Cancer immunology – development of novel anti-cancer therapies
DOI: https://doi.org/10.4414/smw.2015.14066
Sacha I
Rothschild, Daniela S
Thommen, Wolfgang
Moersig, Philipp
Müller, Alfred
Zippelius
Summary
The vast majority of tumours are characterised by high frequencies of genetic and epigenetic alterations resulting in tumour-specific antigens, which may, in principle, be recognised by cytotoxic T cells. Though early clinical immunotherapy trials have yielded mixed results with ambiguous clinical benefit, cancer immunotherapy is now attracting increasing attention as a viable therapeutic option, mainly in melanoma and lung cancer, but increasingly also in other malignancies. In particular, recent therapeutic efforts targeting inhibitory receptors on T cells to overcome tumour-induced immune dysfunction have the potential to reshape current treatment standards in oncology. The clinical development has been pioneered by the antibody ipilimumab, which blocks cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) and has demonstrated survival benefit in two randomised landmark trials in melanoma. Capitalising on this success, the research on the clinical implication of T cell checkpoint inhibition has been boosted. Early clinical trials have demonstrated meaningful response rates, sustained clinical benefits with encouraging survival rates and good tolerability of next-generation checkpoint inhibitors, including programmed death-1 (PD-1) and programmed death ligand 1 (PD-L1) inhibitors, across multiple cancer types. Attractive perspectives include the concurrent blockade of immunological (non-redundant) checkpoints, which has recently been demonstrated using combinations of immune checkpoint modulators themselves or with other therapies, such as chemotherapy, targeted therapy or radiotherapy. This article summarises the mechanism of action and subsequent clinical studies of immune checkpoint antibodies in oncology with a particular focus on melanoma and lung cancer.
Introduction
Though early clinical immunotherapy trials have yielded mixed results with ambiguous clinical benefit [1], cancer immunotherapy is attracting increasing attention as a viable therapeutic option, and is now regarded as the fourth cornerstone of anti-cancer treatment. The increasing knowledge as to how the immune system works, in particular with regard to chronic viral infections and cancer, has paved the way for the rational development of novel treatment strategies. The latter are designed to activate and/or re-activate effector cells of the immune system, in particular tumour reactive T cells.
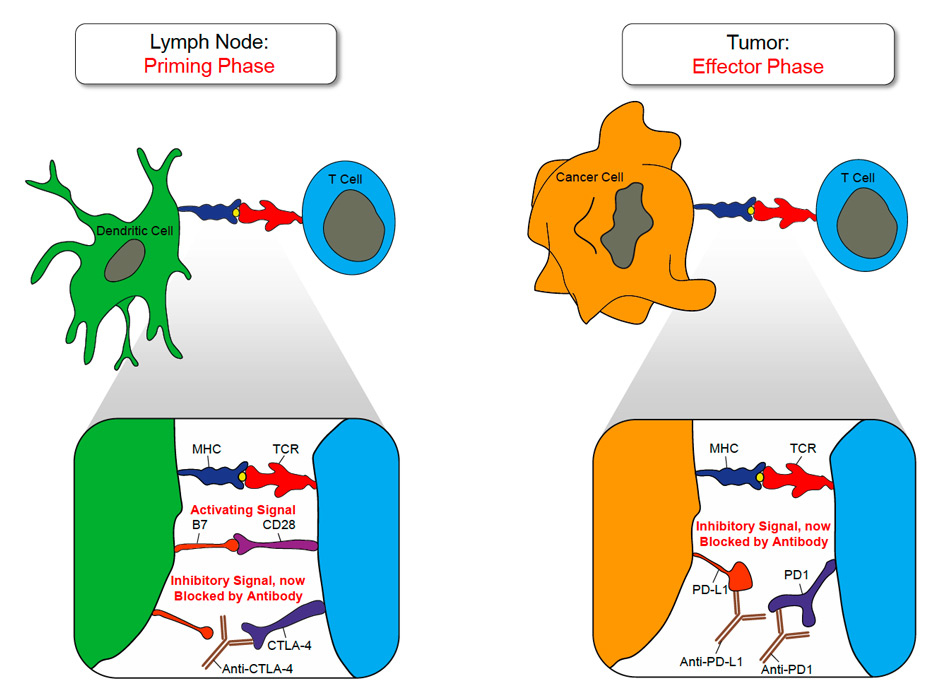
Figure 1
Blockade of CTLA-4 or PD-1 signalling in anti-cancer immunotherapy:
The T cell priming phase is schematically depicted on the left site. T cells engage APCs, such as dendritic cells (DCs) via their t-cell receptor (TCR). Recognition of the cognate MHC/peptide complex by the TCR results in the intracellular transmission of an activating signal in the T cell, which is complemented by a co-activating signal provided by the CD28/B7 interaction. In order to down-modulate T cell priming and expansion CTLA-4 is up regulated on activated T cells after 2–3 days and competes with CD28 for B7. CTLA-4 blocking antibodies can therapeutically inhibit the co-inhibitory signal provided by CTLA-4.
On the right site the effector phase is outlined. During this phase immune effector function can be dampened by PD-1/PD-L1 interaction. PD-1 or PD-L1 blocking antibodies can therapeutically inhibit the co-inhibitory signal provided by the PD-1/PD-L1 interaction and restore effector function in tumour resident effector T cells.
The ability of the immune system to distinguish self from non-self and to mount a vigorous attack against foreign invaders such as viruses and bacteria provides the basis for the rapid and specific clearance of most infections. The concept of cancer immuno-editing [2, 3] postulates that the immune system is able to detect and eliminate most tumours at an early, clinically non-detectable state. However some tumours are not completely destroyed and equilibrium is reached between tumour growth and destruction by the immune system. Though this balanced state can last for years, many, if not most, tumours eventually escape immune-surveillance and become clinically detectable.
