Childhood asthma: causes, risks, and protective factors; a role of innate immunity
DOI: https://doi.org/10.4414/smw.2014.14036
Georgios T
Noutsios, Joanna
Floros
Summary
Childhood asthma is an umbrella of multifactorial diseases with similar clinical features such as mast cell and eosinophil infiltration causing airway hyper responsiveness, inflammation, and airway obstruction. There are various factors that are implicated in childhood asthma pathogenesis. A combined contribution of genetic predisposition, environmental insults, and epigenetic changes account for polarisation of the immune system towards T helper (Th) type 2 cell responses that include production of pro-inflammatory cytokines, IgE, and eosinophil infiltrates, shown to associate with asthma. Environmental cues in prenatal, perinatal, and early childhood seem to determine development of asthma incidence or protection against it. Mode of birth delivery, use of antibiotics, oxidative stress, exposure to tobacco smoke and an industrialised lifestyle are significant contributors to childhood asthma exacerbation. Environmental stimuli such as exposure to maternal antibodies through breast milk, and certain early infections favour Th1 cell responses, leading to the production of anti-inflammatory cytokines that protect from asthma. Aside from the Th cell responses the role of innate immunity in the context of alveolar macrophages, dendritic cells, and surfactant protein A (SP-A) and SP-D is discussed. SP-A and SP-D enhance pathogen phagocytosis and cytokine production by alveolar macrophages, bind and clear pathogens, and interact with dendritic cells to mediate adaptive immunity responses. Further study of the interactions between genetic variants of genes of interest (SP-A and SP-D) and the environment may provide valuable knowledge about the underlying mechanisms of various interactions that differentially affect asthma susceptibility, disease severity, and reveal potential points for therapeutic interventions.
List of abbreviations
Afu aspergillus fumigatus
AhR arylhydrocarbon receptor
AHR bronchial airway hyper-responsiveness
AMs alveolar macrophages
AP-1 activator protein 1
BDNF brain-delivered neurotrophin factor
BPD bronchopulmonary dysplasia
BT bronchial thermoplasty
CS Cesarean section
DCs dendritic cells
DEPs diesel exhaust particles
ETS environmental tobacco smoke
FcεRI-β high affinity receptor IgE
GBS group B streptococcal
GM-CSF granulocyte- macrophage colony-stimulating factor
GSDM Gasdermin-B
GSTM1 glutathione S-transferase M1
GSTP1 glutathione S-transferase pi 1
GWAS genome wide association studies
HLA-G human leukocyte antigen gene
HPA hypothalamic-pituitary-adrenal infantile axis
hTG humanised transgenic
ICAM-1 serum soluble intracellular adhesion molecule 1
IL interleukin
INF-β Interferon-beta
ΙNF-γ interferon-γ
LRIs lower respiratory viral infections
miRNAs microRNAs
MMP-9 matrix metalloproteinase 9
NF-κB nuclear factor kappa-light-chain-enhancer of activated B cells
NQ01 quinoneoxidoreductase 1
ORMDL3 sphingolipid biosynthesis regulator 3
OxS oxidative stress
PAH polyaromatic hydrocarbons
PM particulate matter
RDS respiratory distress syndrome
ROS reactive oxygen species
RSV respiratory syncytial virus
SCIT subcutaneously administered immunotherapy
SNPs single nucleotide polymorphisms
SP-A surfactant protein A
TB tuberculosis
TGF-β transforming growth factor β
Th T helper
TIMP-1 tissue inhibitor of matrix metalloproteinase 1
TLR toll-like receptors
Tregs T regulatory cells
TSLP genes thymic stromal lymphoprotein
WHO World Health Organization
WTC World Trade Center
Definition – problem
Childhood asthma is one of the most common respiratory disorders worldwide with increased prevalence in Western industrialised societies where they inflict a high economic burden. In America more than 7.1 million children have asthma (National Center for Health Statistics, 2011) and this translates to thousands of annual hospitalisations or 774,000 emergency room visits due to severe asthma attacks in children under 15 years old. In 2009, 3,388 people died from asthma and out of these 157 were children under 15 years old. The health care expenditures for asthma are estimated to be more than $50.1 billion per year in the USA, while the impact on the productivity of working parents rises to $5.9 billion loss per year (2007 report) [1]. Also, asthma is responsible for 10 million missed school days per year. According to World Health Organization (WHO) (2011), 235 million people worldwide suffer from asthma, and it is the most common chronic disease among children. The economic cost for the UK is estimated to be $1.8 billion and for Australia $460 million (WHO, 2013). Worldwide, the economic burden of asthma exceeds those of HIV/AIDS and TB combined. Childhood asthma is a complex syndrome composed of different phenotypes (i.e., observable biochemical, clinical, morphological, and physiological characteristics) and endotypes (i.e., subtypes of asthma), each with a distinct pathophysiology. The different phenotypes of childhood asthma, their clinical appearance and symptoms, the current diagnostic procedures and therapeutic regimens have been described previously by Wenzel [2]. Moreover, it has been noted that exhaled nitric oxide can help to distinguish the asthma phenotypes [3]. The five different phenotypes include early-onset allergic, late-onset eosinophilic, exercise induced, obesity related and neutrophilic asthma. The endotypes of childhood asthma define distinct pathophysiological and functional characteristics of the disease. There have been four different endotypes identified: early-on set severe allergic asthma, late-onset persistent eosinophilic asthma, aspirin-exacerbated airway disease, and allergic bronchopulmonary mycosal asthma [4]. The pathogenetic sources of asthma remain unknown and although genetic, environmental, and epigenetic factors have been identified, an effective therapeutic intervention is yet to be established.
