widely used in clinical routine, they are more complex (though more precise) and so far no data have been published on their impact on implementation of secondary prevention and clinical management in clinical routine.
Recently, a new approach [19] was suggested by scientists to how the former group of “cryptogenic strokes” should be defined and treated. Originally, patients were categorised as having “cryptogenic strokes”, a subgroup within the “undetermined” group according to the TOAST criteria, if even after extensive evaluation no aetiology was determined. They proposed that most of these types of stroke are of an embolic nature, they called this embolic stroke of undetermined source (ESUS). Per definition an ESUS has to be a nonlacunar brain infarction without a proximal arterial stenosis or a major-risk cardioembolic source that would present a clear indication for anticoagulation. They furthermore recommended anticoagulation therapy in these patients and currently there are several trials ongoing, which aim to prove that these patients might benefit from the novel anticoagulants.
In the initial phase of atherosclerosis the endothelium is injured. This leads to a dysfunctional endothelium. More and more low density lipoproteins (LDLs) are deposited in the intima part of the artery. The accumulation of LDL induces endothelial cells to produce chemokines. These chemokines (e.g. monocyte chemotactic protein-1 [MCP-1]) attract monocytes and change them into macrophages. The macrophages enter into the intimal part of the vessel. Endothelial cells and macrophages oxidise LDL. Macrophages which take up oxidised LDL then turn into foam cells. The foam cells increase in size, unable to metabolise the lipoproteins, and finally die. This in turn leads to inflammation of the lipid plaques. The inflammatory reaction finally leads to destabilisation of the plaque and potentially to plaque rupture, inducing an occlusion of the blood vessel further upstream, and consequently to ischaemia of brain tissue [20]. Moreover the diameter of the atherosclerotic vessel locally decreases and this stenosis can then lead to hypoperfusion of the upstream cerebrovascular tissue.
According to the TOAST classification, an ischaemic stroke is due to large artery atherosclerosis if either a major brain supplying artery or major cortical artery shows a >50% stenosis or occlusion and the infarction is located in the corresponding vessel territory. Other competing reasons have to be unlikely. These pathological changes (>50% stenosis) in an intra- or extracranial artery has to be clearly shown by ultrasound or imaging to consolidate the diagnosis of ischaemic stroke due to large artery atherosclerosis [16].
The beginning of the pathophysiological process in ischaemic strokes due to large artery atherosclerosis is of an inflammatory nature. As a consequence, blood biomarkers indicating inflammatory processes in vessels might be promising aetiological biomarker candidates.
Virchow’s triad, including hypercoagulability, endothelial injury and stasis of blood flow, is commonly known to lead to blood clotting. Prothrombin is activated by tissue factors and then changed into thrombin. Thrombin cuts fibrinogen into fibrin. The fibrin molecules react with each other and build cross-linked fibrin. Cross-linked fibrin then interacts with thrombocytes, which themselves adhere to the fibrin, building a thrombus. A thrombus moved by blood flow is called an embolus. In ischaemic stroke due to cardioembolism, the source of the ischaemic event is an embolic occlusion of a brain vessel and the origin of the embolus is located in the vicinity of the heart.
There are mainly three mechanisms of embolism coming from the heart (fig. 2):
1. local haemostasis in a cardiac chamber which leads to thrombosis (e.g. left atrial septal aneurysm and left ventricular aneurysm or, most important, atrial fibrillation);
2. material released from an abnormal cardiac surface (e.g. calcific degeneration);
3. abnormal passage between the venous and arterial system in the heart which allows venous material to end up in the arterial circulation (e.g. patent foramen ovale).
Major sources for cardioembolism are atrial fibrillation, mitral stenosis, prosthetic valves, infective endocarditis, marantic endocarditis, atrial myxoma, acute myocardial infarction, left ventricular thrombosis, left ventricular aneurysm and left ventricular failure. Minor sources are patent foramen ovale, atrial septal aneurysm, atrial or ventricular septal defects, calcific aortic stenosis, mitral annular calcification and fibroelastoma [21]. In the case of atrial fibrillation ‒ the most common source of cardioembolism – supraventricular tachyarrhythmia causes uncoordinated muscle contractions leading to a sluggish blood flow in the atrium, with the consequence that coagulation thrombi are produced, consisting of mostly fibrin and platelets [22].
Ischaemic stroke due to cardioembolism usually comes with greater mortality and morbidity because of the occlusion of mainly large intracranial arteries [22]. Moreover the highest recurrent rate in ischaemic stroke among all subgroups is found in cardioembolism [7].
According to the TOAST criteria a cardiac source of embolism must be detected to verify whether it is an ischaemic stroke due to cardioembolism. In case there are no such findings, clinical signs and imaging of the patient are very similar to those patients suffering from large artery atherosclerosis. In that peculiar case blood biomarker may add important information to better identify a cardioembolic source.
Ischaemic stroke due to small vessel disease (SVD) is mainly due to arteriosclerotic SVD and sporadic and hereditary cerebral amyloid angiopathy [23].
Arteriosclerotic SVD can be attributed to several pathomechanisms: fibrinoid necrosis, lipohyalinosis, microatheroma, microaneurysms and segmental arterial disorganisation.
In the case of arteriosclerosis as the cause of small vessel disease, the number of smooth muscle cells in the tunica media is reduced, the blood vessel wall thickens through deposition of fibro-hyaline material and consequently the vessel lumen decreases, leading to an undersupply to the surrounding cerebrovascular tissue. Arteriosclerotic SVD is most often associated with systemic hypertension [23].
In sporadic and hereditary cerebral amyloid angiopathy, a progressive accumulation of βA4 immunoreactive amyloid proteins in the wall of small and medium arteries and arterioles can lead to occlusion of the vessel [23].
The most important risk factors for a small vessel stroke are hypertension, diabetes and smoking [24]. According to the TOAST criteria, a stroke is due to small vessel disease, if a cardioembolic source has been excluded and the absence of a carotid or vertebral stenosis of >50% of the vessel diameter has been shown. Occlusion of small vessels causes small infarcts, termed lacunes, most commonly located in the caudate, putamen, external capsule, internal capsule, corona radiata, pontine tegmentum and thalamus. Furthermore, the diameter of the typical subcortical infarction has to be ≤1.5 cm in diameter [16].
There are several rather rare diseases, which also lead through distinct mechanisms to cerebral ischaemia (table 1). According to the TOAST criteria, they belong to the huge subgroup classified as “other determined aetiologies”. According to the TOAST criteria [16], patients suffering from an ischaemic stroke in this subgroup should have a CT or MRI imaging finding of ischaemic stroke, furthermore the specific cause of the stroke should be revealed by blood tests or arteriography, and cardioembolism or large artery atherosclerosis should be excluded by diagnostic testing. The ischaemic stroke can be of any size and at any location in the brain.
Some examples of this category are strokes due to systemic vasculitis as it is, for example, the case in patients with lupus erythematosus, other causes are cervical artery dissections or some underlying haematological disorders. Since this aetiological subgroup contains a plethora of different diseases, including some very rare causes, we provide the reader with only an overview of some important exponents:
Unfortunately, the aetiology of ischaemic stroke remains undetermined in up to 25–39% of patients [3], thus up to at least ¼ of ischaemic stroke patients lack potential adequate secondary prevention treatment.
According to the TOAST criteria the following patient groups are classified as such when even after extensive evaluation no aetiology is determined (cryptogenic) or the cause has not been found because the evaluation was only cursory or the physician determined two or more possible causes, making a final diagnosis impossible [16].
If we could reduce the proportion of undetermined strokes with the help of biomarkers, recurrence rate might be reduced substantially.