A major hurdle for immunotherapeutic approaches is that a plethora of mechanisms are active at the tumour site, which act in concert to counteract effective anti-tumour immunity. Tumours do exploit similar molecular and cellular mechanisms, which under physiological conditions maintain tolerance and prevent immune-mediated damage. The immune-suppressive armamentarium of the tumour micro-environment encompasses diverse cell types (e.g. regulatory T cells and myeloid derived suppressor cells) as well as molecular mechanisms, such as soluble factors (e.g. TGF-β, IL-10, IL-3, SCF), cell restricted, immuno-suppressive molecules (e.g. arginase, IDO), and the expression of co-inhibitory receptors on tumour resident effector cells (e.g. CTLA-4, PD-1, Tim-3, Lag-3 and BTLA as well as their ligands). The latter are commonly referred to as “immune checkpoints”. The interplay of all these factors and co-inhibitory receptor-ligand interactions leads to a state of functional exhaustion in tumour-specific effector cells, such as T cells, at the tumour site [4]. Hence, treatment strategies which aim at blocking these immune checkpoints may restore immune cell proliferation and effector function and help to overcome immune resistance. Therapeutic inhibition of CTLA-4 has provided the first evidence for the clinical relevance of this strategy [5]. CTLA-4 seems to regulate T cells predominantly during the early phase of activation. T cells do recognise their cognate antigen presented on major histocompatibility complexes (MHC) by antigen presenting cells (APCs). However, in addition to this first signal transmitted via the T cell receptor (TCR) a second, co-stimulatory signal is required to initiate full T cell expansion and activation. This second signal is provided by the B7 co-stimulatory molecules B7–1 (CD80) and B7–2 (CD86), which are expressed on mature APCs [6]. To avoid excessive T cell proliferation, activated T cells up-regulate CTLA-4 following peptide-MHC engagement [7]. By competing with the CD28 co-stimulatory receptor on T cells for binding to B7–1 and B7–2 on APCs, CTLA-4 dampens T cell responses, thereby avoiding tissue damage under physiological conditions. It should be further noted that CTLA-4 is constitutively expressed on tumour resident regulatory T cells, which are part of the immuno-suppressive tumour micro-environment. Mouse models have demonstrated that CTLA-4 targeting antibodies hold the potential to decimate this regulatory T cell subset by antibody-dependent cellular cytotoxicity (ADCC) [8].
During the T cell effector phase, which follows the priming and expansion phase, other immune checkpoint molecules such as PD-1 are up-regulated. PD-1 appears to play a more prominent role in modulating T cell activity in peripheral tissues via interaction with its ligands, PD-L1 (B7–H1, CD274) and PD-L2 (B7–DC, CD273), which are up-regulated under inflammatory conditions, mediated, in part, by type I and type II IFNs. Of note, PD-L1 and to a lesser extent, PD-L2 are expressed on many human solid and haematological tumours [9]. Figure 1 summarises the blockade of PD-1 or CTLA-4 signalling in tumour immunotherapy. Together, these findings suggest that interrupting the PD-1:PD-L1/PD-L2 interaction could be an effective anti-cancer therapy by blunting inhibition of immune responses in the tumour micro-environment. In the following sections current immunotherapeutic approaches are reviewed. As currently the most advanced results from clinical trials including randomised phase III trials have been reported for malignant melanoma and non-small cell lung cancer (NSCLC), we decided to place a particular focus on these two cancer types.
Immunotherapy for melanoma
In 1998 high dose interleukin-2 (IL-2) (Proleukin®) was approved as a first immunotherapeutic agent for the treatment of metastatic melanoma based on its ability to elicit durable responses [10, 11]. However, high toxicity and lack of survival benefit in randomised clinical trials have prevented its general acceptance. Later, the combination of IL-2 with a gp100:209–217(210M) peptide vaccine was shown to increase the clinical response rate, yet no significant benefit in overall survival (OS) has been observed [12].
Vaccines
There are two main mechanisms, by which vaccines exert their activity: (a) tumour cell vaccines or whole-cell vaccines expose the immune system to a variety of tumour antigens; (b) antigen-based vaccines consist of a specific antigen expressed on the tumour cell. The latter technology requires identification of that specific antigen on the host’s tumour to be effective. The vaccines are generally paired with immune – activating adjuvants. Though the potential of vaccines for therapeutic efficacy has been extensively demonstrated in tumour models, their clinical benefit has still not been documented in phase III trials. Along this line, a large phase III randomised, blinded, placebo-controlled trial of the recombinant MAGE-A3 and AS15 adjuvant failed to reach its first co-primary endpoint, which was to significantly extend disease-free survival (DFS) when compared to placebo in stage IIIB/C melanoma patients with macroscopic nodal disease, whose tumours expressed the MAGE-A3 gene and had their tumours removed surgically. Melanoma-associated antigen-A3 (MAGE-A3) is a cancer-testis antigen, with unknown function, but the expression is associated with worse prognosis [13]. Interestingly, promising signs of clinical benefit have been demonstrated in a phase II trial in early metastatic melanoma [14], especially in patients expressing a specific immune gene signature. This apparent association with clinical outcome confirms mechanistic work in mice on the existence of a cancer immune phenotype conducive to immune responsiveness [15]. Other attempts of anti-cancer vaccines included T-VEC or Talimogene laherparepvec, a type of an oncolytic immunotherapy. In a randomised prospective phase III trial (OPTIM), 436 patients with stage IIIB, IIIC or IV melanoma were randomised in a 2:1 ratio to receive either intralesional T-VEC or subcutaneous GM-CSF. T-VEC significantly improved durable response rates (DRR), but failed to extend OS. The objective response rate (ORR) with T-VEC was 26% versus 6% with GM-CSF. A trend towards an improved OS was shown. A tolerable safety profile of the vaccine was observed [16]. Potential combination therapies with immune-checkpoint blockade and rapid progress in technologies that enable better vaccine design raise the expectation that clinically effective vaccine approaches will belong to the immunotherapy armamentarium in the future.
Immune checkpoint inhibitors
Recent therapeutic efforts targeting inhibitory receptors on T cells to overcome tumour-induced immune dysfunction have reshaped current treatment standards in advanced malignant melanoma. The clinical development has been pioneered by the CTLA-4 blocking antibody ipilimumab (Yervoy®), which was approved in 2011 and was the first agent with a documented survival benefit in two randomised landmark trials in melanoma [17, 18]. In a phase III clinical trial 676 heavily pre-treated patients with advanced melanoma received either ipilimumab alone or in combination with a gp100–derived peptide vaccine compared to gp100 alone [18]. Remarkably, an improved median OS was found for patients treated with ipilimumab independent of an additional treatment with the vaccine (10.0 months and 10.1 months in arms with ipilimumab plus gp100 and ipilimumab alone, respectively) compared to 6.4 months with gp100 alone (Hazard ratio, HR 0.68, p <0.001 and HR 0.66, p = 0.003). A second phase III study randomly assigned 502 patients with previously untreated metastatic melanoma and compared ipilimumab plus dacarbazine with dacarbazine plus placebo [17]. Addition of ipilimumab to the chemotherapy led to an increase in OS of 11.2 months versus 9.1 months (HR 0.72, p <0.001). Durability of clinical responses has evolved as a hallmark characteristic of antibodies blocking immune checkpoints. Pooled data from 4846 patients who received ipilimumab within clinical studies or expanded access programmes showed a median OS of 9.5 months. Remarkably, the patients experienced a survival plateau at 20–25% after 3 years that extended through at least 10 years [19]. Furthermore, re-exposition with ipilimumab of patients who progressed upon responding to the first treatment for more than 3 months led to a response rate of almost 20% and >65% disease control rate [20]. Of note, the survival benefit of ipilimumab has been observed across all lines of therapy, treatment regimens and dose levels.
