Advances and challenges in understanding the multifaceted pathogenesis of amyotrophic lateral sclerosis
DOI: https://doi.org/10.4414/smw.2015.14054
Florent
Laferriere, Magdalini
Polymenidou
Summary
Amyotrophic lateral sclerosis (ALS) is an adult-onset neurodegenerative disease, which primarily affects motor neurons leading to progressive paralysis and death within a few years from onset. The pathological hallmark of ALS is the presence of cytoplasmic ubiquitinated protein inclusions in motor neurons and glial cells primarily in the spinal cord. While the vast majority of ALS occurs sporadically (sALS), in ~10% of cases, called familial ALS (fALS), there is clear indication of genetic inheritance. In the last decade, enormous progress was made in unravelling the aetiology of the disease, with the identification of ALS-causing mutations in new genes, as well as key molecular players involved in the origin or progression of ALS. However, much more needs to be done, as the pathogenic mechanisms triggered by a genetic or sporadic event leading to cytotoxicity and neuronal cell death are still poorly understood. The recent discoveries offer new possibilities for devising experimental animal and cellular models, which will hopefully contribute to the development of new techniques for early diagnosis and the identification of therapeutic targets for ALS. Here we review the current understanding of the aetiology, genetics, and pathogenic factors and mechanisms of ALS. We also discuss the challenges in deciphering ALS pathogenesis that result from the high complexity and heterogeneity of the disease.
Introduction
Amyotrophic lateral sclerosis (ALS), also called Charcot’s or Lou Gehrig’s disease, is an adult-onset neurodegenerative disorder. This motor neuron disease is characterised by rapidly progressive paralysis, atrophy, and respiratory failure leading to death typically within 1 to 5 years after symptom onset. The incidence of ALS is approximately 2 to 3 per 100,000 people each year, and the risk of developing the disease peaks between 50 and 75 years of age and is slightly higher in men. At the time of diagnosis, approximately 50% of motor neurons are already lost, highlighting the tremendous necessity to develop early diagnostic tests. There is no cure for ALS and the only effective treatment is riluzole, a glutamate release inhibitor, which was shown to slow the rate of progression and to prolong survival time, albeit only by approximately three months [1, 2].

Figure 1
Known genetic causes in familial and sporadic ALS.
Most ALS cases are sporadic (sALS) and only 10% are inherited, called familial (fALS). (A) 20% of fALS are caused by mutations in SOD1, which is the first known ALS-linked gene, identified in 1993. Mutations in the RNA/DNA-binding proteins TDP-43 and FUS cause a small fraction (~5%) of fALS each. Hexanucleotide repeat expansions in C9ORF72 are the most common genetic cause of ALS accounting for almost 40% of fALS in people of European ancestry. Several other genes have been identified as genetic causes of ALS and collectively today over 60% of fALS can be explained by known mutations. (B) While the aetiology of the vast majority of sALS remains unknown, mutations in genes known to cause fALS were identified in a small fraction of apparently sporadic patients. Most strikingly, hexanucleotide repeat expansions in C9ORF72 are causing approximately 7% of sALS without familial history in people of European ancestry.
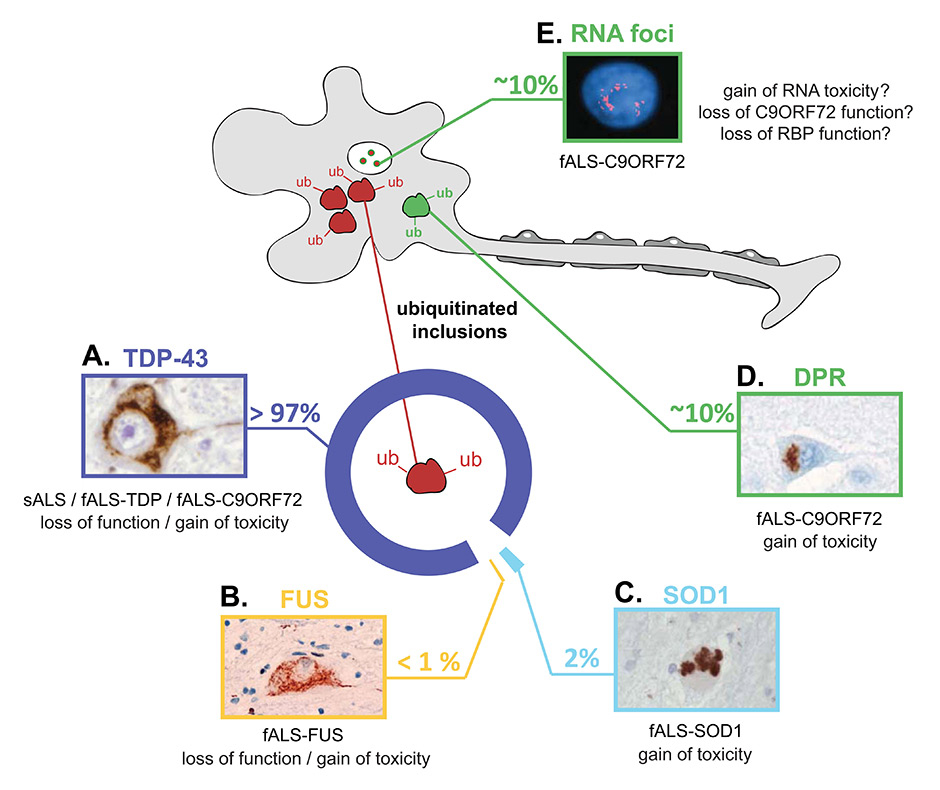
Figure 2
Pathogenic hallmarks of ALS and the underlying factors and mechanisms.
The pathologic hallmark of ALS is the presence of ubiquitinated protein inclusions in motor neurons and glial cells primarily of the spinal cord and motor cortex. The major protein component of these inclusions is different in the various types of ALS, highlighting the heterogeneity of the disease. (A) In the majority of ALS cases, including all sporadic and most familial cases, the ubiquitinated inclusions contain TDP-43. When TDP-43 accumulates in the cytoplasm, it is excluded from the nucleus, where it normally resides in healthy cells. These alterations suggest that either loss of normal TDP-43 function or gain of toxicity (or both) may trigger motor neuron dysfunction and death. (B) In 0.5% of fALS that carry mutations in FUS, the cytoplasmic inclusions do not contain TDP-43, but they contain FUS protein. Similar observations to that of TDP-43, with partial nuclear clearance of FUS suggest that its neurotoxity may be the result of either loss of function or gain of toxicity. (C) In fALS patients with SOD1 mutations, misfolded SOD1 accumulates and becomes toxic to neurons. These patients do not contain any TDP-43 or FUS inclusions. (D) In contrast, patients with hexanucleotide repeat expansions in C9ORF72show typical TDP-43, similar to those found in sALS (A). In addition, C9ORF72 patients accumulate abnormal dipeptide repeat (DPR) proteins, which are produced by an unconventional type of translation of the hexanucleotide repeat RNA, which escapes the nucleus, and binds to ribosomal complexes. In addition to spinal cord motor neurons, DPRs have been observed in the cerebellum, which is typically spared in non-C9ORF72 ALS. (E) In addition to TDP-43 and DPR pathology, C9ORF72 patients accumulate nuclear RNA foci that contain sense and antisense hexanucleotide repeats in neuronal and glial cells. Insert images reproduced with permission from (A) [54], (B) [60], (C) [35], (D) [39], (E) [20].