The final common pathway of all ischaemic strokes is the obstruction of arterial flow to the brain. Brain tissue depends on a constant supply of oxygen and glucose. In the case of ischaemic stroke this supply is decreased or even abolished. The impact on neurones depends on the size, location and duration of the ischaemia. After 5 to 10 minutes of local depletion of glucose and oxygen, neurones are harmed irreversibly. The inadequate supply of energy can lead to a deterioration of the ion gradient. The neuronal and non-neuronal cells depolarise. Thereby the brain cell loses potassium in exchange for sodium, chloride and calcium ions. This electrolyte imbalance leads to a water inflow into the cell and results in rapid swelling of the neurones and glia cells ‒ cytotoxic oedema. The harmed cell now releases excitatory neurotransmitters such as glutamate. Glutamate interacts with several cell membrane proteins and, as a result of excessive cell depolarisation, many enzymes are activated, which now release oxygen free radicals. These oxygen free radicals react with intra- and extracellular proteins and thereby finally lead to cell necrosis. Oxygen free radicals may even induce programed cell death (apoptosis) in damaged cells [25].
| Table 1: Examples of “Other determined ischaemic strokes”. Adapted from Handbook of Clinical Neurology, Volume 93, Pages 1–982 (2008) Stroke Part II: Clinical Manifestations and Pathogenesis, Edited by Marc Fisher (ISBN: 978–0–444–52004–3). | ||
| Haematological disease: | Vasculitis and vasculopathies: | Cervical artery dissection |
| – Protein C, S, Z deficiencies | – Giant cell arteriitis / polymyalgia rheumatica | – Idiopathic |
| – Factor V Leiden mutation | – Kawasaki syndrome | – Traumatic |
| – Plasminogen deficiency | –Takayasu’s arteritis | – Marfan’s syndrom |
| – Essential thrombocythaemia | – Polyarteritis nodosa | – Fibromuscular dysplasia |
| – Von Willebrand’s disease | – Lupus erythematosus | – Ehlers-Danlos syndrome |
| – Antiphospholipid syndrome | – Isolated central nervous system angiitis | |
| – Behçet disease | ||
| Infections: | Drug Abuse: | |
| – Human immunodeficiency virus, varicella zoster virus | – Cocaine | |
| – Lyme disease | – Amphetamine | |
| – Syphilis | – Ephedrine | |
| – Infective endocarditis | – Phenylpropanolamine | |
| – Malaria | ||
| Table 2: Aetiological blood biomarkers of large artery atherosclerosis (LAA) strokes. | |||||
| Blood biomarker LAA marker | Description | Potential clinical application | Study reference | Size | Study design |
| Decorin | Matrix proteoglycan, component of connective tissue, binds to type I collagen fibrils and is involved in matrix assembly (cell cycle) | Diagnosis of LAA in acute ischaemic stroke | Xu et al. 2012 [28] | 65 patients, 23 LAA | Case control study |
| CD62P | P-selectin, stimulated by thrombin, recruitment of leucocytes in inflammation, binding of thrombocytes | Diagnosis of LAA in acute ischaemic stroke | Tsai et al. 2009 [29] | 54 patients, 22 LAA | Case control study |
| CD63 | Cell surface protein, involved in adhesion of blood cells to endothelial cells | Diagnosis of LAA in acute ischaemic stroke | Tsai et al. 2009 [29] | 54 patients, 22 LAA | Cohort study |
| CXCL16 | Scavenger receptor and chemokine, involved in oxLDL uptake in monocytes, enhances atheriosclerosis | Diagnosis of LAA in acute ischaemic stroke | Ma et al. 2014 [37] | 227 patients, 86 LAA | Cohort study |
| LP PLA2 | Platelet activating factor, increases inflammatory reaction | Diagnosis of LAA in acute ischaemic stroke | Katan et al. 2014 [40] | 1,946 patients, 381 LAA | Cohort study |
| Delgado et al. 2012 [41] | 166 TIA patients, 38 LAA | Cohort study | |||
| LIGHT | Tumour necrosis factor ligand, proinflammatory prothrombotic effects on tissue, induces atherosclerosis | Differentiation of LAA in acute stroke compared with healthy controls | Liu et al. 2008 [43] | 62 patients, 20 LAA | Case control study |
| sCD40L | Binds CD40 on APCs, promotes inflammatory action on endothelial cells | Differentiation of vessel disease from CE origin | Oberheiden et al. 2012 [32] | 51 patients, 11 LAA | Cohort study |
| Wang et al. 2013 [31] | 119 patients, 30 LAA | Cohort study | |||
| Total serum cholesterol | Hydrophobic sterol molecule, found in cell membranes, induces atherosclerosis? | Diagnosis of LAA in men in acute ischaemic stroke | Cui et al. 2012 [44] | 33,469 patients, 612 ischaemic stroke, 107 LAA | Cohort study |
| Serum lipoprotein A | Lipoprotein, involved in atherosclerosis | Diagnosis of LAA in ischaemic stroke | Kim et al. 2010 [45] | 1,012 patients, 281 LAA | Cohort study |
| Adiponectin | Adipocytokine of adipose tissue, reduction of atherosclerotic development, improve insulin resistance | Diagnosis of LAA in acute ischaemic stroke | Kuwashiro et al. 2014 [49] | 342 patients, 34 LAA | Cohort study |
| C-reactive protein | Inflammatory marker | Diagnosis of LAA in noncardioembolic ischaemic stroke | Zeng et al. 2013 [46] | 99 patients with noncardioembolic stroke, 52 LAA | Cohort study |
| Ladenvall et al. 2006 [47] | 1,200 patients, 73 LAA | Cohort study | |||
| APC = antigen presenting cell; CD = cluster of differentiation; CXCL16 = C-C-C motif chemokinreceptor 16; LIGHT = lymphotoxin-like inducible protein that competes with glycoprotein D for binding herpesvirus entry mediator on T lymphocytes; LP PLA2 = lipoprotein-associated phospholipase A2; oxLDL = oxidised low density lipoprotein; TIA = transient ischaemic attack | |||||
| Table 3: Table of BNP/NTproBNP studies. | |||||
| Author | Biomarker | Cut-off (pg/ml) | AUC | Sensitivity (%) | Specificty (%) |
| Tamura et al. 2012 [54] | BNP | 90 | 0.892 | 85 | 78 |
| Montaner et al. 2008 [55] | BNP | 76 | 0.75 | 72 | 68 |
| Zhixin et al. 2013 [58] | BNP | 55.50 | 0.896 | 75 | 88.7 |
| Yukiiri et al. 2008 [59] | BNP | 77 | 0.763 | 75.8 | 76.8 |
| Shibazaki et al. 2009 [57] | BNP | 140.0 | 0.87 | 80.5 | 80.5 |
| Fonseca et al. 2011 [61] | NTproBNP | 265 | 0.92 | 94.4 | 72.9 |
| Hajsadeghi et al. 2013 [62] | NTproBNP | 342 | 0.882 | 93 | 75 |
| Rodriguez-Yanez et al. 2009 [63] | NTproBNP | 360 | 0.921 | 87 | 83 |
| AUC = area under the concentration-time curve; BNP = brain natriuretic peptide; NTproBNP = N-terminal probrain natriuretic peptide | |||||
Aetiological blood biomarkers are associated with physiological or pathophysiological processes in the human body leading to an ischaemic stroke. Ideally, they should be reliable and simple to measure; they should help scientists to gather further meaningful information about the aetiology of ischaemic stroke. Specifically, they should improve the identification of patients who need specific secondary preventive treatment and thereby allow faster initiation of treatments, ultimately leading to a better outcome.
The markers we describe in the following paragraphs were identified by a PubMed search. We collected data from studies, written in English, on aetiological blood biomarkers in ischaemic stroke patients, listed in PubMed up to October 2014. Search terms were: stroke / ischaemic stroke, cerebrovascular disease, etiology/aetiology, blood biomarker / biomarker. Among a total of 1,268 manuscripts, we selected 117 possible relevant papers according to their heading in the title. The title of the paper had to be a relevant hint to our paper topic. After reading the abstracts of these 117 papers, we finally selected 31 manuscripts with detailed information about blood biomarkers and their significant association with a specific ischaemic stroke aetiology compared with other aetiologies. We then categorised the biomarkers by each aetiology according to the TOAST classification system. Since the field of biomarkers in the setting of stroke aetiology is rather a new site of interest for scientists, so far there are only a few reviews on this topic. Currently, an interesting, comprehensive review has been published in this field [26].
Table 2 summarises the characteristics of the following aetiological blood biomarkers of LAA strokes.
Decorin is a matrix proteoglycan. It is a component of connective tissue. Decorin binds to type 1 collagen fibrils and is involved in matrix assembly. It is associated with the ability to modify plaque phenotype by reducing inflammation. Decorin reduced the activity of macrophages, the activity of gelatinase and the collagen plaque content in a mouse model [27].
In a small case control study [28], decorin levels in patients with ischaemic stroke (n = 35) were lower compared with healthy controls (n = 30). Moreover, among stroke patients decorin levels were about 1.3 lower in large artery atherosclerosis (n = 23) (average: approximately 7,200 pg/ml), than in other ischaemic stroke subtypes (n = 12) (average: approximately 9,200 pg/ml) when measured <3 days after symptom onset.
Cluster of differentiation (CD) 62P, or P selectin as it is called, is a cell adhesion protein found on blood vessel endothelium and on activated platelets. It plays a role in the initial recruitment of leucocytes in an inflammatory process such as atherosclerosis.