Importance of sex prematurity interactions
Males are more susceptible to asthma in early childhood than females, but later on females are more prone to the disease in conjunction with other contributing factors such as weight gain, obesity, and hormonal changes [5]. It is known that the menstrual cycle, pregnancy, and menopause cause dramatic fluctuation in oestrogen levels and this fluctuation activates proteins that can produce inflammatory responses in the airways and impact the risk of asthma. Male preterm infants have higher rates of asthma incidence and chronic lung diseases than females [6]. The basis for sex-based differences in asthma is not entirely known. However, females appear to have higher airflow rates (i.e., volume of air that passes through lungs per unit time) and forced expiratory volumes (i.e., vital capacity or the volume of air that is expelled from the lung during a maximally forced expiratory effort after taking the deepest possible breath) than males possibly due to differences in airway smooth muscle and wall diameters [7].
Nowadays, the “male disadvantage” hypothesis for the prematurely born infant is well established [8]. Males in comparison to females show higher frequency of RDS, asthma and/or other lung related injuries. Sex differences in gonadal steroid production in uteroand increased cerebrospinal levels of IL-8 and leptin in females in comparison to males [9] have been discussed as plausible reason for increased asthma occurrence in males. Moreover, it has been demonstrated that maternal stress factors during pregnancy (such as maternal smoking and obesity) increase the vulnerability to lung diseases in males compared to females [10]. Also, a difference has been observed in intrauterine fetal growth retardation between sexes, with males showing greater fetal retardation, as well as slower lung maturation than females [11]. In preterm females some in utero prenatal infections have a protective effect against asthma while males tend to be more susceptible to them [12]. These fetal stresses (in utero bacterial infections) in females may result in acceleration of maturation of lung development and immune system responses and this may not happen in males.
Prematurity is often accompanied with therapies such as mechanical ventilation and postnatal corticosteroids, both of which are shown to strongly associate with an increased incidence of childhood asthma. The barotrauma caused by mechanical ventilation and the corticosteroids effect on infantile lung development and bronchial remodelling are most likely the underlying contributors to the increased childhood asthma incidence [13]. Postnatally preterm females in comparison to males have an extremely active thymus [14] that may be particularly responsive to cytokines that modulate immune regulatory functions, thus rendering protection against asthma in females.
The parental contribution to childhood asthma is rather complex and not fully understood. For example in the 1989 Isle of Wight Birth Cohort (n = 1,456) maternal asthma was associated with asthma in girls but not in boys; paternal asthma was associated with asthma in boys but not in girls. Maternal eczema was associated with increased risk of eczema in girls only, whereas paternal eczema did the same for boys [15]. Others have shown that maternal asthma more than paternal asthma is associated with early childhood asthma [16]. However, the prevalence of asthma later in puberty and adolescence seems to depend on the sex of the child, with girls being more susceptible to asthma [17]. These indicate that further studies are needed to unravel the observed complexity of childhood asthma.
Causes and risk factors of childhood asthma
A number of diverse factors have been implicated in asthma pathogenesis in children. These include genetic predisposition (asthmatic parents), environmental stimuli during prenatal and early childhood that include allergens (mite, cat, dog, grass, pollen, and mould) [18], maternal infection and smoking during pregnancy, environmental tobacco smoke, mode of birth delivery (i.e., Casearean section), viral respiratory illnesses, obesity, diet, hygiene, and toxic exposures, fig. 1.