Jean-Martin Charcot first described the clinical presentation and lesion profile of ALS almost 150 years ago. The disease is characterised by upper and lower motor neuron degeneration, with the neurological progressive deterioration of the motor cortex, spinal cord and brainstem. ALS is linked to another neurodegenerative disease called frontotemporal lobar degeneration (FTLD), which is the most common type of dementia below 60 years of age and which is characterised by behavioural and language dysfunction. ALS and FTLD symptoms overlap in a subset of patients or within families [3–6] and the two conditions share common pathological phenotypes and causative gene mutations [7–9].
Approximately 10% of ALS and 30% of FTLD cases, called familial ALS (fALS) or FTLD (fFTLD) respectively, show clear family history indicating genetic inheritance. Superoxide dismutase 1 (SOD1) is the first ALS-linked gene that was identified in 1993 [10] and for almost fifteen years ALS research was focused on mutant forms of this protein [11, 12]. Since 2008, starting with the discovery of ALS-linked mutations in the DNA/RNA-binding proteins, TDP-43 [13–17] and FUS [18, 19], we have witnessed an era of unprecedented genetic discoveries in ALS. In 2011, the most common genetic cause of ALS was identified to be large expansions in an intronic hexanucleotide repeat region of a poorly characterised gene, called C9ORF72 [20, 21]. Moreover, in addition to SOD1, TARDBP, FUS and C9ORF72, many other genes were identified as rare genetic causes of ALS in the last years [22, 23] and collectively today, more than 60% of familial ALS can be explained by known gene mutations (fig. 1A).
In the remaining 90% of ALS, called sporadic ALS (sALS), there is no indication of genetic inheritance. While the cause of most sALS is not understood, recent studies have shown that fALS-linked mutations may also trigger disease in sporadic cases. Indeed a small percentage of sALS patients were shown to carry de novo mutations in known ALS-causing genes [24–27]. C9ORF72 repeat expansions, which were also found in a substantial fraction (~7%) of apparently sporadic ALS patients, most likely are not occurring de novo[28, 29], but rather represent cases with insufficient family history or incomplete penetrance [30]. Taken together, ~10% of apparently sporadic ALS cases are caused by known genetic mutations, while the aetiology of the rest ~90% sALS remains enigmatic (fig. 1B).
The pathological hallmark of ALS is the presence of ubiquitin-positive inclusions consisting of misfolded protein aggregates in the affected motor neurons and glial cells of the spinal cord and motor cortex. The major protein component of these inclusions in most ALS cases is TDP-43 [31, 32]. Notable exceptions are patients with mutations in SOD1 and FUS, which lack TDP-43 inclusions, but harbour misfolded SOD1 [33–35] or FUS [18, 19] protein accumulations, respectively. Moreover, patients with C9ORF72 expansions exhibit the most complex pathology with typical TDP-43 inclusions, atypical TDP-43 negative inclusions [20, 36, 37] that consist of abnormal dipeptide proteins [38–40] as well as nuclear repeat RNA foci [20, 41] (fig. 2).
The discoveries of the past decade have entirely revolutionised our view of ALS by introducing new key molecular players in pathogenesis. These discoveries have also questioned the current disease classifications with the recognition of the clinical, pathological, and genetic spectrum of ALS/FTLD and the unexpected blurring divide between sporadic and genetic disease. In this review, we first present the heterogeneity of ALS by introducing some of the main genes and proteins that play a role in one or several aspects of this complex disease. Then, we describe the current thinking on how these upstream events lead to the onset and rapid progression of ALS.
ALS is a heterogeneous disease with diverse genetic causes and complex pathology
Familial ALS is caused by mutations in many different genes that trigger pathogenesis, potentially through one or more common pathways [9]. In this review we focus on, arguably, the most important ALS-linked genes and proteins, namely SOD1, TDP-43, FUS and C9ORF72, which we discuss in the order of their chronological implication in the pathogenesis of ALS.
SOD1
Mutations in superoxide dismutase 1 (SOD1) was the first described genetic cause of ALS [10]. Today more than 170 mutations in the SOD1 gene have been identified to cause ALS. Interestingly, these mutations span the entirety of the SOD1 protein sequence [9], suggesting that a single amino acid change in any position of SOD1 is sufficient to disrupt its physiological conformation, leading to misfolding and accumulation of abnormal forms of the protein. In total, SOD1 mutations occur in almost 20% of familial and rare sporadic ALS cases (fig. 1).
SOD1 is a 153 amino acid enzyme that is responsible for scavenging free superoxide radicals in the body. Normally present in the cytoplasm or the mitochondrial intermembrane space as a stable homodimer, SOD1 misfolds when it carries ALS-causing mutations [42] or in cell stress conditions [43, 44]. Although the misfolded protein is ubiquitinated, and thus directed to degradation [45], SOD1 escapes the proteolytic process, which results in formation of ubiquitin positive inclusions observed in fALS cases [35].
The crucial role of misfolded SOD1 in the pathogenesis of familial ALS with SOD1 mutations is unequivocal. However, the presence of misfolded SOD1 in sporadic ALS cases remains controversial. While some studies have shown no evidence of misfolded SOD1 in sALS patients [35, 46], others reported a diffuse cytoplasmic misfolded SOD1 in motor neurons in several sALS cases, which were not linked to any SOD1 mutations [47]. This discrepancy may stem from the different anti-misfolded SOD1 antibodies used in these studies, as well as the inherent challenges in detecting misfolded proteins in postmortem autopsy material. Further studies are needed to resolve this issue that is of great importance for our understanding of the commonalities in disease pathogenesis of ALS cases with seemingly distinct origins.
TDP-43
An incredible transformation in our perception and understanding of ALS pathogenesis began with the identification of TAR-DNA-binding protein of 43 kDa (TDP-43) as the major component of ubiquitin positive neuronal cytoplasmic inclusions that are the hallmark of ALS and FTLD [31, 32]. This 414 amino acid RNA/DNA binding protein has two RNA recognition motifs (RRM1 and 2) and a glycine-rich region (GRR) that resembles the prion domain of yeast prion proteins. More than 35 mutations in the TARDBP gene, which encodes TDP-43, are causes of ALS [13–17]. Most of these mutations are clustered in exon 6 of the TARDBP gene encoding the C-terminal GRR [9, 48] and cause approximately 5% of fALS and <1% of sALS (fig. 1).
TDP-43 is a nuclear protein that belongs to the ribonucleoprotein family and is involved in RNA transcription, splicing and transport [9, 48]. It is also able to shuttle between the nucleus and the cytoplasm, where it is associated with granules [49], playing a key role in RNA transport. Under cell stress conditions, TDP-43 translocates to the cytoplasm, where it forms stress granules [50–52]. Stress granules are non-membranous cytoplasmic foci composed of non-translating mRNP that rapidly aggregate in cells exposed to stress conditions such as starvation or oxidation, allowing selective RNA inactivation [53]. The GRR region is necessary for this function through its prion-like ability to change conformation between a globular and an aggregated form [52].