In a case control study [29], the percentage of platelets expressing CD62P in patients with ischaemic stroke of noncardioembolic origin (n = 54, median percentage of platelets expressing P selectin: 5.4%) were compared with healthy controls (n = 28, median number of platelets expressing P selectin: 3.77%). They then compared patients with ischaemic stroke due to large artery atherosclerosis with patients suffering from ischaemic stroke due to small vessel disease. P selectin was measured at three different time-points (<48 h, 7 d, 30 d, 90 d after symptom onset). The percentage of circulating platelets expressing P selectin was increased two-fold (p <0.05) from onset to 3 months after the event in patients with ischaemic stroke due to large artery atherosclerosis (n = 22, median percentage of platelets expressing P selectin: 9.9%) compared with ischaemic stroke due to small vessel disease (n = 32, median percentage of platelets expressing P selectin: 4,9%).
CD63 (cluster of differentiation 63) is a cell surface glycoprotein and marker. CD63 levels are elevated in inflammatory processes. To enable platelets to aggregate, they have to be activated thus CD63 might contribute to atherothrombosis and atherothrombotic stroke [30].
In a small case control study [29] CD63 levels in patients with acute ischaemic stroke of noncardioembolic origin (n = 54, median percentage of platelets expressing CD63: 2.33%) and healthy controls (n = 28, median percentage of platelets expressing CD63: 1.15%) were measured. CD63 levels were 1.52 times higher (p <0.05) in patients with acute ischaemic stroke due to large artery atherosclerosis (n = 22, median percentage of platelets expressing CD63: 3.2%) than in cases due to small vessel disease (n = 32, median percentage of platelets expressing CD63: 2.1%) if measured 48 hours after symptom onset. Afterwards (7, 30 and 90 d after symptom onset) there was no significant difference detectable.
The soluble CD40 ligand belongs to the tumour necrosis factor superfamily (TNFSF) family. It binds to the CD40 receptor on antigen-presenting cells. It promotes inflammatory activation on endothelial cells [31]. Thus it is prominently involved in vessel inflammation [30].
In a small cohort study [32] levels of soluble CD40L were significantly higher in patients with large artery atherosclerotic stroke (n = 11, 165.01 ± 73.76 pg/ml; p <0.047) compared with patients suffering from stroke of cardioembolic origin (n = 17, 75.33 ± 30.72 pg/ml), but levels did not differ significantly between patients suffering from stroke due to large artery atherosclerosis and patients suffering from stroke due to small vessel disease (n = 13).
In another small study [31], scientists compared ischaemic stroke patients (n = 82) with normal healthy individuals (n = 20) and patients who had carotid artery stenosis without stroke (n = 17). Soluble CD40L levels were higher in patients suffering from stroke either because of large artery atherosclerosis (8.41 ± 2.89 ng/ml) or small vessel disease (7.41 ± 3.49 ng/ml), compared with healthy controls (0.56 ± 0.40 ng/ml) and compared with patients with carotid stenosis without stroke (3.71 ± 1.19 ng/ml). In the same study, the mean fluorescence intensity of CD40L expression on peripheral blood monocytes was significantly higher in patients suffering from large artery atherosclerosis (26.87 ± 9.43) compared with all other subgroups (normal controls 7.65 ± 2.64, carotid artery stenosis 14.59 ± 2.18, small vessel disease 7.08 ± 3.09, cardioembolism 7.94 ± 2.46).
CXCL16 (C-C-C motif chemokinreceptor 16), also known as SR-PSOX (scavenger receptor that binds phosphatidylserine and oxidised lipoprotein), is a chemokine and works as a scavenger receptor [33, 34]. In atherosclerotic plaques high levels of CXCL16 were found. It is seen to be involved in the uptake of oxidised low-density lipoprotein (LDL) in monocytes and thereby enhancing atherosclerosis [33, 35, 36].
In a small cohort study [37], scientists compared CXCL16 levels in patients who suffered from ischaemic stroke due to LAA (n = 86) or SVD (n = 67) and healthy controls (n = 74). Mean CXCL16 levels were higher in SVD (c = 2.13 ng/ml, p <0.05) and LAA (c = 2.36 ng/ml, p <0.01) strokes compared to healthy controls(c = 2.04 ng/ml).
When mean CXCL16 levels in LAA and SVD strokes were compared, mean CXCL16 levels were 1.1 times higher in patients suffering from LAA stroke compared with SVD strokes (p <0.05).
Lipoprotein-associated phospholipase A2 (LP PLA2) is an enzyme produced by inflammatory cells. It plays an important role in the development of atherosclerosis. LP PLA2 hydrolyses oxidised phospholipids, thus producing lysophosphatidylcholine and oxidised nonesterified fatty acids (OxNEFA). OxNEFA have chemotactic activity towards monocytes and lysophoshatidylcholine is able to up-regulate inflammatory mediators and thereby enhance the process of evolving atherosclerosis [38].
In a cohort study [39] higher levels of LP PLA2 were associated with carotid stenosis (n = 19) as compared with patients with ischaemic stroke without showing carotid stenosis (n =111) and healthy controls (n = 20).
In a large cohort study [40] (n = 1,946) LP PLA2 was found to be associated with ischaemic stroke due to large artery atherosclerosis in white non-Hispanic men (n = 381, hazard ratio 3.05). This association remained even after adjusting for confounders.
In another study [41], scientists compared patients suffering from transient ischaemic attack (TIA) with healthy controls. Patients suffering from TIA due to large artery atherosclerosis had higher LP PLA2 activity (207 nmol/ml/min) compared with patients suffering from TIA due to non-large artery atherosclerosis (184 nmol/ml/min, p = 0.044).
The lymphotoxin-like inducible protein that competes with glycoprotein D for binding herpesvirus entry mediator on T lymphocytes (LIGHT) is a member of the TNFSF, the tumour necrosis factor super family. LIGHT has proinflammatory and prothrombotic effects. It is an inducer of matrix metalloproteinase (MMP) in macrophages and is involved in platelet adhesion to endothelium. When macrophages are costimulated with oxLDL (oxidised low density lipoproteins) and LIGHT at the same time, the macrophage significantly increases the uptake of neutral lipids (≈20% increase) which results in quicker building of foam cells [42] and thus atherosclerosis.
In a small case control study [43] scientists compared levels of LIGHT in patients suffering from atherosclerotic ischaemic stroke (n = 20) to patients with asymptomatic carotid stenosis (stenosis >50%, n = 19) and healthy controls (n = 23).LIGHT levels were 4.1 times higher in patients suffering from ischaemic stroke due to large artery atherosclerosis (LIGHT: 350 pg/ml, p <0.05) than in normal healthy patients (LIGHT: 85 pg/ml, p <0.05), but LIGHT levels were not significantly different compared with patients with asymptomatic carotid stenosis.
Cholesterol is a hydrophobic sterol molecule. It is the base substance of cell membranes and of many hormones. Particularly high levels of cholesterol in the blood circulation, depending on how they are transported within lipoproteins, are strongly associated with the progression of atherosclerosis.
In a large Japanese cohort study with 612 ischaemic stroke patients, scientists found out that high serum total cholesterol levels in Japanese men (>6.21 mmol/l) were associated with large artery atherosclerosis (multivariable hazard ratio 2.86). They did not find such differences for ischaemic stroke due to large artery atherosclerosis in Japanese women [44].
Serum lipoprotein A is a lipoprotein consisting of apolipoprotein A and apolipoprotein B-100. It belongs to the group of LDL (low density lipoprotein). Serum lipoprotein A might be involved in the process of atherosclerosis.
In a large cohort study [45], serum lipoprotein A levels were measured in patients suffering from ischaemic stroke or TIA (n = 1,012). Highest levels of serum lipoprotein A were found in patients suffering from LAA (n= 281, median c = 34.6 mg/dl) (CE: n = 204, median c = 29.2 mg/dl, SVD: n = 261, median c = 24.2 mg/dl, other determined: n = 74, median c = 24.1, undetermined: n = 192, median c = 27 mg/dl)
C-reactive protein (CRP) is an acute-phase protein produced in the liver. CRP is an inflammation marker. It is part of the innate immune response, and it is capable of several immunological actions that may contribute to the chronic inflammatory process that constitutes atherosclerosis. In the acute phase, the systemic immune-inflammatory reaction is more enhanced in cardioembolic stroke then in other stroke subtypes, which makes CRP an ambivalent marker for strokes.
In a small study [46], scientists examined levels of CRP in noncardioembolic ischaemic strokes (n = 99). They studied the differences in CRP levels between patients suffering from ischaemic stroke due to large artery atherosclerosis (n = 52) and patients suffering from small vessel disease (n = 47). When CRP levels were measured on the day of onset, levels were 1.85 times higher in LAA patients compared with SVD patients (p <0.05) [46].