Capitalising on the success of CTLA-4 blockade, the research on the clinical implication of T cell checkpoint inhibition has been boosted and next-generation agents such as antibodies targeting the PD-1/PD-L1 signalling axis have been developed. Table 1 provides an overview of phase I trials including anti-PD-1 or –PD-L1 monoclonal antibodies (mAbs) in metastatic melanoma. Nivolumab (MDX-1106 or BMS-936558) is a fully human IgG4 PD-1 antibody. In a first phase I dose-escalation trial, nivolumab was investigated in 39 patients with advanced tumours such as metastatic melanoma, colorectal cancer (CRC), castrate-resistant prostate cancer (CRPC), NSCLC, or renal cell carcinoma (RCC) [21]. The antibody was generally well tolerated. Extensions of the phase I study with 296 patients showed an objective response rate (ORR) in 28% of patients with pre-treated melanoma [22]. Notably, objective responses were durable as 19 of 26 reported responses were ongoing at the time of data analysis. In a follow up analysis of this trial the median OS was 16.8 months, and 1- and 2-year survival rates were 62% and 43%, respectively [23]. Very recently, nivolumab has been approved by the Japanese healthcare authorities for treatment of unresectable melanoma. As so far only a limited number of patients have been treated, further follow-up in a post-marketing setting is required.
Pembrolizumab (MK-3475) is a humanised IgG4 monoclonal antibody against PD-1. In a phase I trial, 135 patients with advanced melanoma received pembrolizumab [24]. A clinical response was seen in 38% of patients with most responses ongoing at the time of data cut-off. Remarkably, previous treatment with ipilimumab did not affect the clinical benefit as significant responses were as common and robust as in patients who had not received ipilimumab before. In Switzerland pembrolizumab is currently available through an early access programme (EAP) by MSD Merck Sharp & Dohme. The EAP is open for patient inclusion in selected tumour centres in Switzerland.
Several antibodies are now in clinical development, aimed at blocking PD-L1, the ligand of PD-1. BMS-936559 was shown to be safe and clinically active across multiple tumour types in a phase I trial [25]. From 55 pre-treated patients with advanced melanoma, 9 showed an overall response (3 complete remissions and 6 partial remissions). A total of 5 patients had a response lasting at least 1 year, and 5 were still ongoing at time of data analysis. In addition, 14 of 55 patients (27%) had stable disease (SD) for at least 24 weeks. However, further development of BMS-936559 was stopped. A phase I study investigating MPDL3280A, a genetically engineered IgG1 anti-PD-L1 mAb, in 45 patients with different histological subtypes of melanoma showed encouraging results with an ORR of 28% and a progression-free survival (PFS) rate of 41% at 24 weeks [26].
As a next step, the effect of combination therapies was explored. Concurrent treatment with nivolumab and ipilimumab in pre-clinical models showed encouraging results with synergistic anti-tumour activity [27], providing the rationale for a subsequent phase I dose-escalating study. In the study 69 patients with advanced melanoma who were all previously treated received concurrently nivolumab at doses of 0.3, 1 or 3 mg/kg and ipilimumab at 3 mg/kg. A total of 14 of 37 patients (38%) showed a radiographic response. One third of patients achieved rapid and profound tumour responses with more than 80% tumour reduction at week 12 and rapid resolution of symptoms. At the dose level of 1 mg/kg nivolumab and 3 mg/kg ipilimumab that was chosen for further trials, 100% (9) patients achieved a reduction of tumour volume >80%. Responses were durable for up to 100 weeks at the time of the data cut-off. However, the encouraging clinical results come at a cost of significantly increased immune-related adverse events [28]. Though the latter appear manageable, the safety profile needs be considered in future trials.
Combinatorial approaches other than checkpoint inhibitors have been investigated, such as the combined treatment with MAPK-targeted therapy and immunotherapy. While combined CTLA-4 and BRAF inhibition in BRAFV600 mutated melanoma takes advantage of non-overlapping mechanisms of action and thus appears to be an intuitive and exciting approach, so far phase I data revealed significant dose-limiting hepatotoxicity with the combination of ipilimumab and vemurafenib [29]. A phase I trial of bevacizumab + ipilimumab showed an impressive anti-tumour activity with manageable toxicity [30]. A large variety of other combination strategies are currently explored in early-phase trials, each with a compelling pre-clinical rationale. Future challenges include the proper sequencing, timing and dosing of combination therapies.
|
Table 1: Overview of phase I trials including anti-PD-1 or -PD-L1 monoclonal antibodies in metastatic melanoma. |
|
Target
|
Agent
|
n
|
ORR%
|
CR
|
OS
|
| |
|
|
|
|
1 yr
|
2 yr
|
|
PD-1
|
Pembrolizumab
(10 mg/kg q2, 10 mg/kg q3, 2 mg/kg q3) |
135 |
32% |
9% |
81% |
n.a. |
|
|
Nivolumab + peptide vaccine
(1, 3, 10 mg/kg q2) |
87 |
25% |
2% |
n.a. |
n.a. |
|
|
Nivolumab
(0.1, 1, 3, 10 mg/kg q2) |
107 |
31% |
? |
62% |
43% |
|
|
|
|
|
|
PFS 24 weeks
|
|
PD-L1
|
MPDL3280A
(0.3, 1, 3, 10, 20 mg/kg q2) |
45 |
28% |
? |
41% |
| |
BMS-936559
(0.3, 1, 3, 10 mg/kg q2) |
52 |
17% |
6% |
42% |
Immunotherapy in lung cancer
Historically, lung cancer has not been considered sensitive to immune-based therapies and was believed to be a non-immunogenic tumour [31]. Most lung cancer patients present with metastatic disease and are immune suppressed with decreased peripheral und tumour lymphocyte counts [32, 33]. Regulatory T cells (Tregs-CD4+) play a key role in the suppression of the tumour immune surveillance by suppressing cytotoxic T lymphocytes (CD8+ T cells). Tregs have been found at higher levels in peripheral blood and tumour environment in patients with lung cancer [34]. On the other hand, a high number of tumour-infiltrating Tregs, CD8+ T cells, natural killer cells, and/or dendritic cells has been associated with improved patient survival [35–40]. Based on promising results of immunotherapeutic approaches in other solid tumours and the development of new molecules unleashing the immune system, immunotherapy has again raised interest in the treatment of lung cancer. Strategies to actively enhance the immune response in lung cancer include vaccination to stimulate antibody and T-cell responses to cancer cells and use of immune checkpoint inhibitors to boost T-cell immune responses to lung cancer cells.