In ALS, TDP-43 accumulation and aggregation is accompanied by a disturbance in its normal cellular localisation. In healthy cells, TDP-43 is mainly localised in the nucleus, whereas in affected cells, the misfolded form of the protein is redistributed to the cytoplasmic compartment, which leads to depletion/loss of nuclear TDP-43 [31, 32, 54]. Moreover, TDP-43 undergoes biochemical alterations such as hyperphosphorylation, polyubiquitination and C-terminal fragmentation [31, 32]. The mislocalisation and biochemical signature of pathological TDP-43 is characteristic for almost 97% of ALS cases, including most sporadic and familial ALS, with the exception of fALS caused by SOD1 or FUS mutations (fig. 2). The intracellular accumulation and aggregation of TDP-43 in fALS cases with TARDBP mutations resembles the pathogenic pattern described in sALS in the absence of mutations [17], suggesting that the same downstream pathways are operating in these two types of ALS.
FUS
More recently, Fused in Sarcoma (FUS), also called Translocated in Liposarcoma (TLS), has been identified as another protein involved in ALS. Mutations in the FUSgene have been described as a cause of ~4% of fALS [18, 19] and rare sALS cases (fig. 1). Interestingly, FUS and TDP-43 show significant structural and functional homology [48]. Indeed, FUS is a 526 amino acid protein that has the ability to bind nucleic acids and comprises a prion-like domain. The nucleic acid binding of FUS is due to the presence of glycine-arginine (R/G)-rich motifs, an RNA recognition motif (RRM), and a zinc finger (ZnF) domain. The first half of the protein, which contains a glutamine-glycine-serine-tyrosine (Q/G/S/Y)-rich region and the R/G-rich motifs, is responsible for the high aggregation propensity of FUS [55] and similarly to TDP-43, FUS is predicted to attain prion-like properties [56]. Moreover, like TDP-43, FUS is mainly nuclear, and utilises a transportin shuttle transport to reach the cytoplasm [57], where it partitions in stress granules [58, 59] and forms intracellular aggregates in fALS cases with FUS mutations [60]. In these familial cases, FUS inclusions occur in the absence of TDP-43-positive deposition [18, 19], an observation that in combination with the functional and structural similarities of TDP-43 and FUS [48], suggest that the two proteins trigger ALS by an independent initiating event, but potentially through common downstream pathways. For this reason, we will combine our discussion of TDP-43 and FUS in the rest of this manuscript.
C9ORF72
In 2011, the identification of a massive GGGGCC hexanucleotide repeat expansion in a poorly characterised gene called C9ORF72, as the most common genetic cause of ALS and FTLD [20, 21], was another major breakthrough in the field. In normal healthy controls, this repeat length is shorter than 25 units, whereas in ALS or FTLD patients it can expand up to 800 to 4400 units [20, 61]. In people of European ancestry, C9ORF72 hexanucleotide repeat expansions cause 40% of fALS and 25% of fFTLD (fig. 1), while up to 90% of families with concurrent ALS and FTLD have hexanucleotide repeat expansions in C9ORF72[22, 23, 62]. Unexpectedly, C9ORF72 hexanucleotide repeat expansions were also found in approximately 7% of apparently sporadic ALS cases in people of European ancestry, suggesting that the fraction of ALS with genetic origin may be larger than anticipated.
The normal function of C9ORF72 is not well understood, but recent studies suggest that it may be important for endosomal trafficking [63–65]. C9ORF72 is homologous to proteins related to Differentially Expressed in Normal and Neoplasia (DENN), which is a GDP/GTP exchange factor (GEF) that activates Rab-GTPases [63, 64]. Furthermore, C9ORF72 was found in the extracellular space and in cytoplasmic vesicles, where it colocalises with Rab proteins that are implicated in autophagy and endocytic transport in mouse and human neurons [65].
Patients with C9ORF72 expansions show the most complex pathology of all ALS cases. They not only harbour TDP-43 inclusions, similar to those of sALS patients (fig. 2), but a striking pathological hallmark of C9ORF72 ALS is the presence of atypical TDP-43-negative ubiquitinated protein inclusions in the hippocampus and cerebellum [20, 36, 37], which is usually spared in non-C9ORF72 ALS [66, 67]. These inclusions were recently identified to contain five different aggregating dipeptide-repeat (DPR) proteins [40, 68–70], namely poly-(Gly-Ala), poly-(Gly-Pro), poly-(Gly-Arg), poly-(Pro-Arg) and poly-(Pro-Ala) [38–40]. The latter are generated from the hexanucleotide repeat RNA by an unconventional type of translation, called repeat-associated non-ATG (RAN) translation, which operates in both sense [38, 39] and antisense [40] transcripts. Lastly, the hexanucleotide repeat expansions accumulate in nuclear RNA foci in neurons [20] and glial cells [41] of C9ORF72 ALS patients.
From human pathology and genetics to experimental ALS models
The major advancements in ALS genetics and pathology have revolutionised the way we understand the disease and they are directing research towards unravelling the underlying pathogenic mechanisms. Just like in any other human disease research field, the first step towards identification of molecular mechanisms is the construction of experimental animal and cellular models, either by mutating or depleting genes encoding for ALS-linked proteins.
SOD1
Loss of SOD1 does not appear to cause spontaneous motor neuron degeneration, since homozygote SOD1 knockout mice are viable and develop without obvious motor abnormalities [71]. In contrast, transgenic mice expressing human SOD1 with fALS-linked mutations, such as G93A [72] or G85R [42] were shown to develop late-onset, progressive paralysis and aggressive disease with short duration [42]. Strikingly, the G85R mutant provokes disease at protein expression levels below endogenous mouse SOD1 [73]. Many more mutant SOD1 animal models have been created and were shown to faithfully recapitulate the clinical and pathological features of human ALS, as they develop an adult-onset, rapidly lethal motor neuron disease with gliosis and ubiquitinated misfolded SOD1 deposition (reviewed in [12]). The symptom onset, lifespan and rate of progression of the mutant SOD1 models vary depending on the mutation and expression level of human SOD1.
Early reports on transgenic mice overexpressing wild type (WT) SOD1, which were produced to study Down syndrome – as the SOD1 gene is located on chromosome 21 – showed that increased levels of non-mutated SOD1 protein do not lead to ALS, since they were first described as phenotypically normal, before some complications linked to Down syndrome were identified [74, 75]. Recently, massive overexpression (50–fold) of human WT SOD1 was shown to cause motor neuron degeneration with deposition of misfolded SOD1, reaching terminal stages at ~400 days of age [76], suggesting that SOD1 can become pathologic, even in the absence of mutations.