In another study [47], CRP levels were measured in ischaemic stroke patients (n = 600) and healthy controls (n = 600) at two different time-points, at acute phase and after 3 months. Highest levels of CRP were measured in cardioembolic strokes (n = 98) when measured at acute phase (1.5 times higher in cardioembolism than in LAA strokes) but after a 3-month follow-up the highest levels of CRP were found in the subgroup of LAA (n = 73) (1.3 times higher in LAA than in cardioembolic strokes).
Adiponectin is secreted by the adipose tissue. It is involved in lipid and glucose metabolism, it stimulates glucose uptake and metabolism in muscle cells and decreases glucose production in the liver, and it is involved in energy homeostasis. Adioponectin improves the insulin resistance level of the tissue. Adiponectin might play a protective role in case of atherosclerosis [48].
In a small study [49], adiponectin levels were 0.53 times lower in patients suffering from LAA stroke (n = 34, 2.7 ± 0.5 µg/ml, p = 0.047) compared with patients suffering from cardioembolic stroke (n = 49, 5.3 ± 0.5 µg/ml). Adiponectin levels were also 0.71 times lower in LAA strokes compared with healthy control patients (n = 171, 3.8 ± 0.2 µg/ml).
Brain natriuretic peptide (BNP) is a vasoactive peptide hormone, produced in heart and brain tissue. Its function is of a natriuretic and diuretic nature. BNP is mostly secreted from myocardium in response to increased intracardiac pressure or increased wall tension [50–53]. Thus it represents underlying cardiac disease, which might be associated with thrombus formation.
Several studies [54–59] have showed that high BNP levels in blood are associated with strokes of cardioembolic origin.
In a cohort study with 223 ischaemic stroke patients, a BNP cut off level of 90 pg/ml had a sensitivity 85% and a specificity of 78% to identify a cardioembolic origin, blood samples were taken >7 days after symptom onset (cardioembolic stroke: n = 69).
When blood samples were taken within 24 hours after symptom onset in another cohort study (n = 707), a BNP cut-off of 76 pg/ml identified a cardioembolic stroke with a specificity of 68% and a sensitivity of 72% (cardioembolic stroke: n = 259).
In a further independent cohort of 362 ischaemic stroke patients, those suffering from cardioembolism (n = 144) had the highest median BNP levels (n = 144, 179.5 pg/ml) compared with the median level in other subtypes (57.2 pg/ml) (undetermined n = 144, LAA n = 76, SVD n = 61, other n = 16).
Similar results were found in a study [58] with 142 ischaemic stroke patients. In this study it was possible to distinguish cardioembolicfrom noncardioembolic stroke with a cut-off value of 66.50 pg/ml, with a sensitivity of 75.0% and a specificity of 88.7%.
Finally, in a recent small cohort study [59] scientists compared BNP levels among aetiological subtypes in ischaemic stroke patients (n = 131). Patients suffering from cardioembolism could be identified with a cut-off BNP value of 77 pg/ml, with a sensitivity of 75.8% and a specificity of 76.8%, compared with all other aetiologies.
N-terminal probrain natriuretic peptide (NTproBNP) is a stable fragment of the precursor hormone of BNP. This precursor molecule is divided into two parts. It follows that NTproBNP is produced in an equimolar amount to BNP, but it is more stable, which might be an advantage over BNP as blood biomarker.
In a cohort study of 163 ischaemic stroke patients [60], the highest NTproBNP levels were found in CE stroke patients (mean levels 917.4 ± 82.8 pmol/l, p <0.001, n = 82) compared with other subtypes. (Mean level in ischaemic stroke subtypes: LAA: 717.2 ± 66.6 pmol/l p <0.001, n = 37): SVD: 696.6 ± 67.4 pmol/l, p <0.001; n = 22; other determined: 683.4 ± 69.4 pmol/l, p <0.001, n = 22.)
In a relatively small study [61] (n = 93 patients, 66 with ischaemic stroke [28 due to cardioembolism]) NTproBNP levels were 3.9 times (p <0.001) higher in patients suffering from ischaemic stroke due to cardioembolism (491.6 pg/ml) than noncardioembolic ischaemic stroke (124.7 pg/ml) when measured <72 hours after symptom onset. In this study the cut-off value was 265 pg/ml NTproBNP. It was possible to distinguish cardioembolism from other subtypes with a sensitivity of 94.4%, a specificity of 72.9%, and a positive predictive value of 56.6% and a negative predictive value of 97.2%. Interestingly, NTproBNP levels were not associated with the size of stroke.
In a slightly larger cohort study [62] (n = 125 patients, 57 patients with stroke of cardioembolic origin), a NTproBNP level ≥342 pg/ml, was associated with a cardioembolic stroke with a sensitivity of 93% and a specifity of 75%.
In another study [63] scientists tried to indentify the cause of ischaemic strokes in patients with cryptogenic ischaemic stroke. Among patients with cryptogenic strokes NTproBNP levels above 360 pg/mL were attributed to an underlying cardioembolic source of the stoke with a sensitivity of 87% and a specificity of 83%.
BNP and NTproBNP seem to be quite reliable markers for cardioembolism in ischaemic stroke, measured early after stroke onset. All studies showed that BNP or NTproBNP levels in ischaemic stroke of cardioembolic origin are significantly higher than in ischaemic stroke due of noncardioembolic origin. Ten studies were found showing that BNP and NTproBNP are able to distinguish accurately a cardioembolic stroke from other subtypes. However no direct comparison between BNP and NTproBNP were made, and thus so far we are not able to state which marker might perform better.
A list of the above mentioned studies which contain cut-off values and AUC (area under the curve) (eight out of ten studies) for either BNP or NTproBNP is presented in table 3:
Midregional proatrial natriuretic peptide (MRproANP) is a stable fragment of the precursor hormone of ANP. It is produced in an equimolar amount to ANP. ANP has a natriuretic, diuretic and vasodilating function and thereby is able to regulate blood pressure. It is mainly produced by cardiomyocytes of the atrium and secreted during increased atrial wall tension [64, 65]. The midregional fragment of proANP is more stable than the N- or C-terminal part of the precursor in vivo and in blood ex vivo, which might render it generally more applicable to clinical practice then other fragments of natriuretic peptides [66].
In a prospective observational study [67], MRproANP levels were 1.66 times higher (p <0.0001) in patients with ischaemic stroke due to cardioemolism (n = 131, 206 pmol/l) compared with patients with noncardioembolic strokes (n = 228, 124 pmol/l). If MRproANP levels were >180 pg/ml the ischaemic stroke was most probably due to cardioembolism (specificity 60.3%, sensitivity 71%, positive likelihood ratio 2.0, negative likelihood ratio 0.6). This association was independent of known risk factors for cardioembolic strokes such as known atrial fibrillation or congestive heart failure.
D-dimer is a fraction of fibrin, produced when plasmin is activated. It is a marker of haemostatic imbalance. D-dimers are a product of the degradation of fibrin by plasmin, thus they are involved in thrombus formation. In cases of ischaemic stroke due to cardioembolism, thrombi mainly consist of fibrin and platelets [22]. It follows that the degradation of the thrombus is associated with a higher systemic D-dimer concentration.
Several studies [55, 68, 69] showed that D-dimer levels are significantly higher in ischaemic strokes of cardioembolic origin than in those of noncardioembolic origin.
In a relatively small cohort study [68] (patients n = 98, patients with ischaemic stroke of cardioembolic origin n = 53), D-dimer levels were 1.9 times higher in cardioembolic strokes compared with other subtypes of stroke. When D-dimers were measured within 6 hours after symptom onset, a level below 300 µg/l ruled out a stroke due to cardioembolism (sensitivity 100%, specificy 53%).
Another study [55] with a similar result compared cardioembolic (n = 259; mean value 1.1 µg/ml, p <0.01) to noncardioembolic ischaemic strokes (n = 448; LAA mean value 0.5 µg/ml, SMV mean value 0.6 µg/ml, Undetermined stroke: mean value 0.8 µg/ml). When the D-Dimer level was >0.96 µg/ml (960 µg/l) the corresponding OR was 2.2 for cardioembolism compared to large artery atherosclerosis.
In a further cohort study [69], mean D-dimer levels were 1.7 times higher in patients suffering from cardioembolic ischaemic stroke (1,287.50 ng/ml, n = 34) than patients suffering from noncardioembolic stroke (750.20 ng/ml, n = 42).