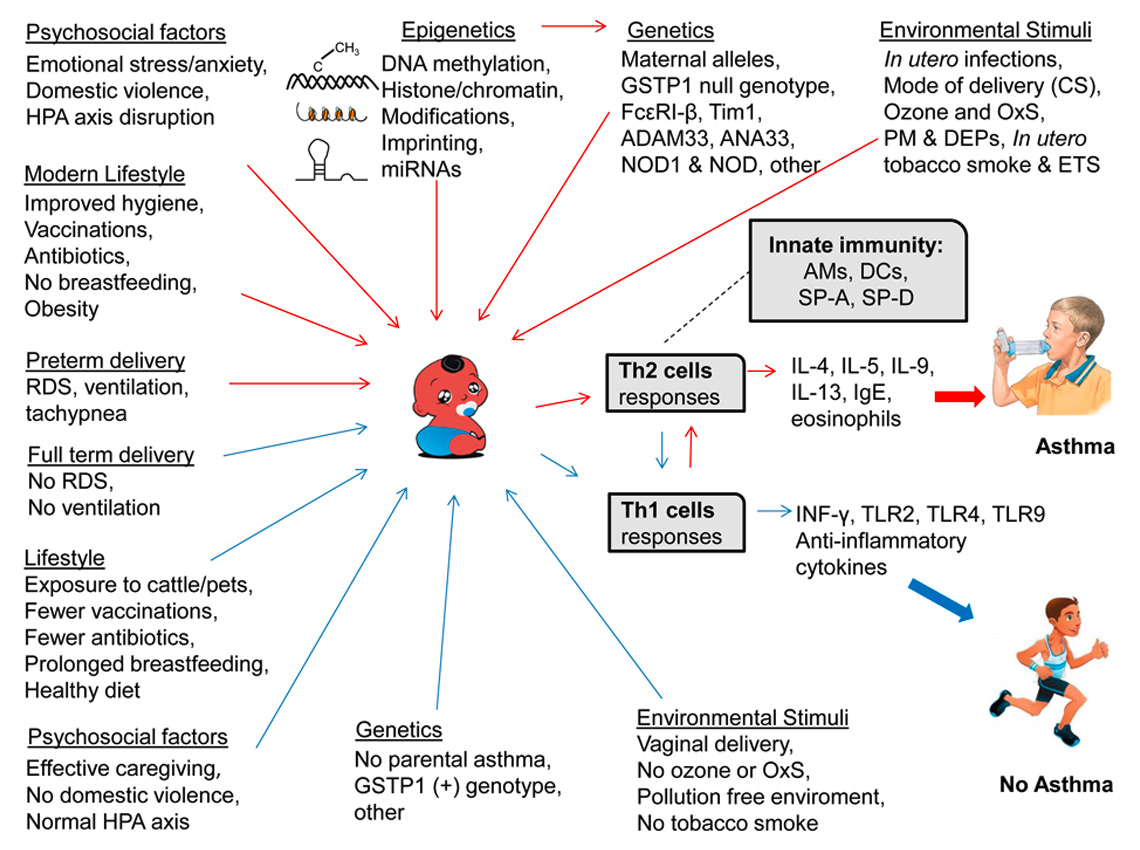
Figure 1
Diagrammatic summary of childhood asthma causes, risks and protective factors. The figure depicts the causes and the risk factors of childhood asthma that include genetics, epigenetics and environmental insults. These favour the Th2 biased cell response and select against Th1 cell response leading to inflammation and exacerbation of asthma (red arrows). The figure shows the factors leading to the Th1 cell response that produces anti-inflammatory cytokines and these in turn protect against asthma (blue arrows). The role of innate immunity in asthma, as exemplified by AMs, DCs, SP-A and SP-D is also noted with a broken line. The human cartoons were taken from Google. (Infant cartoon: http://123freevectors.com/black-baby/#.UVtFcFdyPfo, Asthmatic child: http://krames.sjmctx.com/HealthSheets/3,S,88710, Healthy child: http://www.wernerbaumgartner.info/)

Figure 2
Contributions of different immune system cells and molecules in pathogenesis of childhood asthma. The solid lines describe the T helper cells that have been associated with asthma in humans whereas the broken lines represent asthma studies that have been conducted in animal models and their role in human is yet to be determined. Th2 responses lead to production of IL-4, IL-5, IL-9, IL-13 which are associated with increased levels of IgE and eosinophils leading to asthma. Th1 responses include anti-inflammatory cytokine INF-γ, and early infant immune system development (possibly by TLR-2, TLR-4, TLR-9). Th1 responses render protection against asthma. Tregs inhibit the production of Th2 cells and Th2 cytokines, and are regulators of immune system self-tolerance, prevent autoimmunity and suppress allergies. Th17 cells produce pro-inflammatory cytokines IL-17A, IL-17F, IL-21, IL-22, IL-26 and are associated with an increased number of neutrophils and amount of inflammation. Th22 cells, which produce pro-inflammatory IL-22, are believed to play an important role in asthma pathogenesis. Th9 cells produce IL-9, which stimulates expression of FcεRI-α and mast cell proteases. Th9 cells are also believed to play an important role in asthma disease.
No single factor can account for the rapidly increasing incidence of childhood asthma during the last two decades. Genetic changes alone should be excluded because the gene pool does not change rapidly enough to explain this increase. It is assumed that a combined contribution of environmental and epigenetic (the result of environmental insults) changes, accounts for the increased prevalence of this emerging health risk. Because childhood asthma is correlated with chronic comorbid diseases [19] such as increased respiratory infections, bronchitis, cystic fibrosis, pneumonia, atopic dermatitis, otitis media (middle ear effusion), olfactory disorders and lung cancer, it is possible that prevention and treatment of childhood asthma will eliminate and/or benefit a great number of significant life-long health burdens.