Vaccines
Belagenpumatucel-L (Luxanix, NovaRx Corporation, San Diego, CA, USA) is an allogeneic tumour cell vaccine composed of four different NSCLC cell lines (H460, H520, SKLU-1, and RH2), which are genetically modified to suppress the expression of transforming growth factor-beta (TGF-β), an important cytokine associated with local immunosuppression in lung cancer [41]. In a randomised phase II trial 75 patients with stage II-IV NSCLC pretreated with chemotherapy were enrolled and treated with three different doses of belagenpumatucel-L [41]. A survival advantage for patients treated with higher doses was reported. The safety and tolerability of the vaccine was acceptable. Based on these results, belagenpumatucel-L was tested as maintenance treatment in a randomised phase III trial (STOP) in patients with stage IIIA, IIIB, and IV NSCLC, whose disease was stable or responding to first-line platinum-based chemotherapy. A total of 532 patients were randomised to belagenpumatucel-L or placebo. Belagenpumatucel-L was given by one monthly and two quarterly intradermal injections. This trial did not meet its predefined primary end point in the entire patient population; median OS was 20.3 months with the vaccine compared to 17.8 months with placebo (HR 0.94; p = 0.594) [42]. Significantly prolonged OS with belagenpumatucel-L was however demonstrated in patients who started vaccination within 12 weeks of the completion of front-line chemotherapy, had prior radiotherapy and with non-adenocarcinoma histology. These data, along with its safety profile, support the continued development of belagenpumatucel-L for this indication.
MAGE-A3 is expressed in 35% of early and 55% of advanced stages of NSCLC. The MAGE-A3 vaccine was evaluated in a randomised phase II trial in patients with surgically resected NSCLC stage Ib-II [43]. In total, 182 patients were randomised between MAGE-A3 (five intramuscular injections every three weeks followed by eight injections once every three months) and placebo. With a median follow-up time of 28 months, the tumour recurrence rate was 30.6% in the vaccine-treated patients versus 43.3% in the placebo-treated patients. While there was a non-significant difference in the DFS in the entire population, an exploratory analysis of a sub-population of patients with an immunogenic gene signature showed a significantly reduced risk of cancer recurrence [44]. The randomised phase III MAGRIT trial randomised patients with resected stage IB-IIIA MAGE-A3–positive NSCLC, to either vaccination or placebo after adjuvant chemotherapy. For this trial more than 11’000 patients were screened. Ultimately 2270 patients were enrolled. In a press release in March 2014 the company GSK announced that the trial did not meet its first co-primary endpoint of DFS.
Mucinous glycoprotein-1 (MUC-1) is a tumour-associated antigen associated with oncogenesis and resistance to chemotherapy [45]. Tumour-associated MUC-1 is aberrantly glycosylated [46] and over-expressed in approximately 60% of lung cancers [47]. L-BLP25 (tecemotide, Stimuvax, Oncothyreon, Seattle, WA, USA) incorporates a synthetic MUC-1 lipopeptide and monophosphoryl lipid A within a liposomal delivery system. In a randomised phase II trial 171 patients with stage IIIB/IV NSCLC were randomly assigned to best supportive care or vaccine with cyclophosphamide. The authors observed a pronounced trend in 2–year OS in favour of L-BLP25, in particular in patients with stage IIIB disease [48]. Recently, the results of the START trial, a randomised phase III trial with 1239 patients with unresectable stage IIIA/IIIB NSCLC non-progressive following chemo- and radiotherapy have been presented [49]. OS was not significantly prolonged with L-BLP25 versus placebo (median OS 25.6 vs. 22.3 months, HR 0.88, p = 0.123). However, in the large sub-group of patients receiving concomitant chemoradiotherapy there was a statistically significant OS benefit for the vaccination group (median OS 30.8 vs. 20.6 months, HR 0.78, p = 0.016).
TG4010 contains a genetically modified virus that expresses MUC-1 and IL-2 [50]. This vaccine was evaluated in patients with stage IIIB/IV MUC-1–positive NSCLC in addition to chemotherapy (cisplatin/gemcitabine) in a randomised phase II trial [50]. There was no difference in the outcome parameter of both arms of the trial. In an exploratory sub-group analysis, benefit was reported in the TG4010–treated group with normal levels of activated natural killer cells at baseline. The ongoing randomised phase III TIME trial includes patients with stage IV NSCLC and adds TG4010 or placebo to standard first-line chemotherapy (NCT01383148).
Immune checkpoint inhibitors
In contrary to vaccines, immune checkpoint inhibitors have so far only been investigated in advanced/metastatic lung cancer and mainly in pre-treated patients.
The activity of ipilimumab (10 mg/kg) in combination with paclitaxel and carboplatin was evaluated in a randomised phase II trial in patients with chemotherapy-naïve stage IIIB/IV NSCLC [51]. Patients were randomly assigned to a concurrent ipilimumab regimen, a phased ipilimumab regimen, or a control regimen. Patients without progression received maintenance treatment with either ipilimumab or placebo (control arm). Anti-tumour responses were highest in the phased ipilimumab group. The immune-related PFS (ir-PFS) as primary endpoint was significantly prolonged in patients receiving the phased schedule of ipilimumab versus chemotherapy alone (median ir-PFS 5.7 vs. 4.6 months, HR 0.72, p = 0.05). Combining ipilimumab with paclitaxel and carboplatin did not increase overall treatment-related toxicities. In a pre-specified subset analysis ipilimumab significantly improved the activity of chemotherapy in patients with squamous histology. Currently, a phase III study of phased ipilimumab with chemotherapy is accruing patients with squamous cell lung carcinoma (NCT01285609).
In a parallel phase II trial for patients with extensive small-cell lung cancer with the same three treatment arms, a significant benefit of the phased schedule of ipilimumab and chemotherapy has been reported (median ir-PFS 6.4 vs. 5.3 months, HR 0.64, p = 0.03) [52].
In a phase I trial nivolumab monotherapy was administered to patients with various solid tumours, including 129 patients with NSCLC [22, 53]. Patients received one of five different nivolumab doses, ranging from 0.1 mg/kg to 10 mg/kg every two weeks. In NSCLC patients, objective responses were observed in patients across doses of 1–10 mg/kg and in all histological subtypes. With extended treatment, the median response duration for the 3 mg/kg dose had not been reached at the time of analysis (range, 16.1+ to 133.9+ weeks). The dose of 3 mg/kg has been selected for further trials. Currently, nivolumab is evaluated in several phase III trials. The CheckMate 017 trial (NCT01642004) for squamous and the CheckMate 057 trial (NCT01673867) for non-squamous NSCLC completed recruitment of patients after failure of platinum-based chemotherapy, which were randomised between nivolumab and docetaxel. The CheckMate 026 trial is a randomised trial for the first-line setting comparing nivolumab to investigator’s choice chemotherapy in PD-L1 positive NSCLC (NCT02041533). Additional trials are ongoing, including a combination trial with ipilimumab and nivolumab in advanced or metastatic solid tumours, including NSCLC (NCT01928394).