Soluble thrombomodulin (sTM), or CD141 antigen as it is called, is an endothelial-specific type 1 membrane receptor. When bound to thrombin protein C is activated. Protein C degrades clotting factors Va and VIIIa and this leads to a reduction of coagulation [70]. After an ischaemic stroke soluble thrombomodulin is most likely upregulated to balance blood clotting homeostasis
In a small cohort study [71], scientists investigated soluble thrombomodulin levels in patients suffering from ischaemic stroke (n = 93). Mean levels of sTM were highest (3.79 ± 1.26 ng/ml) in patients suffering from ischaemic stroke due to cardioembolism and lower in the LAA subgroup (2.38 ± 0.68 ng/ml, p <0.009) as well as in the SVD group (2.38 ± 0.44 ng/ml, p <0.05).
All of these three molecules are inflammatory markers. Inflammation plays a role in thrombus formation or degradation. An alternative association with cardioembolic stroke might be explained by the fact that the systemic immune-inflammatory reaction is more pronounced in cardioembolic stroke then in other stroke subtypes.
In a small study [72] (ischaemic stroke patients n = 120, cardioembolism n = 20), mean levels of tumour necrosis factor-alpha (TNF-α), interleukin-6 (IL-6) and interleukin-1beta (IL-1β) in patients with ischaemic stroke due to cardioembolism were significantly higher than in patients suffering from ischaemic stroke of noncardioembolic origin.
Aetiological blood biomarkers of SVD stroke are listed in table 5.
Homocysteine is an amino acid that is produced via demethylation of methionine [73]. Higher levels of homocysteine can lead to endothelial cell injury [74]. Evidence suggests that homocysteine may further promote fatty deposits in blood vessels by damaging the inner lining of arteries and promoting blood clots.
In a case control study [75] with 172 patients suffering from SVD and 172 healthy controls, mean homocysteine levels were 1.21 times higher in patients with ischaemic stroke due to SVC compared with healthy controls (p <0.0005). However the study did not compare homocysteine levels with other subtypes such as LAA or CE.
After platelet activation, platelet CD40 ligand (CD40L) is presented on the cell surface. It then binds CD40 on endothelial cells and thereby induces the production of adhesion molecules, inflammatory cytokines and procoagulant tissue factor [76], all factors with a causal relevance to atherogenesis, most probably also in small vessels [30].
When comparing platelet CD40L expression [32], patients suffering from small vessel disease (n = 13, mean fluorescence intensity: 2.08 ± 0.16) had significantly (p = 0.029) higher levels compared with patients suffering from cardioembolic ischaemic stroke (n = 17, mean fluorescence intensity: 1.81 ± 0.16), but there was no significant difference between platelet CD40L in SVD and LAA.
Intercellular adhesion molecule 1 (ICAM1) is a glycoprotein expressed on the surface of endothelial cells, macrophages and lymphocytes. ICAM1 is induced by IL-1 and TNF. ICAM 1 is involved in adhesion and transendothelial migration of lymphocytes [77], which may contribute to small vessel atherosclerosis.
In a cohort study [78], scientists followed up 267 healthy individuals and compared baseline ICAM1 levels in subjects who developed white matter lesions on MRI with subjects who did not develop white matter lesions after 3 and 6 years. Patients who showed MRI findings of white matter lesions after 3 years (n = 23, mean ICAM1 levels: 372.6 ng/ml) had 1.19 times higher ICAM1 levels in their blood compared with patients who had no such findings (n = 219, mean ICAM1 level: 312.8 ng/ml; p = 0.005).
When comparing MRI findings and ICAM1 levels after 6 years, ICAM1 levels in patients with MRI findings (white matter lesions, n = 32, mean ICAM1 levels: 347.4 ng/ml) were still 1.12 times higher in these patients than in healthy subjects (n = 152, mean ICAM1 levels: 309.0 ng/ml).
| Table 4: Aetiological blood biomarkers of cardioembolic strokes. | |||||
| Blood biomarker CE marker | Description | Potential clinical Application | Study of Reference | Size | Study Design |
| NTproBNP | N-terminal pr-brain natriuretic peptide, vasoactive peptide hormone, natriuretic and diuretic properties | Diagnosis of CE in acute ischaemic stroke | Fonseca et al. 2011 [61] | 92 patients (66 ischaemic stroke, 28 CE) | Cohort study |
| Rodriguez-Yanez et al. 2009 [63] | 262 patients, 100 CE | Cohort study | |||
| Hajsadeghi et al. 2013 [62] | 125 patients, 57 CE | Cohort study | |||
| Cojocaru et al. 2009 [60] | 163 patients, 81 CE | Cohort study | |||
| BNP | V Brain natriuretic peptide, vasoactive peptide hormone, natriuretic and diuretic properties, heart and brain origin | Diagnosis of CE in acute ischaemic stroke | Montaner et al. 2008 [55] | 707 patients, 259 CE | Cohort study |
| Yukiiri et al 2008 [59] | 131 patients, 62 CE | Cohort study | |||
| Tamura et al. 2012 [54] | 223 patients, 69 CE | Cohort Study | |||
| Nigro et al. 2014 [56] | 342 patients, 144 CE | Cohort Study | |||
| Zhixin et al. 2013 [58] | 142 patients, 36 CE | Cohort Study | |||
| Shibazaki et al. 2009 [57] | 200 patients, 82 CE | Cohort study | |||
| D-dimer | Fibrin fractions due to plasmin activation, marker of haemostatic imbalance | Diagnosis of CE in acute ischaemic stroke | Isenegger et al. 2009 [68] | 98 patients, 53 CE | Cohort study |
| Montaner et al. 2008 [55] | 707 patients, 259 CE | Cohort study | |||
| Tombul et al. 2005 [69] | 76 patients, 34 CE | Cohort study | |||
| MRproANP | Midregional proatrial natriuretic peptide, secreted during atrial wall tension stress, natriuretic, diuretic, vasoactive peptide hormone | Diagnosis of CE in acute ischaemic stroke | Katan et al. 2010 [67] | 362 patients, 131 CE | Cohort study |
| sTM | Soluble thrombomodulin, builds complex with thrombin to activate C-reactive protein | Diagnosis of CE in acute ischaemic stroke | Dharmasaroja et al. 2012 [71] | 93 patients 41 CE | Cohort study |
| TNF-α | Tumour necrosis factor alpha Inflammatory marker | Diagnosis of CE in acute ischaemic stroke | Licata et al. 2009 [72] | 120 patients, 20 CE | Cohort study |
| IL-6 | Interleukin-6, proinflammatory cytokine | Diagnosis of CE in acute ischaemic stroke | Licata G. et al. 2009 [72] | 120 patients, 20 CE | Cohort study |
| IL-1β | Interleukin-1β, inflammatory marker | Diagnosis of CE in acute ischaemic stroke | Licata G. et al. 2009 [72] | 120 patients, 20 CE | Cohort study |
| CE = cardioembolism | |||||
| Table 5: Aetiological blood biomarkers of small vessel disease (SVD) stroke. | |||||
| Blood biomarker SVD Marker | Description | Potential clinical application | Study reference | Size | Study design |
| Homocysteine | Amino acid, high concentrations linked to cardiovascular disease | Diagnosis of SVD | Hassan et al. 2003 [75] | 344 patients, 172 with SVD | Cohort study |
| CD40 ligand on platelet surface | Expressed on platelets, when bound to CD40 on endothelial cell activates procoagulant tissue factor, adhesion molecules and inflammatory cytokines | Diagnosis of SVD | Oberheiden et al. 2012 [32] | 51 patients, 13 SVD | Cohort study |
| ICAM1 | Intracellular adhesion molecule1, improves adhesion and transendothelial migration of lymphocytes | Diagnosis of SVD | Markus et al. 2005 [78] | 242 patients 23 SVD (3-year follow up) 184 patients 32 SVD (6-year follow up) | Cohort study |
The use of rapidly measurable blood biomarkers for aetiological assessment on admission may advance the identification of patients who need specific secondary preventive treatment such as oral anticoagulation, and thus accelerate the onset of optimal secondary prevention, improve patient outcome and reduce subsequent costs. However, as the research interest in blood biomarkers in the field of stroke aetiology has evolved only over the last few years, most of the above data on aetiological markers reside still in an exploratory phase. The studies are rather small and only few are controlled for potential confounding factors. A new aetiological blood biomarker, before it can be used in clinical routine, generally needs to provide additional information to established clinical features. But most of those studies which have been published so far were very small, did not include all statistical analyses that have been newly proposed to assess the additive discriminatory value of blood markers in vascular research, and they mostly involved post-hoc analyses. There is clearly a need for validation of these results in larger well-designed studies. Only few markers were evaluated and validated in rather large, well-designed studies, and the most promising potential was shown for the family of the natriuretic peptides in identifying cardioembolic strokes.