Genetic and epigenetic factors
Increasing evidence supports the notion that in children, an early onset of allergic immune response might be a triggering factor for the physiological lung remodelling process. This in turn may lead to a lower lung function, bronchial airway hyper-responsiveness (AHR), and persistent asthma into adolescence. In addition, childhood asthma has been implicated in a predisposition toward developing certain allergic hypersensitivity reactions. Asthma and allergies run in parallel under certain genetic (i.e., asthmatic parents) and perinatal influences [20] (such as in utero infections [12], antibiotics, and tobacco smoke exposure [21]) rather than being the stepping stones for a progressive atopic march [19]. Atopy is defined as a genetic predisposition toward the development of immediate hypersensitivity reactions against common environmental antigens.
Genetics: It has been found that children whose parents are asthmatic are more likely to have asthma at school age. Children sensitised to allergens, with strong family history of asthma, by the age of three years have significantly lower airway conductance (i.e., lower instantaneous rates of gas flow in the airway per unit of pressure difference between the mouth, the nose, and the alveoli) [22]. Specific asthma-associated gene polymorphisms have been shown to be more likely to be passed down to children from the mother than the father. For example polymorphisms in the high affinity receptor IgE (FcεRI-β) (L181I and L183V on exon 6 and E237G on exon 7) [23] that increased asthma risk, were transmitted from mother to offspring and resulted in greater IgE levels and positive allergic skin tests. Another example is that of glutathione S-transferase pi 1 (GSTP1) locus polymorphisms [24], where genotypes Val105/Val105 and Ala114/Val114 were associated with greater lung function and their transmission was maternal rather than paternal, fig 1.
Fetal programming of gene expression during development is critical to the formation of a normal lung. Two large genome wide association studies (GWAS) in European [25] and diverse US populations [26] produced remarkably similar results. Single nucleotide polymorphisms (SNPs) in or near seven loci were associated with asthma in both studies, and SNPs in or near four of these loci had p-values at or near genome wide levels of significance in both studies, with contributions from ethnically diverse samples. Variation at the 17q21 asthma locus that encodes the GSDML [GSDML gene is predicted to be generated due to a duplication of Gasdermin-B (GSDM) gene] and ORMDL3 (sphingolipid biosynthesis regulator 3) genes, is specifically associated with childhood onset asthma [27]. Moreover, variations have been found in epithelial cell-derived cytokines genes, thymic stromal lymphoprotein (TSLP), IL-33, IL1RL1 (that encodes the receptor of IL-33) and ST2 (the receptor for IL-33 on mast cells, Th2 cells, Tregs and macrophages) [27]. All these associations highlight the importance of epithelial cell-derived cytokines that promote differentiation and activation of Th2 cells and their receptors. Despite the success of GWAS for discovering common risk alleles for many complex diseases and quantitative phenotypes, only a small proportion of the heritability is accounted for by these variations, and is now apparent for asthma. This has been termed as “the dark matter” or “missing heritability”, and has been discussed extensively in the literature [28, 29].
Epigenetics:The variability of asthma symptoms (i.e., incidence and remittance) can be the result of epigenetic influences induced by early (perinatal) or later (infantile) environmental exposures. Epigenetics gene expression regulation is a heritable change of gene expression that happens without any alteration in the DNA sequences and involves i) histone and chromatin modifications, ii) DNA methylation in the promoter regions, iii) imprinting (methylation of DNA sequences that alter the binding of specific transcription factor and/or enhancer elements), and iv) micro-RNA (miRNA) changes in conjunction with environmental stimuli.
In vitro studies have demonstrated that environmental insults such as in utero exposure to maternal tobacco smoke, environmental tobacco smoke (ETS), oxidative stress, diesel exhaust particles (DEPs), polyaromatic hydrocarbons (PAH), either during pregnancy or shortly after birth, mediate histone modification, and methylation of DNA sequences in T-helper cell genes that lead to induction of polarisation towards allergic type 2 (Th2) immune responses associated with asthma versus Th1 responses that are involved in atopic protection [30], fig. 1. T lymphocytes expressing CD4 glycoproteins on their surface are called T helper (Th) cells and are the main factories of pro- and anti- inflammatory cytokines; these cytokines are the hormonal messengers responsible for cell mediated immunity and allergic responses. Th1 pro-inflammatory responses mainly include production of interferon-γ (ΙNF-γ) (which inhibits the synthesis of IgE and eosinophil degranulation) and are mostly associated with intracellular elimination of parasites. Th2 responses mainly include the production of interleukin (IL)-4, IL-5, IL-9, and IL-13, and are associated with increased levels of IgE and eosinophil production leading to atopy [31], fig. 1. Thus, the Th1 response is considered protective for asthma and the Th2 response is associated with severe airway inflammation and asthma [32]. However, other studies have demonstrated contradicting results. In particular, when Th1 cells were transferred into naïve mouse recipients, the Th1 cells migrated to lungs and although they secreted INF-γ, they failed to counterbalance the Th2–cell-induced AHR and caused severe airway inflammation [30]. It is postulated that environmental stimuli may render protection against asthma by additional non Th1–Th2 regulatory mechanisms.