In the KEYNOTE-001 trial treatment-naïve NSCLC patients with PD-L1 expression were randomised between two schedules of pembrolizumab (10 mg/kg every two or every three weeks) [54]. The ORR was 26% and the rate of disease-stabilisation was 64% (independent review). All responses are ongoing at the time of data-cut-off. The KEYNOTE-010 trial randomises NSCLC patients to docetaxel or pembrolizumab in the second-line setting (NCT01905657). The KEYNOTE-024 trial compares pembrolizumab to platinum-based doublet chemotherapy in treatment-naïve patients with PD-L1 positive metastastic NSCLC (NCT02142738).
The first in human trial of the anti-PD-L1 monoclonal antibody (BMS-936559) also included patients with advanced NSCLC [25]. The maximum-tolerated dose was not reached. In an expansion cohort including 75 patients with pre-treated metastatic NSCLC objective responses and long lasting disease stabilisations were seen irrespective of the histological subtype.
In an interim analysis of the phase I dose escalation/expansion study in patients with solid tumours treated with MPDL3280A, the ORR in the 40 patients with pre-treated NSCLC was 23%, and all responses were ongoing or improving at data cut-off (range, 1+ to 214+ days). The rate of PFS at 24 weeks was 46% [55]. Two phase II trials (FIR, NCT01846416 and BIRCH, NCT02031458) in PD-L1 positive NSCLC investigated ORR and safety. The randomised phase III trial OAK (NCT02008227) compares MPDL3280A to docetaxel in the second-line setting. As early trials have shown higher response rates in patients with PD-L1 positive tumours as determined by immunohistochemistry, patients have been selected on the basis of PD-L1 expression. However, it needs to be emphasised that patients with PD-L1 negative tumours showed clinically important responses. Therefore, the role of PD-L1 immunohistochemistry as a predictive marker for anti-PD-(L)1 antibodies has not yet been established. An additional issue which currently complicates the introduction of PD-L1 status is that the immunohistochemical assay has not been standardised. Important questions such as the specificity of antibodies used and the role of PD-L1 expression in tumours versus non-tumour stromal cells need to be solved in the near future.
In a Phase I dose-escalation study (Study 1108) of the anti-PD-L1 antobody MEDI4736 in 27 patients with advanced solid tumours including NSCLC, reduction of tumour burden was seen at multiple dose levels as early as six weeks after treatment initiation. Clinical activity was maintained for at least one year, with 19% of patients achieving a partial response and 39% of patients achieving disease control [56].
|
Table 2:Frequent immune-related adverse events reported with immune checkpoint inhibitors. Adapted from Davies et al. [69]. |
|
Organ
|
Adverse events |
|
Dermatological
|
Pruritus, rash, vitiligo, urticaria, alopecia, pruritic rash, macular rash, hypopigmentation, erythema, erythematous rash |
|
Endocrine
|
Hypothyroidism, hyperthyroidism, hypopituitarism, hypophysitis, adrenal insufficiency |
|
Gastrointestinal
|
Diarrhoea, colitis, hepatitis, pancreatitis |
|
Pulmonary
|
Pneumonitis, pulmonary oedema |
|
Ocular
|
Uveitis, episcleritis |
Immunotherapy in other solid tumours
Numerous vaccines and immune checkpoint inhibitors are under investigation in various solid tumours. However, the only approved drug is sipuleucel-T (provenge®, Dendron) for the treatment of metastatic CRPC (mCRPC). Sipuleucel-T is a type of therapeutic cancer vaccine consisting of autologous peripheral-blood mononuclear cells (PBMCs), including antigen-presenting cells (APCs), that have been activated ex vivo with a recombinant fusion protein (PA2024) [57]. PA2024 consists of a prostate antigen, prostatic acid phosphatase that is fused to granulocyte–macrophage colony-stimulating factor, an immune-cell activator. Up to now, three phase III trials with sipuleucel-T have been published. The D9901 trial randomised 127 patients with asymptomatic mCRPC to sipuleucel-T or placebo[58]. Time to progression (TTP) was the primary endpoint of the trial and was not different in the two treatment arms. However, OS as the secondary endpoint in the trial was significantly prolonged in patients treated with sipuleucel-T (median OS 25.9 vs. 21.4 months, p = 0.01). Similarly, a second trial (D9902A) showed an improved OS for sipuleucel-T (median OS 19.9 vs. 15.7 months). However, this result was not statistically significant (p = 0.331). A combined analysis of both trials showed a relative risk reduction in mortality of 33% for patients treated with sipuleucel-T [59]. The IMPACT trial (immunotherapy for prostate adenocarcinoma treatment) randomised 512 patients to sipuleucel-T or placebo [60]. OS as the primary endpoint was significantly prolonged (median OS 25.8 vs. 21.7 months, p = 0.03). The most common adverse events of sipuleucel-T are fever, chills and headache. Sipuleucel-T is approved in the US. The promising results of immuntherapeutic approaches in melanoma and lung cancer gave rise to an enormous scientific interest so that these agents are currently investigated in nearly all types of solid tumours.
Toxicities of immunotherapies
Some of the toxicities seen with immunotherapeutic agents are directly linked to their mode of action as activators of the immune system. Inhibition of immune checkpoints such as CTLA-4 and PD-1/PD-L1 will lead to profound activation of T-cells, which may recognise self antigens [61]. Table 2 summarises the most common types of immune-related adverse events reported with immune checkpoint inhibitors [18, 22, 24, 25, 28, 51, 62]. Most of the so far published trials only have a short follow-up. There is, however, insufficient knowledge on potential chronic toxicities of these drugs, particularly in patients with long-term remissions. In the light of clinical trials investigating these drugs in the adjuvant setting, the safety profile needs to be carefully monitored.
CTLA-4 antibody ipilimumab
In a pooled analysis of 325 patients treated with 10 mg/kg ipilimumab every three weeks for four doses, irAEs of any grade were observed in 72.3%. G3/4 irAEs were observed in 25.2%, mainly in the gastrointestinal tract (12%), liver (7%), skin (3%), and endocrine system (3%) [63]. The spectrum of endocrine irAEs includes hypopituitarism caused by hypophysitis and, more rarely, thyroid disease or abnormalities in thyroid function tests. Primary adrenal insufficiency has been reported occasionally as well. For the management of irAEs specific guidelines have been established [62, 64]. These include administration of systemic glucocorticoids or other immunosuppressive agents. Retrospective analysis suggests that patients who experience G3/4 irAEs may be more likely to benefit from anti-CTLA-4 therapy [65–67].