TDP-43 and FUS
The effects of mutation or depletion of TDP-43 and FUS have been studied in various models. In mice, homozygous deletion of TARDBP causes early embryonic lethality due to defective outgrowth of the inner cell mass prior to implantation [77–79]. Interestingly, aged heterozygous TDP-43 mice develop mild motor dysfunction, despite apparently unchanged TDP-43 protein levels [79]. Two independent studies have shown that selective deletion of TDP-43 from motor neurons induces adult-onset muscle weakness and atrophy [80, 81]. Importantly, in both studies the mice were not reported to develop paralysis, despite motor neuron loss. More recently, constitutive expression of an artificial micro RNA (amiR), targeting TDP-43 resulted in its partial loss, especially in astrocytes [82]. These mice showed a complex phenotype, starting with transient hyperactivity at 40 days, which subsided by 70 days, after which they developed weakness, which progressed to paralysis and death at approximately 150 days. In contrast to the conditional motor neuron TDP-43 depletion [80], which showed more prominent phenotype in males, the amiR-expressing mice showed no gender disparities [82]. Taken together the above studies suggest that while loss of TDP-43 may contribute to motor neuron degeneration, it is not possible to mimic the full spectrum of the ALS clinical presentation by mere depletion of TDP-43.
The alternative hypothesis that mutant or aggregated wild type TDP-43 acquires a new toxic function was tested in a series of transgenic mice and rats. Overexpression of mutant [83–87] or wild type [88] TDP-43 in rodent models induced various abnormalities with aspects of motor neuron disease, but failed to clearly reproduce the ALS symptom spectrum. Disappointingly, in sharp contrast with the mutant SOD1 models, none of the existing transgenic TDP-43 overexpressing rodents developed progressive paralysis or TDP-43 protein aggregation and nuclear clearance.
Disruption of the Fus gene in inbred mice is lethal due to major defects in B-lymphocyte development [89], whereas similar disruption in an outbred background produces mice that survive until adulthood and develop male sterility [90]. While the cause of the discrepancy in the phenotypes of these two Fus knockout lines is not known, the fact that outbred mice without Fus reach adulthood without developing motor neuron dysfunction speaks against the contribution of FUS loss of function in ALS. In rats, overexpression of human FUS carrying the R521C mutation, resulted in loss of cortical, hippocampal and motor neurons, accompanied by denervation and paralysis [91]. Transgenic mice overexpressing human wild type FUS show no overt phenotype, unless they are bred to homozygocity, in which case they develop aggressive motor neuron degeneration and death at ~100 days of age [92]. Interestingly, the homozygous mice that express ~2–fold above endogenous FUS levels develop skein-like FUS inclusions in motor neurons.
Nevertheless, the apparent inability of these models to reproduce an ALS-like disease and mimic the associated pathology, questions their relevance for ALS research. The lack of faithful TDP-43 and FUS disease models is currently one of biggest challenges in ALS research and alternative models that mimic all aspects of the disease are urgently needed.
C9ORF72
The identification of C9ORF72 hexanucleotide repeat expansions in ALS is very recent, and the generation of relevant animal models are currently underway in several labs worldwide. First reports on mice with heterozygous disruption of the mouse homologue of C9orf72, suggested that the mice are born without obvious abnormalities [93, 94], but no data on the consequences of the complete loss of C9orf72 or expression of expanded hexanucleotide repeats is available at the time of writing. Nevertheless, cellular models based on human motor neurons differentiated from induced pluripotent stem cells (iPSCs) from ALS patients carrying the C9ORF72 repeat expansion were shown to recapitulate the main pathological aspects, including repeat RNA foci and dipeptide protein inclusions [41, 95–98]. We anticipate that these models will become extremely valuable in understanding C9ORF72-ALS pathogenesis.
From experimental models to molecular ALS pathogenesis
The current experimental models of ALS, allow us to investigate the molecular mechanisms of disease pathogenesis. So, how do those distinct and very diverse genetic mutations and protein misfolding events cause selective motor neuron cytotoxicity and death, leading to the degeneration of an entire part of the central nervous system? And most importantly, what are – if any – the common downstream events that give rise to the characteristic ALS clinical presentation?
SOD1
Studies on the SOD1 animal models revealed that compromising the enzymatic activity of SOD1 has little or no effect on ALS, but that a toxic property of misfolded SOD1 drives disease. Unexpectedly, this toxic property does not simply operate within the vulnerable motor neurons. Neuronal mutant SOD1 – which is sufficient to cause ALS-like symptoms in mice [99] – rather directs the onset and early disease [100, 101], while SOD1 expression in glial cells, such as microglia [101] or astrocytes [102], accelerates disease progression. Most recently, mutant SOD1 within oligodendroglia has been shown to cause their dysfunction, which in turn, substantially accelerated disease onset and survival of mutant SOD1 mice [103]. In conclusion, mutant SOD1 toxicity is non-cell autonomous [12] and the convergence of toxicity within motor neurons and their neighbouring glial cells triggers disease.
While the non-cell autonomous nature of ALS is one of the major lessons learned from SOD1 animal models, a statement of concern is that 20 years after the initial discovery, no consensus has emerged as to the exact pathogenic mechanism(s) triggered by misfolded SOD1. Nevertheless, multiple mechanisms have been described, many or all of which are operating in different cell types and are thereby contributing to different aspects of disease (for detailed review see [12]). One of the first identified cytotoxic pathways of mutant SOD1 is excitotoxicity, due to failure in clearing synaptic glutamate, the neurotransmitter that triggers motor neurons to fire. Excitotoxicity in mutant SOD1 patient samples [104] and animal models [42, 105] was shown to originate from deficiency in the glutamate transporter EAAT2, due to decrease in the exosomal transfer of neuronal miR-124a into astrocytes [106]. Mutant SOD1 motor neurons from transgenic mice [107] or human patients [98] also show intrinsic membrane hyperexcitability, which was suggested to contribute to the selective vulnerability of motor neurons in ALS [108]. Mutant SOD1 induces endoplasmic reticulum (ER) stress through multiple mechanisms. Accumulation of misfolded SOD1 aggregates bind to the ER chaperone BiP (Binding immunoglobulin Protein) [109], which mediates the activation of ER stress transducers, such as IRE1, PERK and ATF6. Moreover, mutant SOD1 binds to Derlin-1 [110], a protein that dislocates misfolded proteins from the ER to the cytosol and whose inhibition triggers ER stress. Indeed, ER-related genes are upregulated at pre-symptomatic stages of mutant SOD1 mice [111], as well as in human iPSC-derived motor neurons [112]. Mutant SOD1 was also shown to impair the proteasome [113] and block autophagy [114] leading to dysregulation of protein levels, and to decreased clearance of mutant SOD1. In addition, misfolded SOD1 directly damages mitochondria through deposition on the cytoplasmic face of the outer membrane of the organelles [115, 116]. Other mutant SOD1-mediated neurotoxic mechanisms include extracellular toxicity from aberrant SOD1 secretion [117], excessive production of extracellular superoxide [118], axonal structure and transport dysregulation [119], or even spinal cord microhaemorrhages [120]. The challenge in ALS research is now to define which of the above mechanisms are merely downstream consequences of neuronal dysfunction and which ones occur early in disease, thereby determining the onset or progression of ALS.