Aside from the well-established Th1–Th2 paradigm, numerous studies show that the actual situation is much more complicated and other Th cells such as Th17, Th22, Th9, and T regulatory cells (Tregs) also play key roles in the pathogenesis of childhood asthma [33] (fig. 2). For example Th17 cells are identified as a new subset of Th cells that are characterised mainly by the production of IL-17A and other well-known pro-inflammatory mediators such as IL-17F, IL-21, IL-22, and IL-26 that play a role in the inflammatory processes [34, 35]. These together, along with the observation that increased IL-17A mRNA is correlated with an increased number of neutrophils in asthmatic patients [36], suggest that Th17 plays an important role in asthma aggravation by recruiting neutrophils to the already inflamed bronchial airways (fig. 2). Moreover, in the past decade a small subpopulation of Th cells that produce IL-22 but lack the production of IL-17A has been identified as new subset of cells, namely Th22 cells [37]. These are believed to play an important role in asthma pathogenesis albeit their function in asthmatic patients has to be elucidated. Another distinct subset of Th cells involved in asthma pathogenesis is Th9 cells, which produce large amounts of IL-9 [38]. This cytokine is known to stimulate the expression of FcεRI-α (in Th cells and neutrophils) and mast cell proteases, and it has been suggested that IL-9 primes mast cells to respond to allergen challenge with increased FcεRI-α and proteases. However, all studies on Th9 cells have been conducted in mouse models and the existence of Th9 in humans remains to be determined. The Tregs have been suggested to play an important role in the immunopathogenesis of allergic asthma. Tregs are regulators of the immune system’s self-tolerance, can prevent autoimmunity, and suppress allergies. In several in vitrostudies Tregs have been reported to inhibit the production of Th2 cells and the Th2 cytokines, and to suppress the development of asthma [39]. The number of Tregs in allergic subjects appears to be significantly decreased in the lung airway tissue, BAL, and peripheral bood [40]. All the subsets of T helper cells described above could help to explain the existence and treatment of the different phenotypes and endotypes of childhood asthma.
MicroRNAs (miRNAs) constitute one of the epigenetic mechanisms regulating gene expression. These are a recently discovered short (~22 nucleotide) non-coding RNA sequences that bind to complementary sequences in the 3’UTR of multiple mRNAs, usually resulting in gene silencing or suppression of gene expression. Differential expression of miRNAs has been shown to contribute to asthma and other diseases and hold the potential as valuable diagnostic markers, fig. 1. For example, a single nucleotide polymorphism in the 3’UTR of the asthma-susceptibility human leukocyte antigen gene (HLA-G) affects the binding of three miRNAs. The binding of these regulatory miRNAs most probably blocks the HLA-G mRNA translation or mediates the degradation of HLA-G mRNA [41]. HLA-G, which is expressed in the placental cells that constitute the interface between the fetus and the mother, displays immunosuppressive properties. Reduced levels of HLA-G have been associated with asthma.
In addition, it has been shown that the inheritance of childhood asthma can take place across multiple generations. Grand maternal smoking has been linked with grandchild asthma vulnerability independent of maternal smoking [42], supporting further the notion that environmental challenges mediate heritable epigenetic modifications to important genes that can be passed on to the second offspring generation.
Environmental stimuli
Infection and Immunity:During pregnancy in utero exposures to infections, such as maternal vulvovaginitis (Candida albicans), chorioamnionitis (bacterial infection of the foetal membranes, amnion, and chorion), and Group B streptococcal (GBS) bacterial infection, negatively affect foetal pulmonary and immunological development and have been shown to associate with bronchopulmonary dysplasia [43] (BPD) and potentially childhood asthma [12], fig. 1. Furthermore, prescribed perinatal antibiotics to fight maternal infections are known to cross the placenta, enter the foetal bloodstream and cause alterations in the neonatal microflora including the proliferation of resistant bacteria and induction of immune developmental modifications [44] that may lead to asthma. Also, a wide variety of insults can alter or interfere with the foetal respiratory and/or immune system maturation and thus can significantly impact the risk of asthma, respiratory distress syndrome (RDS), and/or other airway related diseases.
The mode of child delivery has been associated with risk of childhood asthma. Over the past two decades in the western industrialised societies the prevalence of asthma runs in parallel with increased rates of Cesarean section (CS) [45], fig. 1. There are two plausible biological explanations for this correlation. In the CS the newborn does not get exposed to the maternal birth canal and perineum microbial flora, and thus the infant’s sterile gut does not stimulate the maturation of the immune system as would have been the case in a normal vaginal delivery. Another possible biological reason is that the CS in newborns may lead to RDS and transient tachypnoea and both of these factors are related to increased asthma risk in the early childhood. A limited number of studies have demonstrated that preterm deliveries accompanied with neonatal respiratory morbidity are associated with increased risk of childhood asthma [46].