Anti-PD-1/-PD-L1 agents
In the initial phase I trial with nivolumab including 39 patients, no maximum tolerated dose (MTD) was identified and most AEs were immune-related [21]. No G3/4 irAEs were observed in the 4 weeks following the first dose of nivolumab. One serious treatment-related AE of inflammatory colitis occurred and improved after administration of infliximab and steroids. No patient developed anti-nivolumab antibodies during the study period. In the expanded cohort with 304 patients G3/4 drug-related AEs occurred in 14% of patients, including three deaths from pneumonitis [22]. The most common drug-related AEs, however, included pruritus, rash, fatigue, nausea, decreased appetite, and diarrhoea; most of these (166/207 across grades) were of low grade. AEs with possible immune-related causes occurred in 41% of patients and included vitiligo, hepatitis, pneumonitis, colitis, thyroiditis, and hypophysitis. However, the overall frequency and intensity of autoimmune toxicities seems to be lower as with ipilimumab.
In the initial phase I trial with pembrolizumab including 17 patients, no dose limiting toxicities were reported [68]. In this initial cohort there were no drug-related AEs of grade 3 or higher. It should be noted, however, that one case of pneumonitis was treated with corticosteroids (the patient improved clinically but was subsequently taken off study). The most common drug-related AEs were pruritus, fatigue, and nausea. This favourable toxicity profile was confirmed in the expansion cohort of melanoma, NSCLC and head and neck cancer patients.
The fact that pneumonitis is one of the side effects of PD-1 antibodies raised some concerns especially for its use in lung cancer patients. Preliminary results from PD-L1 antibodies suggest that the rate of pneumonitis is lower with these agents.
In various malignancies combination trials of different immune checkpoint inhibitors or immune checkpoint inhibitors with other targeted agents are ongoing. Although early clinical results are encouraging, the additional toxicity needs to be considered.
Conclusions
A better understanding of the role of the immune system in tumour immunosurveillance has resulted in the development of a new generation of immunotherapeutic agents. In particular, it is now recognised that tumours can evade immune destruction via the dysregulation of co-inhibitory or checkpoint signals. Results from early phase studies of immune checkpoint modulators such as CTLA-4, PD-1 and PD-L1 inhibitors in a range of solid tumours including melanoma and NSCLC are highly promising and may provide new therapeutic options for the treatment of those cancers. Future challenges include incorporating immunotherapeutic agents either in combination or in sequence with cytotoxic chemotherapy, molecular targeted therapy and radiotherapy, its administration in early stage disease and the identification of predictive biomarkers.
References
1 Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. [Internet]. 2004;10:909–915. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15340416.
2 Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science (80–. ). [Internet]. 2011;331:1565–1570. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=21436444.
3 Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases – elimination, equilibrium and escape. Curr Opin Immunol. [Internet]. 2014 [cited 2014 Jun 3];27:16–25. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24531241.
4 Gajewski TF, Woo S-R, Zha Y, Spaapen R, Zheng Y, Corrales L, et al. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr Opin Immunol. [Internet]. 2013 [cited 2014 May 29];25:268–276. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23579075.
5 Sharma P, Wagner K, Wolchok JD, Allison JP. Novel cancer immunotherapy agents with survival benefit: recent successes and next steps. Nat Rev Cancer [Internet]. 2011 [cited 2014 Jun 25];11:805–812. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3426440&tool=pmcentrez&rendertype=abstract.
6 Chambers CA, Allison JP. Costimulatory regulation of T cell function. Curr Opin Cell Biol. [Internet]. 1999 Apr [cited 2014 Jun 28];11:203–210. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10209159.
7 Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity [Internet]. 1994 [cited 2014 May 27];1:405–413. Available from: http://www.ncbi.nlm.nih.gov/pubmed/7882171.
8 Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. [Internet]. 2013 [cited 2014 May 27];210:1695–1710. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3754863&tool=pmcentrez&rendertype=abstract.
9 Callahan MK, Wolchok JD. At the bedside: CTLA-4– and PD-1–blocking antibodies in cancer immunotherapy. J Leukoc Biol. [Internet]. 2013 [cited 2014 Jun 17];94:41–53. Available from: http://www.jleukbio.org/search?submit=yes&pubdate_year=&volume=&firstpage=&author1=.
10 Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol [Internet]. 1999 [cited 2014 Jun 6];17:2105–2116. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10561265.
11 Rosenberg SA, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, Parkinson DR, et al. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA [Internet]. [cited 2014 Jun 5];271:907–913. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8120958.
12 Schwartzentruber DJ, Lawson DH, Richards JM, Conry RM, Miller DM, Treisman J, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. [Internet]. 2011 [cited 2014 Jun 11];364:2119–2127. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3517182&tool=pmcentrez&rendertype=abstract.
13 Gure AO, Chua R, Williamson B, Gonen M, Ferrera CA, Gnjatic S, et al. K. Cancer-testis genes are coordinately expressed and are markers of poor outcome in non-small cell lung cancer. Clin Cancer Res. [Internet]. 2005 [cited 2014 Jun 8];11:8055–8062. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16299236.
14 Ulloa-Montoya F, Louahed J, Dizier B, Gruselle O, Spiessens B, Lehmann FF, et al. Predictive gene signature in MAGE-A3 antigen-specific cancer immunotherapy. J Clin Oncol. [Internet]. 2013 [cited 2014 May 29];31:2388–2395. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23715562.
15 Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci. Transl. Med. [Internet]. 2013 [cited 2014 May 27];5:200ra116. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23986400.
16 Andtbacka RHI, Collichio FA, Amatruda T, Senzer NN, Chesney J, Delman KA, et al. OPTiM: A randomized phase III trial of talimogene laherparepvec (T-VEC) versus subcutaneous (SC) granulocyte-macrophage colony-stimulating factor (GM-CSF) for the treatment (tx) of unresected stage IIIB/C and IV melanoma. J Clin Oncol. 2013;31(suppl; abstr LBA9008). .
17 Robert C, Thomas L, Bondarenko I, O’Day S, M DJ, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med [Internet]. 2011;364:2517–2526. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=21639810.
18 Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med [Internet]. 2010;363:711–723. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=20525992.
19 Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in metastatic or locally advanced, unresectable melanoma. Eur Cancer Congr. 2013. p. abstract 24. .
20 Robert C, Schadendorf D, Messina M, Hodi FS, O’Day S. Efficacy and safety of retreatment with ipilimumab in patients with pretreated advanced melanoma who progressed after initially achieving disease control. Clin Cancer Res. [Internet]. 2013 [cited 2014 Jun 28];19:2232–2239. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23444228.
21 Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol [Internet]. 2010;28:3167–3175. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=20516446.