TDP-43 and FUS
The pathogenic mechanisms of TDP-43 and FUS may result 1) from loss of their normal function due to their nuclear depletion in ALS, 2) from gain of toxic function due to their cytoplasmic aggregation, or 3) from a combination of the two mechanisms that are not mutually exclusive.
The motor neuron degeneration developed after in vivo depletion of TDP-43 in mouse models [80–82] supports the notion that TDP-43 loss of function contributes to ALS pathogenesis. TDP-43 [121, 122] and FUS [123, 124] bind thousands of RNA and are involved in multiple steps of RNA transcription and processing [48]. The two proteins bind mostly to distinct RNAs, but also share a set of approximately 45 common targets that depend on the function of both proteins and whose disturbance contributes to neuronal dysfunction and death in ALS [121, 123]. Most recently, TDP-43 was shown to form cytoplasmic mRNP granules that deliver target mRNAs to distal neuronal compartments, a function that is impaired by ALS-linked mutations [49], suggesting another potential pathogenic mechanism in ALS. Taken together, these findings indicate that alterations in RNA homeostasis and transport play a key role in the pathogenesis of ALS.
The participation of TDP-43 [50–52] and FUS [58, 59] in stress granules, suggest a possible gain of toxic function mechanism in ALS, as these RNA granules might be precursors of the pathological cytoplasmic mislocalisation and aggregation seen in patients. TDP-43 intrinsic aggregative properties have been described, and its propensity for toxic misfolding and deposition is accentuated by ALS-linked mutations [125]. This cytotoxic deposition could sequester RNA and RNA binding proteins, thereby contributing to pathogenesis. While the observation that RNA binding of TDP-43 [126, 127] and FUS [55] is required for their cytotoxicity supports this view, the exact RNAs and/or RNA-binding proteins that are sequestered in the cytoplasmic TDP-43 and FUS aggregates of human neurons remain unknown. Further evidence that stress granule formation is intimately linked to pathogenesis came from a recent study, which reported that genes modulating stress granules are strong modifiers of TDP-43 toxicity and that prolonged stress granule dysfunction in human ALS spinal cord neurons is likely a direct contributor to pathogenesis [128].
C9ORF72
Several cytotoxic mechanisms have been described for C9ORF72 hexanucleotide repeat expansions and their relative contribution to ALS pathogenesis is currently under investigation. First of all, patients with C9ORF72 expansions show typical TDP-43 cytoplasmic inclusions with nuclear clearance [20, 36, 37, 61], suggesting that all mechanisms described above are relevant for this type of ALS. Moreover, patients carrying hexanucleotide repeat expansions were shown to produce reduced levels of C9ORF72 RNA [20, 61], due to the inactivation of the repeat-containing allele via promoter hypermethylation [129], or transcriptional abortion of repeat RNAs [130]. Reduced C9ORF72 RNA and, by extension, protein may not be enough to perform its normal function, a mechanism known as haploinsufficiency. The long-term phenotype (or lack thereof) of mice with heterozygous disruption of the mouse C9orf72homologue [93, 94], could provide a definitive answer for the contribution of haploinsufficiency in C9ORF72–ALS pathogenesis. Another possibility is that the repeat RNA is toxic per se. Indeed, the DNA and RNA hexanucleotide repeats form complex structures, which result in transcriptional hindrance and in accumulation of abortive transcripts that contain the hexanucleotide repeats [20, 40, 41, 130]. Those abortive transcripts form G-quadruplexes and hairpins and bind essential proteins, primarily nucleolin, whose sequestration leads to nucleolar stress and other downstream defects [130]. Moreover, the repeat-containing transcripts can also escape the nucleus and associate with ribosomal complexes in the cytoplasm, where they are subjected to the repeat-associated non-ATG (RAN) translation [38–40], leading to production and accumulation of abnormal dipeptide proteins. Originally, poly-GA aggregates were shown to be the prominent DPRs in patients [69], and in rat primary neurons [68], suggesting a direct role in neurodegeneration. Poly-GA DPRs were indeed very recently shown to cause neurotoxicity in vitro by inducing ER stress [131]. However, conflicting results showed that the most common DPRs present in both upper and lower motor neurons are the poly-GP [40], which were most recently shown to be present in the cerebrospinal fluid of C9ORF72 patients [132]. Moreover, the relatively short synthetic sense poly-GR and antisense poly-PR DPRs were reported to be highly toxic, when exogenously applied to human astrocytes, since they entered the nucleus, bound to nucleoli and killed the cells in culture [133]. Finally, expression of “protein-only” poly-GR and poly-PR constructs in Drosophila was associated with progressive neuronal death in vivo, which is perhaps attributable to the basic nature or a common structural motif of these DPRs [70]. Altogether, DPR toxicity is a central pathogenic mechanism in C9ORF72-ALS.
Does “prion-like” propagation of ALS proteins drive disease progression?
The physiological function of a protein is often linked to a unique structure and stable three-dimensional conformation, as a result of a normal folding pathway. Some proteins can gain toxic properties upon a disturbance of their structure that can stably exist and be transferred to homologous proteins if their structure allows this pathological conformation. This mechanism underlies the pathogenesis of prion diseases [134], and is suggested to contribute to the progression of many neurodegenerative diseases [135], such as Alzheimer’s (amyloid-β [136] and Tau [137]) and Parkinson’s (α-synuclein) [138] diseases. We recently proposed that a similar mechanism may occur in ALS [56], and many ongoing studies aim now at describing the prion-like pathogenesis of ALS, and the underlying prion-like seeding and spreading mechanisms of the proteins implicated in the disorder. Those could lead, from a single protein-misfolding event, to an actual neurodegenerative disease, following an autocatalytic protein-protein transfer of structural and pathological information.
SOD1
SOD1 spontaneously forms fibrils or aggregates in vitro and ALS-linked mutations enhance this phenomenon [139, 140]. In vitro, the destabilisation of SOD1 structure under experimental specific conditions led to its aggregation, and even triggered a prion-like seeding and increase of aggregation [140]. Temperature increase, reducing conditions, addition or disruption of disulfide-bonds seem to be the destabilising factors that can initiate the seeded misfolding of SOD1 [141–143]. A major step in that field was the finding that SOD1 misfolding can be transmitted, not only from protein to protein, but also from cell to cell following a prion-like propagation mechanism [142, 144, 145]. In vitro formed SOD1 aggregates were actively integrated in neuronal cells through macropinocytosis [142], and triggered aggregation of endogenously expressed SOD1. Once SOD1 aggregation occurs in a cell, the aggregates can be released to the extracellular space and then transferred from cell to cell in a persistent way, heritable after cell passages, supporting a prion-like propagation of aggregation.
TDP-43 and FUS
The aggregation properties of TDP-43 and FUS are associated with their physiological role in the formation of stress granules [50–52, 57, 58]. The existence of two distinct conformational states – one of which is highly aggregative – depends on the presence of GRR, also called prion-like or low complexity domain. Indeed, like SOD1, TDP-43 is able to form aggregates in vitro [146, 147] and its C-terminal region, where the prion-like domain resides, shows the highest aggregation propensity, while ALS-linked mutations enhance this phenomenon [125, 147]. Compared to TDP-43 and SOD1, FUS shows the highest aggregation potential, as it forms aggregates even in native conditions, without any experimental procedure, such as shaking or sonication [55], but this property is not altered by the presence of ALS-causing mutations [55, 57]. Instead, ALS-linked mutations residing in the nuclear localisation signal of FUS increase its cytoplasmic retention, thereby, possibly facilitating the initiation of cytoplasmic FUS aggregation [57].