Early transient infant wheezing induced by lower respiratory viral infections (LRIs) such as bronchiolitis appear to stimulate the children’s immune system and protect them against asthma, whereas persistent wheezing later in childhood is strongly associated with asthma and allergies [47]. Although it is known that respiratory syncytial virus (RSV) is the most common trigger of bronchiolitis, it is well established that rhinovirus is the bio-agent that is mostly associated with childhood asthma. Upregulation of ICAM-1 observed in children diagnosed with allergic asthma, facilitates the translocation of rhinovirus into epithelial cells [18]. The subsequent upregulation of interferon-beta (INF-β) that would normally induce rhinovirus clearance, via apoptosis, fails in asthmatic children allowing the virus to further replicate leading to cell lysis and viral propagation though the airways.
Nutrition also plays a role in childhood asthma pathogenesis. Especially vitamin D and folate (as methyl group donors) seem to be important. Ten different large studies reported conflicting results on the association of maternal folate levels with childhood asthma risk [48–57]. The majority of studies reported no association albeit a few reported a transient in nature, early-onset childhood asthma risk related with folic acid supplementation in late pregnancy. Maternal folate and childhood asthma have been recently reviewed [58].
Ozone: Exposure of children to ozone has been linked with significantly increased levels of eosinophils, production of pro-inflammatory cytokines IL-1, IL-6, IL-8, granulocyte-macrophage colony-stimulating factor (GM-CSF), and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). These lead to airway inflammation, childhood asthma, and other deleterious effects on human lung [59]. Oxidative stress differentially affects individuals probably due to differences in genetic predisposition to asthma. Ozone disrupts epithelial integrity, compromises mucociliary clearance, impairs effective phagocytosis, oxidises biomolecules in the lung, generates free radicals and activates inflammatory lung cells. Oxidative stress activates redox-sensitive transcription factors, such NF-κB and activator protein 1 (AP-1), mediating thus inflammatory cytokine production, and exacerbates asthma and allergic disease. Major polymorphisms of the quinoneoxidoreductase 1 (NQ01) and glutathione S-transferase M1 (GSTM1) genes have been shown to differentially affect asthma risk in children under oxidative stress [60], fig. 1.
Ozone and oxidative stress
(OxS): OxS affects the function of alveolar macrophages (AMs) that serve as the front line of the innate lung cellular immune defense against respiratory pathogens. Moreover, AMs’ dysfunction has been correlated with asthma in a rat model [61]. Specifically, OxS affects protein expression and compromises the function of specific proteins via oxidation. This happens by a variety of mechanisms that usually involve oxidation of specific reactive cysteines and/or modification of amino acids. Carbonylation, one of the more widely characterised modification of proteins under OxS, has been implicated in the pathogenesis and progression of a variety of diseases including asthma [62]. Oxidation of proteins can interfere with their function and their metabolism by either promoting degradation or by forming protein aggregates that are not readily degraded. In vivo mouse model studies have demonstrated that OxS-dependent changes have an impact on epithelial permeability, inflammatory mediators, and lead to pneumonia vulnerability.
Surfactant Protein A (SP-A), an innate immunity molecule and a component of bronchoalveolar lavage, is an example of a protein that its function is affected by OxS [63–65]. Oxidised SP-A may negatively impact the function of AMs and this has been shown by several studies. For example, our in vitro studies have shown that the ability of SP-A to enhance phagocytosis by AMs is significantly reduced after ozone-induced oxidation of SP-A [64, 66, 67]. Moreover, ozone-induced oxidation of SP-A in vivo altered the proteomic expression profile of the bronchoalveolar lavage proteins [68]. This included a decrease in the levels of proteins involved in the redox balance and an increase in the levels of proteins involved in metabolism, protein modification and chaperons. Several important innate immune functions have been ascribed to SP-A [69, 70]: i) recognition and opsonisation of some pathogens, ii) inflammatory mediator expression (TNF-α and interleukins) by some immune cells, iii) maturation of dendritic cells, and iv) regulation of reactive oxygen species (ROS) production.
Particulate matter
(PM): Many bio-agents such as carbon, iron, nickel, copper, organic residues and endotoxins are included in PM. In addition to PM, Diesel Exhaust Particles (DEPs) contain polyaromatic hydrocarbons (PAH), which are one of the ligands of the cytoplasmic arylhydrocarbon receptor (AhR). Upon binding the AhR is activated, and is translocated to the nucleus and this results into changes of transcriptional programmes [71]. DEPs increase eosinophil degranulation and eosinophil adhesion to nasal epithelial cells. DEPs have also been shown to i) induce CD80 expression in AMs, ii) augment histamine release, and iii) shift (in children) the primary immune system responses to Th2 phenotype (and thus select against the Th1 protective responses) [72], fig. 1. As noted above the Th2 phenotype involves production of cytokines (IL-4, IL-5, GM-CSF) and IgE known to associate with asthma.