To find the ideal marker, the pathological mechanism underlying the stroke should be well understood. In the case of cardioembolic strokes, biomarkers that have shown associations with cardiac dysfunction, specifically atrial fibrillation, are the most promising. In this case, natriuretic peptides show a great deal of potential for aetiological relevance. In terms of large artery atherosclerosis, inflammatory markers, especially those known to be associated with carotid atherosclerosis, intima media thickness or intracerebral atherosclerosis, are interesting candidates. LP PLA2 seems thus to be an interesting candidate marker, it is involved in the process of atherosclerosis via hydroxylation of oxidised phospholipids and thereby starts the process of attraction of inflammatory cells on the atherosclerotic site. Currently no marker has yet been identified specifically for small vessel disease in a large enough stroke cohort, thus for this aetiological subtype of stroke, further derivation studies are needed before a marker for further validation can be recommended. For example, markers reflecting a state of chronic high blood pressure or markers related to amyloid deposition in small cerebral vessels would be promising candidates for further evaluation. However, since there are overlapping pathophysiological mechanisms for each aetiology, such as inflammation and thus inflammatory markers, it is most likely that panels of blood biomarkers will be used in the future since no single marker will be sensitive and specific enough.
Newer studies should provide the answer to the question as to whether some aetiological biomarkers will help to improve identification of patients for known effective secondary prevention treatments after acute ischaemic stroke, and if patients with abnormal levels of the selected biomarkers are at increased risk for recurrent events as a hard and clinically relevant endpoint. If one or more of the selected biomarkers are confirmed to be associated with stroke aetiology and recurrent stroke, interventional studies may be undertaken to apply in these patients secondary preventive therapies guided by clinical information and biomarker levels. These markers might in the future even serve as surrogate endpoint for new secondary prevention measures.
To specifically address the question on aetiological biomarkes in acute stroke, the BIOmarker SIGNAture of Stroke AetioLogy Study (BIOSIGNAL) – a multicenter international study funded by the Swiss National Science Foundation (PZ00P3_142422) – was designed and has begun enrolling patients across Europe.
(http://www.research-projects.uzh.ch/p19799.htm https://clinicaltrials.gov/ct2/show/NCT02274727?term=BIOSIGNAL&rank=1)
1 Hankey GJ, Warlow CP. Treatment and secondary prevention of stroke: evidence, costs, and effects on individuals and populations. Lancet. 1999;354(9188):1457–63. doi: 10.1016/S0140–6736(99)04407–4. PubMed PMID: 10543686.
2 Fluri F, Morgenthaler NG, Mueller B, Christ-Crain M, Katan M. Copeptin, procalcitonin and routine inflammatory markers-predictors of infection after stroke. PLoS One. 2012;7(10):e48309. doi: 10.1371/journal.pone.0048309. PubMed PMID: 23118979; PubMed Central PMCID: PMC3485149.
3 Amarenco P, Bogousslavsky J, Caplan LR, Donnan GA, Hennerici MG. Classification of stroke subtypes. Cerebrovasc Dis. 2009;27(5):493–501. doi: 10.1159/000210432. PubMed PMID: 19342825.
4 Schweizerische Aerztezeitung. 2000;81:835–40
5 Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, et al. Heart disease and stroke statistics – 2014 update: a report from the American Heart Association. Circulation. 2014;129(3):e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. PubMed PMID: 24352519.
6 Grysiewicz RA, Thomas K, Pandey DK. Epidemiology of ischemic and hemorrhagic stroke: incidence, prevalence, mortality, and risk factors. Neurol Clin. 2008;26(4):871–95, vii. doi: 10.1016/j.ncl.2008.07.003. PubMed PMID: 19026895.
7 Kolominsky-Rabas PL, Weber M, Gefeller O, Neundoerfer B, Heuschmann PU. Epidemiology of ischemic stroke subtypes according to TOAST criteria: incidence, recurrence, and long-term survival in ischemic stroke subtypes: a population-based study. Stroke. 2001;32(12):2735–40. PubMed PMID: 11739965.
8 The effect of low-dose warfarin on the risk of stroke in patients with nonrheumatic atrial fibrillation. The Boston Area Anticoagulation Trial for Atrial Fibrillation Investigators. New Engl J Med. 1990;323(22):1505–11. doi: 10.1056/NEJM199011293232201. PubMed PMID: 2233931.
9 Warfarin versus aspirin for prevention of thromboembolism in atrial fibrillation: Stroke Prevention in Atrial Fibrillation II Study. Lancet. 1994;343(8899):687–91. PubMed PMID: 7907677.
10 Connolly SJ, Ezekowitz MD, Yusuf S, Eikelboom J, Oldgren J, Parekh A, et al. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361(12):1139–51. doi: 10.1056/NEJMoa0905561. PubMed PMID: 19717844.
11 Patel MR, Mahaffey KW, Garg J, Pan G, Singer DE, Hacke W, et al. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med. 2011;365(10):883–91. doi: 10.1056/NEJMoa1009638. PubMed PMID: 21830957.
12 Granger CB, Alexander JH, McMurray JJ, Lopes RD, Hylek EM, Hanna M, et al. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2011;365(11):981–92. doi: 10.1056/NEJMoa1107039. PubMed PMID: 21870978.
13 Connolly SJ, Eikelboom J, Joyner C, Diener HC, Hart R, Golitsyn S, et al. Apixaban in patients with atrial fibrillation. N Engl J Med. 2011;364(9):806–17. doi: 10.1056/NEJMoa1007432. PubMed PMID: 21309657.
14 Rerkasem K, Rothwell PM. Carotid endarterectomy for symptomatic carotid stenosis. The Cochrane database of systematic reviews. 2011(4):CD001081. doi: 10.1002/14651858.CD001081.pub2. PubMed PMID: 21491381.
15 Brott TG, Hobson RW, 2nd, Howard G, Roubin GS, Clark WM, Brooks W, et al. Stenting versus endarterectomy for treatment of carotid-artery stenosis. N Engl J Med. 2010;363(1):11–23. doi: 10.1056/NEJMoa0912321. PubMed PMID: 20505173; PubMed Central PMCID: PMC2932446.
16 Adams HP, Jr., Bendixen BH, Kappelle LJ, Biller J, Love BB, Gordon DL, et al. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke. 1993;24(1):35–41. PubMed PMID: 7678184.
17 Ay H, Benner T, Arsava EM, Furie KL, Singhal AB, Jensen MB, et al. A computerized algorithm for etiologic classification of ischemic stroke: the Causative Classification of Stroke System. Stroke. 2007;38(11):2979–84. doi: 10.1161/STROKEAHA.107.490896. PubMed PMID: 17901381.
18 Amarenco P, Bogousslavsky J, Caplan LR, Donnan GA, Hennerici MG. New approach to stroke subtyping: the A-S-C-O (phenotypic) classification of stroke. Cerebrovasc Dis. 2009;27(5):502–8. doi: 10.1159/000210433. PubMed PMID: 19342826.
19 Hart RG, Diener HC, Coutts SB, Easton JD, Granger CB, O’Donnell MJ, et al. Embolic strokes of undetermined source: the case for a new clinical construct. Lancet Neurol. 2014;13(4):429–38. doi: 10.1016/S1474–4422(13)70310–7. PubMed PMID: 24646875.
20 Böcker W, Heitz PU, Moch H. Pathologie. 5., vollst. überarb. Aufl. ed. München: Elsevier; 2012. Online-Ressource p.
21 Weir NU. An update on cardioembolic stroke. Postgrad Med J. 2008;84(989):133–42; quiz 9–40. doi: 10.1136/pgmj.2007.066563. PubMed PMID: 18372484.
22 English J, Smith W. Chapter 36 Cardio-embolic stroke. In: Marc F, editor. Handbook of Clinical Neurology. Volume 93: Elsevier; 2008. p. 719–49.
23 Pantoni L. Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010;9(7):689–701. doi: 10.1016/S1474–4422(10)70104–6. PubMed PMID: 20610345.
24 Donnan GA, Norrving B. Chapter 27 Lacunes and lacunar syndromes. In: Marc F, editor. Handbook of Clinical Neurology. Volume 93: Elsevier; 2008. p. 559–75.
25 Deb P, Sharma S, Hassan KM. Pathophysiologic mechanisms of acute ischemic stroke: An overview with emphasis on therapeutic significance beyond thrombolysis. Pathophysiology. 2010;17(3):197–218. doi: 10.1016/j.pathophys.2009.12.001. PubMed PMID: 20074922.
26 Llombart V, Garcia-Berrocoso T, Bustamante A, Fernandez-Cadenas I, Montaner J. Cardioembolic stroke diagnosis using blood biomarkers. Curr Cardiol Rev. 2013;9(4):340–52. PubMed PMID: 24527683; PubMed Central PMCID: PMC3941099.