A wide step in deciphering prion-like spreading of ALS proteins was made with the very recent description of prion-like properties of TDP-43 extracted from ALS patient brains [148]. In this study, insoluble TDP-43 from ALS brains was introduced as seeds into cells expressing human wild type TDP-43. It induced phosphorylated and ubiquitinated TDP-43 aggregation in a self-templating manner. Furthermore, C-terminal fragments of the insoluble protein acted as seeds, and were shown to be sufficient for templating its self-aggregation. All together, these results indicate that TDP-43 and FUS have prion-like properties that may play a key role in the progression of the respective proteinopathy.
An important contributor to self-propagation of pathologic conformations of TDP-43 and FUS stems from their well-documented autoregulation pathways. Indeed, both TDP-43 [121, 149] and FUS [123, 150] proteins were shown to bind their own RNA in the nucleus, thereby autoregulating their level of expression. This may possibly create a vicious circle of more and more protein substrate production, as a result of sequestration of newly produced TDP-43 and FUS into the cytoplasmic aggregates.
C9ORF72
All above mechanisms may contribute to C9ORF72 disease and the spreading of TDP-43 pathology in patients with hexanucleotide repeat expansions. Moreover, the abnormal dipeptide products [38–40], once made by RAN-translation, may undergo seeded aggregation.
Conclusions and open questions
In conclusion, ALS is complex and heterogeneous disease that is caused by many, seemingly diverse, upstream events. The great discoveries that we discussed have not only advanced our understanding of ALS, but they have also, inevitably, raised one major question: is ALS one disease, or does each type of ALS described above represent a distinct disease with independent aetiology and pathogenic mechanisms? Alternatively, do these diverse genetic causes of ALS trigger common downstream mechanisms that lead to selective motor neuron degeneration? And if yes, which are these common mechanisms? As we move forward, the answers to these questions will be instrumental, for devising reliable diagnostic tests and most importantly for the identification of therapeutic targets for this devastating and still untreatable disease.
Uncategorised references
Acknowledgements:The authors thank Ms Zuzanna Maniecka and Dr. Marian Hruska-Plochan for critical comments on the manuscript. FL is the recipient of aMilton Safenowitz Postdoctoral Fellowship for ALS Research Award from the ALS Association. MP has been awarded an Swiss National Science Foundation Professorship and a Career Development Award from the Human Frontier Science Program. This work was supported by the Clinical Research Priority Program Small RNAs of the University of Zurich.
1 Lacomblez L, et al. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet. 1996;347(9013):1425–31.
2 Diagnosis ETFo, et al. EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS) – revised report of an EFNS task force. Eur J Neurol. 2012;19(3):360–75.
3 Caselli RJ, et al. Rapidly progressive aphasic dementia and motor neuron disease. Ann Neurol. 1993;33(2):200–7.
4 Neary D, Snowden JS, Mann DM. Cognitive change in motor neurone disease/amyotrophic lateral sclerosis (MND/ALS). J Neurol Sci. 2000;180(1–2):15–20.
5 Lomen-Hoerth C, Anderson T, Miller B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology. 2002;59(7):1077–9.
6 Ringholz GM, et al. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology. 2005;65(4):586–90.
7 Mackenzie IRA, Rademakers R. The molecular genetics and neuropathology of frontotemporal lobar degeneration: recent developments. Neurogenetics. 2007;8(4):237–48.
8 Polymenidou M, et al. Misregulated RNA processing in amyotrophic lateral sclerosis. Brain Res. 2012;1462:3–15.
9 Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79(3):416–38.
10 Rosen DR, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62.
11 Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006;7(9):710–23.
12 Ilieva H, Polymenidou M, Cleveland DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol. 2009;187(6):761–72.
13 Gitcho MA, et al. TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol. 2008;63(4):535–8.
14 Kabashi E, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40(5):572–4.
15 Yokoseki A, et al. TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann Neurol. 2008;63(4):538–42.
16 Sreedharan J, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319(5870):1668–72.
17 Van Deerlin VM, et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7(5):409–16.
18 Vance C, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323(5918):1208–11.
19 Kwiatkowski TJ, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323(5918):1205–8.
20 DeJesus-Hernandez M, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–56.
21 Renton AE, et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21–Linked ALS-FTD. Neuron. 2011: p. 1–19.
22 Rademakers R, van Blitterswijk M. Motor neuron disease in 2012: Novel causal genes and disease modifiers. Nat Rev Neurol. 2013;9(2):63–4.
23 Renton AE, Chio A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014;17(1):17–23.
24 Alderman EM, et al. Angiogenic activity of human tumor plasma membrane components. Biochemistry. 1985;24(27):7866–71.
25 Chio A, et al. A de novo missense mutation of the FUS gene in a “true” sporadic ALS case. Neurobiol Aging. 2011;32(3):553 e23–6.
26 Dejesus-Hernandez M, et al. De novo truncating FUS gene mutation as a cause of sporadic amyotrophic lateral sclerosis. Hum Mutat. 2010;31(5):E1377–E1389.
27 Lattante S, et al. Contribution of major amyotrophic lateral sclerosis genes to the etiology of sporadic disease. Neurology. 2012.
28 Pamphlett R, et al. Transmission of C9orf72 hexanucleotide repeat expansions in sporadic amyotrophic lateral sclerosis: an Australian trio study. Neuroreport, 2012.
29 van der Zee J, et al. A pan-European study of the C9orf72 repeat associated with FTLD: geographic prevalence, genomic instability, and intermediate repeats. Hum Mutat. 2013;34(2):363–73.
30 Cruts M, et al. Current insights into the C9orf72 repeat expansion diseases of the FTLD/ALS spectrum. Trends Neurosci. 2013;36(8):450–9.
31 Neumann M, et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science. 2006;314(5796):130–3.
32 Arai T, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351(3):602–11.
33 Tan C-F, et al. TDP-43 immunoreactivity in neuronal inclusions in familial amyotrophic lateral sclerosis with or without SOD1 gene mutation. Acta Neuropathol. 2007;113(5):535–42.
34 Mackenzie IRA, et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol. 2007;61(5):427–34.
35 Kerman A, et al. Amyotrophic lateral sclerosis is a non-amyloid disease in which extensive misfolding of SOD1 is unique to the familial form. Acta Neuropathol. 2010;119(3):335–44.
36 Al-Sarraj S, et al. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72–linked FTLD and MND/ALS. Acta Neuropathol. 2011;122(6):691–702.
37 Troakes C, et al. An MND/ALS phenotype associated with C9orf72 repeat expansion: abundant p62–positive, TDP-43–negative inclusions in cerebral cortex, hippocampus and cerebellum but without associated cognitive decline. Neuropathology. 2012;32(5):505–14.