The example of World Trade Center (WTC) towers collapse on 9/11 highlights the importance of the environmental exposure in the development of asthma. It is now known that WTC responders suffered from asthma at more than twice the rate of the general U.S. population as a result of their exposure to the toxic dust [73]. Multiple airborne pollutants increased lung inflammation and AHR, and induced acute lung function reduction.
Tobacco smoke: In utero foetal exposure to maternal tobacco smoke is associated with increased rates of childhood asthma and wheezing, elevated IgE levels, and increased bronchial activity [74]. It is noteworthy mentioning that a null GSTM1 genotype (a mutant copy of GSTM1 that completely lacks function) in conjunction with maternal smoking during pregnancy and/or involuntary infantile passive smoking is strongly correlated with asthma prevalence, while children with GSTM1 (+) genotype are less likely to develop asthma [24], fig. 1. The GSTM1 enzyme is involved in detoxification of the ROS and the tobacco metabolic intermediates. While the gene is polymorphic, in smokers the very common null genotype is shown to be associated with asthma, lung cancer, and DNA damage.
Not only maternal smoking affects fetal lung development and function but also postnatal Environmental Tobacco Smoke (ETS) increases the child’s risk of pulmonary malfunction [59], fig. 1. ETS includes both side stream tobacco smoke and exhaled mainstream smoke. High concentration of more than 3,800 toxic substances such as carbon monoxide, carbon dioxide, acrolein, ammonia, sulfur dioxide, crotonaldehyde, formaldehyde, hydrogen cyanide, and PAH negatively affect the mucociliary function, damage lung cells and tissues, and especially the Clara cells, thus inhibiting lung cell proliferation/regeneration [75]. In addition, it has been demonstrated that a single cigarette puff generates the stunning potent oxidant mixture of 1015toxic free radicals [76], resulting in activation of transcriptional factors [77] that regulate the Th2 immune responses.
Studies have shown that children exposed to mothers smoking more than 10 cigarettes per day have a 63% increased likelihood of developing asthma [42]. ETS has been identified as an asthma risk factor for both children and adults. Tobacco allergenic glycoproteins survive combustion and remain immunologically active influencing the immune system of children and adults [78]. Moreover, it appears that there is a gene-environment synergistic interaction between genetic susceptibility and ETS. Genes in chromosomal regions 1p, 5q, and 7p were shown to interact with ETS contributing to asthma risk, while genes in regions 1q and 9q are probably related to asthma predisposition via a pathway independent of ETS infant exposure [74]. Hence, childhood asthma appears to be the consequence of the interplay between genes and ETS. Exposure to ETS results in elevated IgE, Th2 immune responses, histamine release from mast cells, and influx of eosinophils into the lungs that account for asthma and other systemic inflammatory diseases. Tobacco smoke is by far the most important environmental risk factor for asthma and allergic diseases. Despite the well-established detrimental health effects of ETS in children it is surprising that there are no guidelines to regulate ETS exposure of children within households.
The modern industrialised environmental impact
Approximately 12 polymorphic genes that have been found to regulate childhood asthma, control airway function and remodelling, mediate inflammatory responses, regulate IgE, and control cytokine and chemokine production [79]. Despite the fact that none of these genes have changed over the past two decades, a dramatic increased incidence of asthma is observed.
The modern industrialised lifestyle appears to be a significant contributor to asthma exacerbation rather than the genetic factors, fig. 1. In modern societies the improved hygiene in combination with vaccination and early use of antibiotics [80], which alter the gastrointestinal flora, result in a reduced incident of infection that would normally stimulate the infant’s immune system and hence these events collectively exacerbate asthma incidents [81]. Furthermore, a number of studies indicate that specific respiratory viruses may be implicated in childhood wheezing and asthma [82], while gastrointestinal exposure to bacteria has a beneficial effect on the maturation of immune system and protection against asthma [79].
Several studies have demonstrated that while children are born immunologically naïve and bear several genetic predispositions towards asthma [i.e., these include polymorphisms in Tim1 [83], ADAM33, ANAN33 [84], NOD1 and NOD2 [85], and others in choromosomes 5, 6, 11, 12, 14. Most likely, it is the early postnatal environmental factors that determine either the development of asthma or the immune protection against it. The latter includes environmental stimuli such as prevalence of colonisation of gastrointestinal tract with Gram-positive bacteria (i.e., commensal lactobacillus and bifidobacteria), exposure to insults from daycare or older siblings, contact with farm animals and dog/cat allergens that mediate the early development of the infant immune system possibly by Toll-like receptors (TLR-2, TLR-4, TLR-9), which induce Th1 responses [79], fig. 1. On the other hand, lack of environmental stressors, such as no other siblings, a germ-free environment, lack of prolonged breastfeeding, low lactobacillus, vaccination, and early use of antibiotics drive development of Th2 immune response and over production of cytokines (i.e., IL-4, IL-5, IL-9 and IL-13) that are associated with asthma, fig. 1.