22 Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med [Internet]. 2012;366:2443–2454. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=22658127.
23 Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. [Internet]. 2014 Apr [cited 2014 May 23];32:1020–1030. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24590637.
24 Hamid O, Robert C, Daud A, Hodi FS, Hwu W-J, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. [Internet]. 2013 [cited 2014 May 24];369:134–144. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23724846.
25 Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med [Internet]. 2012;366:2455–2465. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=22658128.
26 Sosman JA, Hamid O, Lawrence D, Sullivan RJ, Ibrahim N, Kluger HM, et al. A study of MPDL3280A (engineered anti-PDL1): activity, safety and characterization of immune response in pre- and on-treatment tumors in metastatic melanoma (mM) pts. Soc Melanoma Res. 2013 Congr. 2013. .
27 Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. [Internet]. 2010 [cited 2014 May 30];107:4275–4280. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2840093&tool=pmcentrez&rendertype=abstract.
28 Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. [Internet]. 2013 [cited 2014 May 30];369:122–133. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23724867.
29 Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. [Internet]. 2013 [cited 2014 Jun 7];368:1365–1366. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23550685.
30 Hodi FS, Lawrence D, Lezcano C, Wu X, Zhou J, Sasada T, et al. Bevacizumab plus Ipilimumab in Patients with Metastatic Melanoma. Cancer Immunol Res. [Internet]. 2014 [cited 2014 May 27]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/24838938.
31 Holt GE, Podack ER, Raez LE. Immunotherapy as a strategy for the treatment of non-small-cell lung cancer. Therapy [Internet]. 2011 [cited 2014 Jun 28];8:43–54. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3042692&tool=pmcentrez&rendertype=abstract.
32 Sato Y, Mukai K, Watanabe S, Gotoh M, Matsuno Y, Furuya S, et al. Lymphocyte subsets in pulmonary venous and arterial blood of lung cancer patients. Jpn. J Clin Oncol. [Internet]. 1989 [cited 2014 Jun 28];19:229–236. Available from: http://www.ncbi.nlm.nih.gov/pubmed/2478739.
33 Wesselius LJ, Wheaton DL, Manahan-Wahl LJ, Sherard SL, Taylor SA, Abdou NA. Lymphocyte subsets in lung cancer. Chest [Internet]. 1987 [cited 2014 Jun 28];91:725–729. Available from: http://www.ncbi.nlm.nih.gov/pubmed/3032522.
34 Woo EY, Yeh H, Chu CS, Schlienger K, Carroll RG, Riley JL, et al. Cutting edge: Regulatory T cells from lung cancer patients directly inhibit autologous T cell proliferation. J Immunol. [Internet]. 2002 [cited 2014 Jun 28];168:4272–4276. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11970966.
35 Al-Shibli KI, Donnem T, Al-Saad S, Persson M, Bremnes RM, Busund L-T. Prognostic effect of epithelial and stromal lymphocyte infiltration in non-small cell lung cancer. Clin Cancer Res. [Internet]. 2008 [cited 2014 Jun 6];14:5220–5227. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18698040.
36 Dieu-Nosjean M-C, Antoine M, Danel C, Heudes D, Wislez M, Poulot V, et al. Long-term survival for patients with non-small-cell lung cancer with intratumoral lymphoid structures. J Clin Oncol. [Internet]. 2008 [cited 2014 Jun 11];26:4410–4417. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18802153.
37 Hiraoka K, Miyamoto M, Cho Y, Suzuoki M, Oshikiri T, Nakakubo Y, et al. Concurrent infiltration by CD8+ T cells and CD4+ T cells is a favourable prognostic factor in non-small-cell lung carcinoma. Br J Cancer [Internet]. 2006 [cited 2014 Jun 17];94:275–280. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2361103&tool=pmcentrez&rendertype=abstract.
38 Zhuang X, Xia X, Wang C, Gao F, Shan N, Zhang L, Zhang L. A high number of CD8+ T cells infiltrated in NSCLC tissues is associated with a favorable prognosis. Appl. Immunohistochem. Mol. Morphol. [Internet]. 2010 [cited 2014 Jun 28];18:24–28. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19713832.
39 Takanami I, Takeuchi K, Giga M. The prognostic value of natural killer cell infiltration in resected pulmonary adenocarcinoma. J Thorac Cardiovasc Surg. [Internet]. 2001 [cited 2014 Jun 28];121:1058–1063. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11385371.
40 Villegas FR, Coca S, Villarrubia VG, Jiménez R, Chillón MJ, Jareño J, et al. Prognostic significance of tumor infiltrating natural killer cells subset CD57 in patients with squamous cell lung cancer. Lung Cancer [Internet]. 2002 [cited 2014 Jun 28];35:23–28. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11750709.
41 Nemunaitis J, Dillman RO, Schwarzenberger PO, Senzer N, Cunningham C, Cutler J, et al. Phase II study of belagenpumatucel-L, a transforming growth factor beta-2 antisense gene-modified allogeneic tumor cell vaccine in non-small-cell lung cancer. J Clin Oncol. [Internet]. 2006 [cited 2014 Jun 28];24:4721–4730. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16966690.
42 Giaccone G, Bazhenova L, Nemunaitis J, Juhasz E, Ramlau R, van den Heuvel MM, et al. A phase III study of belagenpumatucel-L therapeutic tumor cell vaccine for non-small cell lung cancer (NSCLC). Eur Cancer Congr. 2013. 2013. p. LBA2. .
43 Vansteenkiste J, Zielinski M, Linder A, Dahabreh J, Gonzalez EE, Malinowski W, et al. Adjuvant MAGE-A3 immunotherapy in resected non-small-cell lung cancer: phase II randomized study results. J Clin Oncol. [Internet]. 2013 Jul [cited 2014 Jun 3];31:2396–2403. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23715567.
44 Vansteenkiste JF, Zielinski M, Dahabreh IJ, Linder A, Lehmann F, Gruselle O, et al. Association of gene expression signature and clinical efficacy of MAGE-A3 antigen-specific cancer immunotherapeutic (ASCI) as adjuvant therapy in resected stage IB/II non-small cell lung cancer (NSCLC). J Clin Oncol Off J Am Soc Clin Oncol. 2008;26:abstract 7501. .
45 Sangha R, Butts C. L-BLP25: a peptide vaccine strategy in non small cell lung cancer. Clin Cancer Res. [Internet]. 2007 [cited 2014 May 29];13:s4652–4. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17671159.
46 Gendler SJ, Lancaster CA, Taylor-Papadimitriou J, Duhig T, Peat N, Burchell J, et al. Molecular cloning and expression of human tumor-associated polymorphic epithelial mucin. J Biol Chem. [Internet]. 1990 [cited 2014 Jun 28];265:15286–15293. Available from: http://www.ncbi.nlm.nih.gov/pubmed/1697589.