38 Ash PEA, et al. Unconventional Translation of C9ORF72 GGGGCC Expansion Generates Insoluble Polypeptides Specific to c9FTD/ALS. Neuron. 2013;77(4):639–46.
39 Mori K, et al. The C9orf72 GGGGCC Repeat Is Translated into Aggregating Dipeptide-Repeat Proteins in FTLD/ALS. Science, 2013.
40 Zu T, et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci U S A. 2013;110(51):E4968–77.
41 Lagier-Tourenne C, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A. 2013;110(47):E4530–9.
42 Bruijn LI, et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1–containing inclusions. Neuron. 1997;18(2):327–38.
43 Rakhit R, et al. Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis. J Biol Chem. 2002;277(49):47551–6.
44 Furukawa Y, et al. Complete loss of post-translational modifications triggers fibrillar aggregation of SOD1 in the familial form of amyotrophic lateral sclerosis. J Biol Chem. 2008;283(35):24167–76.
45 Basso M, et al. Insoluble mutant SOD1 is partly oligoubiquitinated in amyotrophic lateral sclerosis mice. J Biol Chem. 2006;281(44):33325–35.
46 Brotherton TE, et al. Localization of a toxic form of superoxide dismutase 1 protein to pathologically affected tissues in familial ALS. Proc. Natl Acad Sci U S A. 2012;109(14):5505–10.
47 Bosco DA, et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci. 2010;13(11):1396–403.
48 Lagier-Tourenne C, Polymenidou M, Cleveland DW. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Human Molecular Genetics. 2010;19(R1):R46–R64.
49 Alami NH, et al. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron. 2014;81(3):536–43.
50 Wang I-F, et al. TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J Neurochem. 2008;105(3):797–806.
51 Colombrita C, et al. TDP-43 is recruited to stress granules in conditions of oxidative insult. J Neurochem. 2009;111(4):1051–61.
52 Dewey CM, et al. TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol Cell Biol. 2011;31(5):1098–108.
53 Anderson P, Kedersha N. RNA granules: post-transcriptional and epigenetic modulators of gene expression. Nat Rev Mol Cell Biol. 2009;10(6):430–6.
54 Igaz LM, et al. Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am J Pathol. 2008;173(1):182–94.
55 Sun Z, et al. Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol. 2011;9(4):e1000614.
56 Polymenidou M, Cleveland DW. The seeds of neurodegeneration: prion-like spreading in ALS. Cell. 2011;147(3):498–508.
57 Dormann D, et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. The EMBO Journal. 2010;29(16):2841–57.
58 Bentmann E, et al. Requirements for stress granule recruitment of fused in Sarcoma (FUS) and TAR DNA binding protein of 43 kDa (TDP-43). J Biol Chem. 2012.
59 Dormann D, et al. Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. The EMBO Journal. 2012: p. 1–18.
60 Mackenzie IRA, et al. Pathological heterogeneity in amyotrophic lateral sclerosis with FUS mutations: two distinct patterns correlating with disease severity and mutation. Acta Neuropathol. 2011;122(1):87–98.
61 Gijselinck I, et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 2012;11(1):54–65.
62 Majounie E, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012;11(4):323–30.
63 Zhang D, et al. Discovery of Novel DENN Proteins: Implications for the Evolution of Eukaryotic Intracellular Membrane Structures and Human Disease. Front Genet. 2012;3:283.
64 Levine TP, et al. The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics. 2013;29(4):499–503.
65 Farg MA, et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet. 2014.
66 Geser F, et al. Evidence of multisystem disorder in whole-brain map of pathological TDP-43 in amyotrophic lateral sclerosis. Arch Neurol. 2008;65(5):636–41.
67 Geser F, et al. Clinical and pathological continuum of multisystem TDP-43 proteinopathies. Arch. Neurol. 2009;66(2):180–9.
68 May S, et al. C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol. 2014.
69 Mori K, et al. hnRNP A3 binds to GGGGCC repeats and is a constituent of p62–positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol. 2013;125(3):413–23.
70 Mizielinska S, et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science. 2014.
71 Reaume AG, et al. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet. 1996;13(1):43–7.
72 Gurney ME, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264(5166):1772–5.
73 Bruijn LI, et al. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281(5384):1851–4.
74 Epstein CJ, et al. Transgenic mice with increased Cu/Zn-superoxide dismutase activity: animal model of dosage effects in Down syndrome. Proc Natl Acad Sci U S A. 1987;84(22):8044–8.
75 Schickler M, et al. Diminished serotonin uptake in platelets of transgenic mice with increased Cu/Zn-superoxide dismutase activity. EMBO J. 1989;8(5):1385–92.
76 Graffmo KS, et al. Expression of wild-type human superoxide dismutase-1 in mice causes amyotrophic lateral sclerosis. Hum Mol Genet. 2013;22(1):51–60.
77 Wu L-S, et al. TDP-43, a neuro-pathosignature factor, is essential for early mouse embryogenesis. Genesis. 2010;48(1):56–62.
78 Sephton CF, et al. TDP-43 is a developmentally regulated protein essential for early embryonic development. J Biol Chem. 2010;285(9):6826–34.
79 Kraemer BC, et al. Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathol. 2010;119(4):409–19.
80 Wu LS, Cheng WC, Shen CK. Targeted Depletion of TDP-43 Expression in the Spinal Cord Motor Neurons Leads to the Development of Amyotrophic Lateral Sclerosis (ALS)-like Phenotypes in Mice. Journal of Biological Chemistry. 2012.
81 Iguchi Y, et al. Loss of TDP-43 causes age-dependent progressive motor neuron degeneration. Brain. 2013.
82 Yang C, et al. Partial loss of TDP-43 function causes phenotypes of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2014;111(12):E1121–9.
83 Wegorzewska I, et al. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A. 2009;106(44):18809–14.
84 Zhou H, et al. Transgenic rat model of neurodegeneration caused by mutation in the TDP gene. PLoS Genet. 2010;6(3):e1000887.
85 Igaz LM, et al. Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J Clin Invest. 2011;121(2):726–38.
86 Huang C, et al. Mutant TDP-43 in motor neurons promotes the onset and progression of ALS in rats. J Clin Invest. 2012;122(1):107–18.
87 Arnold ES, et al. ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc Natl Acad Sci U S A. 2013;110(8):E736–45.
88 Wils H, et al. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A. 2010;107(8):3858–63.
89 Hicks GG, et al. Fus deficiency in mice results in defective B-lymphocyte development and activation, high levels of chromosomal instability and perinatal death. Nat Genet. 2000;24(2):175–9.
90 Kuroda M, et al. Male sterility and enhanced radiation sensitivity in TLS(-/-) mice. The EMBO Journal. 2000;19(3):453–62.
91 Huang C, et al. FUS transgenic rats develop the phenotypes of amyotrophic lateral sclerosis and frontotemporal lobar degeneration. PLoS Genet. 2011;7(3):e1002011.
92 Mitchell JC, et al. Overexpression of human wild-type FUS causes progressive motor neuron degeneration in an age- and dose-dependent fashion. Acta Neuropathol. 2012.