27 Al Haj Zen A, Caligiuri G, Sainz J, Lemitre M, Demerens C, Lafont A. Decorin overexpression reduces atherosclerosis development in apolipoprotein E-deficient mice. Atherosclerosis. 2006;187(1):31–9. doi: 10.1016/j.atherosclerosis.2005.08.023. PubMed PMID: 16183063.
28 Xu YZ, Yang ZG, Zhang YH, Zhang YW, Hong B, Liu JM. Dynamic reduction of plasma decorin following ischemic stroke: a pilot study. Neurochem Res. 2012;37(9):1843–8. Epub 2012/06/09. doi: 10.1007/s11064–012–0787–0. PubMed PMID: 22678721.
29 Tsai NW, Chang WN, Shaw CF, Jan CR, Chang HW, Huang CR, et al. Levels and value of platelet activation markers in different subtypes of acute non-cardio-embolic ischemic stroke. Thrombo Res. 2009;124(2):213–8. Epub 2009/02/24. doi: 10.1016/j.thromres.2009.01.012. PubMed PMID: 19233449.
30 Duerschmied D, Bode C, Ahrens I. Immune functions of platelets. Thromb Haemost. 2014;112(4):678–91. doi: 10.1160/TH14–02–0146. PubMed PMID: 25209670.
31 Wang JH, Zhang YW, Zhang P, Deng BQ, Ding S, Wang ZK, et al. CD40 ligand as a potential biomarker for atherosclerotic instability. Neurological Res. 2013;35(7):693–700. doi: 10.1179/1743132813Y.0000000190. PubMed PMID: 23561892; PubMed Central PMCID: PMC3770830.
32 Oberheiden T, Nguyen XD, Fatar M, Elmas E, Blahak C, Morper N, et al. Platelet and monocyte activation in acute ischemic stroke – is there a correlation with stroke etiology? Clin Appl Thromb Hemost. 2012;18(1):87–91. Epub 2011/07/08. doi: 10.1177/1076029611412359. PubMed PMID: 21733938.
33 Shimaoka T, Kume N, Minami M, Hayashida K, Kataoka H, Kita T, et al. Molecular cloning of a novel scavenger receptor for oxidized low density lipoprotein, SR-PSOX, on macrophages. J Biol Chem. 2000;275(52):40663–6. doi: 10.1074/jbc.C000761200. PubMed PMID: 11060282.
34 Matloubian M, David A, Engel S, Ryan JE, Cyster JG. A transmembrane CXC chemokine is a ligand for HIV-coreceptor Bonzo. Nat Immunol. 2000;1(4):298–304. doi: 10.1038/79738. PubMed PMID: 11017100.
35 Minami M, Kume N, Shimaoka T, Kataoka H, Hayashida K, Yonehara S, et al. Expression of scavenger receptor for phosphatidylserine and oxidized lipoprotein (SR-PSOX) in human atheroma. Ann N Y Acad Sci. 2001;947:373–6. PubMed PMID: 11795294.
36 Aslanian AM, Charo IF. Targeted disruption of the scavenger receptor and chemokine CXCL16 accelerates atherosclerosis. Circulation. 2006;114(6):583–90. doi: 10.1161/CIRCULATIONAHA.105.540583. PubMed PMID: 16880330.
37 Ma A, Pan X, Xing Y, Wu M, Wang Y, Ma C. Elevation of serum CXCL16 level correlates well with atherosclerotic ischemic stroke. Arch Med Sci. 2014;10(1):47–52. doi: 10.5114/aoms.2013.39200. PubMed PMID: 24701213; PubMed Central PMCID: PMC3953970.
38 Carlquist JF, Muhlestein JB, Anderson JL. Lipoprotein-associated phospholipase A2: a new biomarker for cardiovascular risk assessment and potential therapeutic target. Expert Rev Mol Diagn. 2007;7(5):511–7. doi: 10.1586/14737159.7.5.511. PubMed PMID: 17892360.
39 Tsai TH, Chen YL, Lin HS, Liu CF, Chang HW, Lu CH, et al. Link between lipoprotein-associated phospholipase A2 gene expression of peripheral-blood mononuclear cells and prognostic outcome after acute ischemic stroke. J Atheroscler Thromb. 2012;19(6):523–31. PubMed PMID: 22447189.
40 Katan M, Moon YP, Paik MC, Wolfert RL, Sacco RL, Elkind MS. Lipoprotein-associated phospholipase A2 is associated with atherosclerotic stroke risk: the Northern Manhattan Study. PloS one. 2014;9(1):e83393. Epub 2014/01/15. doi: 10.1371/journal.pone.0083393. PubMed PMID: 24416164; PubMed Central PMCID: PMCPmc3886969.
41 Delgado P, Chacon P, Penalba A, Pelegri D, Garcia-Berrocoso T, Giralt D, et al. Lipoprotein-associated phospholipase A(2) activity is associated with large-artery atherosclerotic etiology and recurrent stroke in TIA patients. Cerebrovas Dis. 2012;33(2):150–8. Epub 2011/12/20. doi: 10.1159/000334193. PubMed PMID: 22178747.
42 Scholz H, Sandberg W, Damas JK, Smith C, Andreassen AK, Gullestad L, et al. Enhanced plasma levels of LIGHT in unstable angina: possible pathogenic role in foam cell formation and thrombosis. Circulation. 2005;112(14):2121–9. doi: 10.1161/CIRCULATIONAHA.105.544676. PubMed PMID: 16186421.
43 Liu GZ, Fang LB, Hjelmstrom P, Gao XG. Enhanced plasma levels of LIGHT in patients with acute atherothrombotic stroke. Acta Neurol Scand. 2008;118(4):256–9. Epub 2008/04/04. doi: 10.1111/j.1600–0404.2008.01013.x. PubMed PMID: 18384455.
44 Cui R, Iso H, Yamagishi K, Saito I, Kokubo Y, Inoue M, et al. High serum total cholesterol levels is a risk factor of ischemic stroke for general Japanese population: the JPHC study. Atherosclerosis. 2012;221(2):565–9. Epub 2012/02/22. doi: 10.1016/j.atherosclerosis.2012.01.013. PubMed PMID: 22341595.
45 Kim BS, Jung HS, Bang OY, Chung CS, Lee KH, Kim GM. Elevated serum lipoprotein(a) as a potential predictor for combined intracranial and extracranial artery stenosis in patients with ischemic stroke. Atherosclerosis. 2010;212(2):682–8. doi: 10.1016/j.atherosclerosis.2010.07.007. PubMed PMID: 20691971.
46 Zeng L, He X, Liu J, Wang L, Weng S, Wang Y, et al. Differences of circulating inflammatory markers between large- and small vessel disease in patients with acute ischemic stroke. Int J Med Sci. 2013;10(10):1399–405. doi: 10.7150/ijms.6652. PubMed PMID: 23983602; PubMed Central PMCID: PMC3753418.
47 Ladenvall C, Jood K, Blomstrand C, Nilsson S, Jern C, Ladenvall P. Serum C-reactive protein concentration and genotype in relation to ischemic stroke subtype. Stroke. 2006;37(8):2018–23. doi: 10.1161/01.STR.0000231872.86071.68. PubMed PMID: 16809555.
48 Rabe K, Lehrke M, Parhofer KG, Broedl UC. Adipokines and insulin resistance. Mol Med. 2008;14(11–12):741–51. doi: 10.2119/2008–00058.Rabe. PubMed PMID: 19009016; PubMed Central PMCID: PMC2582855.
49 Kuwashiro T, Ago T, Kamouchi M, Matsuo R, Hata J, Kuroda J, et al. Significance of plasma adiponectin for diagnosis, neurological severity and functional outcome in ischemic stroke – Research for Biomarkers in Ischemic Stroke (REBIOS). Metabolism. 2014;63(9):1093–103. doi: 10.1016/j.metabol.2014.04.012. PubMed PMID: 24929894.
50 Davis M, Espiner E, Richards G, Billings J, Town I, Neill A, et al. Plasma brain natriuretic peptide in assessment of acute dyspnoea. Lancet. 1994;343(8895):440–4. PubMed PMID: 7905953.
51 Maisel AS, Krishnaswamy P, Nowak RM, McCord J, Hollander JE, Duc P, et al. Rapid measurement of B-type natriuretic peptide in the emergency diagnosis of heart failure. N Engl J Med. 2002;347(3):161–7. doi: 10.1056/NEJMoa020233. PubMed PMID: 12124404.
52 Lainchbury JG, Campbell E, Frampton CM, Yandle TG, Nicholls MG, Richards AM. Brain natriuretic peptide and n-terminal brain natriuretic peptide in the diagnosis of heart failure in patients with acute shortness of breath. J Am Coll Cardiol. 2003;42(4):728–35. PubMed PMID: 12932611.