47 Rochlitz C, Figlin R, Squiban P, Salzberg M, Pless M, Herrmann R, et al. Phase I immunotherapy with a modified vaccinia virus (MVA) expressing human MUC1 as antigen-specific immunotherapy in patients with MUC1–positive advanced cancer. J Gene Med. [Internet]. 2003 [cited 2014 Jun 15];5:690–699. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12898638.
48 Butts C, Murray N, Maksymiuk A, Goss G, Marshall E, Soulières D, et al. Randomized phase IIB trial of BLP25 liposome vaccine in stage IIIB and IV non-small-cell lung cancer. J Clin Oncol. [Internet]. 2005 [cited 2014 Jun 28];23:6674–6681. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16170175.
49 Butts CA, Socinski MA, Mitchell P, Thatcher N, Havel L, Krzakowski MJ, et al. START: A phase III study of L-BLP25 (Tecemotide) cancer immunotherapy for unresectable stage III non-small cell lung cancer. J Clin Oncol. 2013;31:abstr 7500. .
50 Quoix E, Ramlau R, Westeel V, Papai Z, Madroszyk A, Riviere A, et al. Therapeutic vaccination with TG4010 and first-line chemotherapy in advanced non-small-cell lung cancer: a controlled phase 2B trial. Lancet Oncol. [Internet]. 2011 Nov [cited 2014 Jun 10];12:1125–1133. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22019520.
51 Lynch TJ, Bondarenko I, Luft A, Serwatowski P, Barlesi F, Chacko R, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study. J Clin Oncol [Internet]. 2012 Jun [cited 2014 Jun 28];30:2046–2054. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22547592.
52 Reck M, Bondarenko I, Luft A, Serwatowski P, Barlesi F, Chacko R, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line therapy in extensive-disease-small-cell lung cancer: results from a randomized, double-blind, multicenter phase 2 trial. Ann Oncol [Internet]. 2013 [cited 2014 Jun 28];24:75–83. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22858559.
53 Brahmer JR, Horn L, Antonia SJ, Spigel D, Gandhi L, Sequist L V, et al. NIVOLUMAB (ANTI-PD-1; BMS-936558; ONO- 4538) IN PATIENTS WITH NON-SMALL CELL LUNG CANCER (NSCLC): OVERALL SURVIVAL AND LONG-TERM SAFETY IN A PHASE 1 TRIAL. J Thorac Oncol. 2013;8:abstract MO18.03. .
54 Rizvi, Naiyer A, Garon EB, Patnaik A, Gandhi L, Leighl NB, et al. Safety and clinical activity of MK-3475 as initial therapy in patients with advanced non-small cell lung cancer (NSCLC). J Clin Oncol. 2014;32:abstr 8007. .
55 Horn L, Herbst RS, Spigel D, Gettinger SN, Gordon MS, Hollebecque A, et al. N ANALYSIS OF THE RELATIONSHIP OF CLINICAL ACTIVITY TO BASELINE EGFR STATUS, PD- L1 EXPRESSION AND PRIOR TREATMENT HISTORY IN PATIENTS WITH NON-SMALL CELL LUNG CANCER (NS- CLC) FOLLOWING PD-L1 BLOCKADE WITH MPDL3280A (ANTI-PDL1. J Thorac Oncol. 2013;8:abstract MO18.01. .
56 Lutzky J, Antonia SJ, Blake-Haskins A, Li X, Robbins PB, Shalabi AM, et al. A phase 1 study of MEDI4736, an anti–PD-L1 antibody, in patients with advanced solid tumors. J Clin Oncol. 2014;32:abstr 3001.
57 Goldman B, DeFrancesco L. The cancer vaccine roller coaster. Nat Biotechnol. [Internet]. 2009 [cited 2014 Jun 28];27:129–139. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19204689.
58 Small EJ, Schellhammer PF, Higano CS, Redfern CH, Nemunaitis JJ, Valone FH, et al. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J Clin Oncol [Internet]. 2006;24:3089–3094. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16809734.
59 Higano CS, Schellhammer PF, Small EJ, Burch PA, Nemunaitis J, Yuh L, et al. Integrated data from 2 randomized, double-blind, placebo-controlled, phase 3 trials of active cellular immunotherapy with sipuleucel-T in advanced prostate cancer. Cancer [Internet]. 2009;115:3670–3679. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=19536890.
60 Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med [Internet]. 2010;363:411–422. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=20818862.
61 Pardoll D, Drake C. Immunotherapy earns its spot in the ranks of cancer therapy. J Exp Med [Internet]. 2012;209:201–209. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=22330682.
62 Weber JS, Kähler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. [Internet]. 2012 Jul [cited 2014 Jun 4];30:2691–2697. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22614989.
63 Lebbé C, O’Day S, Chiarion Sileni V, Al. E. Analysis of the onset and resolution of immune-related adverse events during treatment with ipilimumab in patients with metastatic melanoma. Proc. from Perspect. Melanoma XII; Oct. 2–4, 2008. 2008. p. Abstract O–015. .
64 Rubin KM. Managing immune-related adverse events to ipilimumab: a nurse’s guide. Clin J Oncol Nurs. [Internet]. 2012 [cited 2014 Jun 28];16:E69–75. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22459539.
65 Di Giacomo AM, Biagioli M, Maio M. The emerging toxicity profiles of anti-CTLA-4 antibodies across clinical indications. Semin Oncol. [Internet]. 2010 [cited 2014 Jun 28];37:499–507. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21074065.
66 Attia P, Phan GQ, Maker A V, Robinson MR, Quezado MM, Yang JC, et al. Autoimmunity correlates with tumor regression in patients with metastatic melanoma treated with anti-cytotoxic T-lymphocyte antigen-4. J. Clin. Oncol. [Internet]. 2005 [cited 2014 Jun 12];23:6043–6053. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1473965&tool=pmcentrez&rendertype=abstract.
67 Agarwala SS, Ribas A. Current experience with CTLA4–blocking monoclonal antibodies for the treatment of solid tumors. J Immunother. [Internet]. [cited 2014 Jun 28];33:557–569. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20551840.
68 Patnaik A, Kang SP, Tolcher AW, Rasco DW, Papadopoulos KP, Beeram M, et al. Phase I study of MK-3475 (anti-PD-1 monoclonal antibody) in patients with advanced solid tumors. J Clin Oncol. 2012;30:abstr 2512. .
69 Davies M. New modalities of cancer treatment for NSCLC: focus on immunotherapy. Cancer Manag Res. [Internet]. 2014 [cited 2014 Jun 28];6:63–75. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3917949&tool=pmcentrez&rendertype=abstract.