93 Suzuki N, et al. The mouse C9ORF72 ortholog is enriched in neurons known to degenerate in ALS and FTD. Nat Neurosci. 2013;16(12):1725–7.
94 Panda SK, et al. Highly efficient targeted mutagenesis in mice using TALENs. Genetics. 2013;195(3):703–13.
95 Donnelly CJ, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80(2):415–28.
96 Sareen D, et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med. 2013;5(208):208ra149.
97 Almeida S, et al. Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol. 2013;126(3):385–99.
98 Wainger BJ, et al. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 2014;7(1):1–11.
99 Jaarsma D, et al. Neuron-specific expression of mutant superoxide dismutase is sufficient to induce amyotrophic lateral sclerosis in transgenic mice. J Neurosci. 2008;28(9):2075–88.
100 Ralph GS, et al. Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat Med. 2005;11(4):429–33.
101 Boillee S, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312(5778):1389–92.
102 Yamanaka K, et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11(3):251–3.
103 Kang SH, et al. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat Neurosci. 2013;16(5):571–9.
104 Rothstein JD, et al. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38(1):73–84.
105 Howland DS, et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci U S A. 2002;99(3):1604–9.
106 Morel L, et al. Neuronal exosomal miRNA-dependent translational regulation of astroglial glutamate transporter GLT1. J Biol Chem. 2013;288(10):7105–16.
107 van Zundert B, et al. Neonatal neuronal circuitry shows hyperexcitable disturbance in a mouse model of the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J Neurosci. 2008;28(43):10864–74.
108 Saxena S, Caroni P. Selective neuronal vulnerability in neurodegenerative diseases: from stressor thresholds to degeneration. Neuron. 2011;71(1):35–48.
109 Kikuchi H, et al. Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc Natl Acad Sci U S A. 2006;103(15):6025–30.
110 Nishitoh H, et al. ALS-linked mutant SOD1 induces ER stress- and ASK1–dependent motor neuron death by targeting Derlin-1. Genes Dev. 2008;22(11):1451–64.
111 Saxena S, Cabuy E, Caroni P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nature Publishing Group. 2009;12(5):627–36.
112 Kiskinis E, et al. Pathways Disrupted in Human ALS Motor Neurons Identified through Genetic Correction of Mutant SOD1. Cell Stem Cell. 2014.
113 Bendotti C, et al. Dysfunction of constitutive and inducible ubiquitin-proteasome system in amyotrophic lateral sclerosis: implication for protein aggregation and immune response. Progr Neurobiol. 2012;97(2):101–26.
114 Chen S, et al. Autophagy Dysregulation in Amyotrophic Lateral Sclerosis. Brain Pathology. 2011;22(1):110–6.
115 Liu J, et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron. 2004;43(1):5–17.
116 Vande Velde C, et al. Selective association of misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria. Proc Natl Acad Sci U S A. 2008;105(10):4022–7.
117 Zhao W, et al. Extracellular mutant SOD1 induces microglial-mediated motoneuron injury. Glia. 2010;58(2):231–43.
118 Harraz MM, et al. SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J Clin Invest. 2008;118(2):659–70.
119 Williamson TL, Cleveland DW. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci. 1999;2(1):50–6.
120 Zhong Z, et al. ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat Neurosci. 2008;11(4):420–2.
121 Polymenidou M, et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci. 2011;14(4):459–68.
122 Tollervey JR, et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci. 2011;14(4):452–8.
123 Lagier-Tourenne C, et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci. 2012;15(11):1488–97.
124 Rogelj B, et al. Widespread binding of FUS along nascent RNA regulates alternative splicing in the brain. Sci Rep. 2012;2:603.
125 Johnson BS, et al. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem. 2009;284(30):20329–39.
126 Elden AC, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466(7310):1069–75.
127 Voigt A, et al. TDP-43-mediated neuron loss in vivo requires RNA-binding activity. PLoS ONE. 2010;5(8):e12247.
128 Kim HJ, et al. Therapeutic modulation of eIF2alpha phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat Genet. 2014;46(2):152–60.
129 Xi Z, et al. Hypermethylation of the CpG island near the G4C2 repeat in ALS with a C9orf72 expansion. Am J Hum Genet. 2013;92(6):981–9.
130 Haeusler AR, et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 2014;507(7491):195–200.
131 Zhang YJ, et al. Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol. 2014.
132 Su Z, et al. Discovery of a Biomarker and Lead Small Molecules to Target r(GGGGCC)-Associated Defects in c9FTD/ALS. Neuron. 2014.
133 Kwon I, et al. Poly-dipeptides encoded by the C9ORF72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science. 2014.
134 Aguzzi A, Polymenidou M. Mammalian prion biology: one century of evolving concepts. Cell. 2004;116(2):313–27.
135 Aguzzi A. Cell biology: Beyond the prion principle., in Nature. 2009; p. 924–5.
136 Meyer-Luehmann M, et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006;313(5794):1781–4.
137 Clavaguera F, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11(7):909–13.
138 Luk KC, et al. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338(6109):949–53.
139 Prudencio M, et al. Variation in aggregation propensities among ALS-associated variants of SOD1: correlation to human disease. Human Molecular Genetics. 2009;18(17):3217–26.
140 Chia R, et al. Superoxide dismutase 1 and tgSOD1 mouse spinal cord seed fibrils, suggesting a propagative cell death mechanism in amyotrophic lateral sclerosis. PLoS ONE. 2010;5(5):e10627.
141 Hayward LJ, et al. Decreased metallation and activity in subsets of mutant superoxide dismutases associated with familial amyotrophic lateral sclerosis. J Biol Chem. 2002;277(18):15923–31.
142 Münch C, J. O'Brien, Bertolotti A. Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc Natl Acad Sci U S A. 2011;108(9):3548–53.
143 Furukawa Y, Torres AS, O’Halloran TV. Oxygen-induced maturation of SOD1: a key role for disulfide formation by the copper chaperone CCS. EMBO J. 2004;23(14):2872–81.
144 Grad LI, et al. Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. Proc Natl Acad Sci U S A. 2011;108(39):16398–403.
145 Furukawa Y, et al. Intracellular seeded aggregation of mutant Cu,Zn-superoxide dismutase associated with amyotrophic lateral sclerosis. FEBS Lett. 2013;587(16):2500–5.
146 Furukawa Y, et al. A seeding reaction recapitulates intracellular formation of Sarkosyl-insoluble transactivation response element (TAR) DNA-binding protein-43 inclusions. J Biol. Chem. 2011;286(21):18664–72.
147 Guo W, et al. An ALS-associated mutation affecting TDP-43 enhances protein aggregation, fibril formation and neurotoxicity. Nat Struct Mol Biol. 2011;18(7):822–30.
148 Nonaka T, et al. Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 2013;4(1):124–34.
149 Ayala YM, et al. TDP-43 regulates its mRNA levels through a negative feedback loop. The EMBO Journal. 2011;30(2):277–88.
150 Zhou Y, et al. ALS-associated FUS mutations result in compromised FUS alternative splicing and autoregulation. PLoS Genet. 2013;9(10):e1003895.