53 Wright SP, Doughty RN, Pearl A, Gamble GD, Whalley GA, Walsh HJ, et al. Plasma amino-terminal pro-brain natriuretic peptide and accuracy of heart-failure diagnosis in primary care: a randomized, controlled trial. J Am Coll Cardiol. 2003;42(10):1793–800. PubMed PMID: 14642690.
54 Tamura H, Watanabe T, Nishiyama S, Sasaki S, Wanezaki M, Arimoto T, et al. Elevated plasma brain natriuretic peptide levels predict left atrial appendage dysfunction in patients with acute ischemic stroke. J Cardiol. 2012;60(2):126–32. Epub 2012/04/25. doi: 10.1016/j.jjcc.2012.02.010. PubMed PMID: 22525966.
55 Montaner J, Perea-Gainza M, Delgado P, Ribo M, Chacon P, Rosell A, et al. Etiologic diagnosis of ischemic stroke subtypes with plasma biomarkers. Stroke. 2008;39(8):2280–7. Epub 2008/06/07. doi: 10.1161/strokeaha.107.505354. PubMed PMID: 18535284.
56 Nigro N, Wildi K, Mueller C, Schuetz P, Mueller B, Fluri F, et al. BNP but Not s-cTnln is associated with cardioembolic aetiology and predicts short and long term prognosis after cerebrovascular events. PLoS One. 2014;9(7):e102704. doi: 10.1371/journal.pone.0102704. PubMed PMID: 25072816; PubMed Central PMCID: PMC4114527.
57 Shibazaki K, Kimura K, Iguchi Y, Okada Y, Inoue T. Plasma brain natriuretic peptide can be a biological marker to distinguish cardioembolic stroke from other stroke types in acute ischemic stroke. Intern Med. 2009;48(5):259–64. PubMed PMID: 19252345.
58 Zhixin W, Lianhong Y, Wei H, Lianda L, Longyuan J, Yingjian Z, et al. The value of the use of plasma B-type natriuretic peptide among acute ischemic stroke patients in a Chinese emergency department. Clin Neurol Neurosurg. 2013;115(9):1671–6. doi: 10.1016/j.clineuro.2013.02.021. PubMed PMID: 23518421.
59 Yukiiri K, Hosomi N, Naya T, Takahashi T, Ohkita H, Mukai M, et al. Plasma brain natriuretic peptide as a surrogate marker for cardioembolic stroke. BMC Neurol. 2008;8:45. Epub 2008/12/17. doi: 10.1186/1471–2377–8–45. PubMed PMID: 19077217; PubMed Central PMCID: PMCPmc2621245.
60 Cojocaru IM, Cojocaru M, Sapira V, Ionescu A, Barlan S, Tacu N. Could pro-BNP, uric acid, bilirubin, albumin and transferrin be used in making the distinction between stroke subtypes? Rom J Intern Med . 2013;51(3–4):188–95. PubMed PMID: 24620632.
61 Fonseca AC, Matias JS, Pinho e Melo T, Falcao F, Canhao P, Ferro JM. N-terminal probrain natriuretic peptide as a biomarker of cardioembolic stroke. Int J Stroke. 2011;6(5):398–403. Epub 2011/06/08. doi: 10.1111/j.1747–4949.2011.00606.x. PubMed PMID: 21645267.
62 Hajsadeghi S, Kashani Amin L, Bakhshandeh H, Rohani M, Azizian AR, Jafarian Kerman SR. The diagnostic value of N-terminal pro-brain natriuretic peptide in differentiating cardioembolic ischemic stroke. J Stroke Cerebrovasc Dis. 2013;22(4):554–60. Epub 2013/02/21. doi: 10.1016/j.jstrokecerebrovasdis.2013.01.012. PubMed PMID: 23422348.
63 Rodriguez-Yanez M, Sobrino T, Blanco M, de la Ossa NP, Brea D, Rodriguez-Gonzalez R, et al. High serum levels of pro-brain natriuretic peptide (pro BNP) identify cardioembolic origin in undetermined stroke. Dis Markers. 2009;26(4):189–95. Epub 2009/09/05. doi: 10.3233/dma-2009–0630. PubMed PMID: 19729800; PubMed Central PMCID: PMCPmc3833699.
64 Wang TJ, Larson MG, Levy D, Benjamin EJ, Leip EP, Omland T, et al. Plasma natriuretic peptide levels and the risk of cardiovascular events and death. N Engl J Med. 2004;350(7):655–63. doi: 10.1056/NEJMoa031994. PubMed PMID: 14960742.
65 Wang TJ, Gona P, Larson MG, Tofler GH, Levy D, Newton-Cheh C, et al. Multiple biomarkers for the prediction of first major cardiovascular events and death. N Engl J Med. 2006;355(25):2631–9. doi: 10.1056/NEJMoa055373. PubMed PMID: 17182988.
66 Morgenthaler NG, Struck J, Thomas B, Bergmann A. Immunoluminometric assay for the midregion of pro-atrial natriuretic peptide in human plasma. Clin Chem. 2004;50(1):234–6. doi: 10.1373/clinchem.2003.021204. PubMed PMID: 14709661.
67 Katan M, Fluri F, Schuetz P, Morgenthaler NG, Zweifel C, Bingisser R, et al. Midregional pro-atrial natriuretic peptide and outcome in patients with acute ischemic stroke. J Am Coll Cardiol. 2010;56(13):1045–53. Epub 2010/09/18. doi: 10.1016/j.jacc.2010.02.071. PubMed PMID: 20846604.
68 Isenegger J, Meier N, Lammle B, Alberio L, Fischer U, Nedeltchev K, et al. D-dimers predict stroke subtype when assessed early. Cerebrovasc Dis. 2010;29(1):82–6. Epub 2009/11/13. doi: 10.1159/000256652. PubMed PMID: 19907168.
69 Tombul T, Atbas C, Anlar O. Hemostatic markers and platelet aggregation factors as predictive markers for type of stroke and neurological disability following cerebral infarction. J Clin Neurosci. 2005;12(4):429–34. Epub 2005/06/01. doi: 10.1016/j.jocn.2004.06.013. PubMed PMID: 15925775.
70 Esmon CT. Thrombomodulin as a model of molecular mechanisms that modulate protease specificity and function at the vessel surface. FASEB J. 1995;9(10):946–55. PubMed PMID: 7615164.
71 Dharmasaroja P, Dharmasaroja PA, Sobhon P. Increased plasma soluble thrombomodulin levels in cardioembolic stroke. Clin Appl Thromb Hemost. 2012;18(3):289–93. Epub 2012/01/26. doi: 10.1177/1076029611432744. PubMed PMID: 22275395.
72 Licata G, Tuttolomondo A, Di Raimondo D, Corrao S, Di Sciacca R, Pinto A. Immuno-inflammatory activation in acute cardio-embolic strokes in comparison with other subtypes of ischaemic stroke. Thromb Haemost. 2009;101(5):929–37. Epub 2009/05/01. PubMed PMID: 19404547.
73 Schalinske KL, Smazal AL. Homocysteine imbalance: a pathological metabolic marker. Adv Nutr. 2012;3(6):755–62. doi: 10.3945/an.112.002758. PubMed PMID: 23153729; PubMed Central PMCID: PMC3648699.
74 Wall RT, Harlan JM, Harker LA, Striker GE. Homocysteine-induced endothelial cell injury in vitro: a model for the study of vascular injury. Thromb Res. 1980;18(1–2):113–21. PubMed PMID: 7404495.
75 Hassan A, Hunt BJ, O’Sullivan M, Bell R, D'Souza R, Jeffery S, et al. Homocysteine is a risk factor for cerebral small vessel disease, acting via endothelial dysfunction. Brain. 2004;127(Pt 1):212–9. Epub 2003/11/11. doi: 10.1093/brain/awh023. PubMed PMID: 14607791.
76 Schonbeck U, Libby P. CD40 signaling and plaque instability. Circ Res. 2001;89(12):1092–103. PubMed PMID: 11739273.
77 Rothlein R, Dustin ML, Marlin SD, Springer TA. A human intercellular adhesion molecule (ICAM-1) distinct from LFA-1. J Immunol. 1986;137(4):1270–4. PubMed PMID: 3525675.
78 Markus HS, Hunt B, Palmer K, Enzinger C, Schmidt H, Schmidt R. Markers of endothelial and hemostatic activation and progression of cerebral white matter hyperintensities: longitudinal results of the Austrian Stroke Prevention Study. Stroke. 2005;36(7):1410–4. doi: 10.1161/01.STR.0000169924.60783.d4. PubMed PMID: 15905468.
Funding: Swiss National Science Foundation (PZ00P3_142422).