There is a weak but still significant link between childhood obesity and asthma, fig. 1. In the USA 32% of children are either obese or overweight and this is attributed to the Western industrialised diet pattern that includes mainly obesogenic high density energy foods such as processed sugars, animal trans fatty acids, and low whole unprocessed plant foods. The most prevalent hypothesis is that high body weight in children is a state of chronic low grade inflammation, exacerbating AHR and contributing to the development of asthma. Indeed in children a high body mass index is frequently associated with insulin resistance, hyperglyceridaemia, hypercholesterolaemia, and induction of production of leukotrienes and other pro-inflammatory factors known to be involved in respiratory tract infection and airway inflammation. Moreover, studies have shown that it is the non-allergic asthma that correlates the strongest with obesity because it involves high concentration of neutrophils, perturbation of adipokines levels (adiponectin is an anti-inflammatory and leptin is a pro-inflammatory adipokine) and IL-8 inflammatory pathways [86].
Psychosocial factors
Aside from the genetic, epigenetic, and environmental factors also psychosocial factors such as emotional stress of both mother and infant, maternal anxiety or depression, high levels of domestic and community violence, and ineffective maternal responsive caregiving in early infancy are strongly associated with childhood asthma and allergies [87], fig. 1. There is accumulating evidence unveiling that stressful events affect the buffering reactivity of hypothalamic-pituitary-adrenal infantile axis (HPA axis) causing low levels of endogenous glycocorticoids, which in turn lead to allergic inflammation in airway responses that may contribute to the onset of childhood asthma [88].
Race
Studies have shown that asthma is more prevalent in African American children than other ethnic populations, whereas among the culturally diverse Hispanic populations, the Puerto Rican ancestry has the highest rate of mortality and morbidity while Mexican Americans have the lowest rates. However, Chinese Americans have the lowest rates of all (American Lung Association State of Lung Disease in Diverse Communities 2010).
Pathology of childhood asthma
Over the past decade it has been realised that the severity and the chronicity of persistent asthma cannot be explained only by airway inflammation. Airway remodelling is another contributing factor [89]. Bronchial biopsies revealed inflammatory infiltrates dominated by eosinophils, mast cells, lymphocytes, and bronchial goblet cell hyperplasia. In severe disease predominantly raised levels of neutrophils were associated with increased levels of IL-4, IL-5, IL-8, IL-13, matrix metalloproteinase 9 (MMP-9), tissue inhibitor of matrix metalloproteinase 1 (TIMP-1), serum soluble intracellular adhesion molecule 1 (ICAM-1), and transforming growth factor β (TGF-β) [90]. These cytokines seem to correlate with lung tissue remodelling as these lead to vascular changes, thickening of lamina reticularis, increased collagen deposition within the lamina propia, smooth muscle hypertrophy and generation of myofibroblasts [89]. For example, TGF-β stimulates collagen deposition in the airway wall and the subepithelial connective tissue. Collagen deposition is controlled by MMP-9 which has collagen-degrading properties and TIMP-1 which is an of inhibitor metalloproteases. The balance of these two cytokines is thought to be disrupted in asthma, and this has dire consequences on airway remodelling. Briefly, inflammatory infiltrates result in altered airway structures, changes in functional lung muscle, and thickening of all compartments of airway walls. The functional consequences of these events are airway narrowing (obstruction) and progression of asthma [18]. On the other hand TGF-β in addition to its pro-asthmatic contribution due to collagen deposition in lung airways has been known to have anti-inflammatory properties by inhibiting differentiation of immune cells (T cells, B cells, Th1, and Th2) and inhibiting production of INF-γ and IL-2 [91]. It is well established that TGF-β is a critical differentiating factor that exerts potent immunosuppressive effects on Treg cells [92]. Also during inflammation and infection TGF-β helps to convert naïve T cells into Tregs and Th17 to combat infection, while at the same time the same cytokine (TGF-β) protects and maintains Tregs against apoptosis during inflammation. TGF-β is believed to promote immune tolerance, maintain lung homeostasis and regulate its host defence against insults [93]. The above show that the contribution of TGF-β in childhood asthma is very complex and still a matter of debate.
Beside cytokines and TGF-β, also growth factors contribute to the complex pathogenesis of childhood asthma. Recently the family of growth factors called neurotrophins has been shown to have an important role in the lung. In specific the brain-delivered neurotrophin factor (BDNF) is emerging as an important contributor in the early airway and lung development. Disruption of BDNF expression has been reported in premature neonates and during lung infection and inflammation. This may serve as an example of the brain-lung axis in the pathogenesis of various complex lung diseases [94].
Asthma, lung innate immunity and surfactant proteins A and D
The dogma of childhood asthma association with Th2 inflammation has recently been under question because i) Th2 response patterns that mediate AHR and lung tissue remodelling are not clearly linked to inflammation, and ii) the Th2–based therapies for childhood asthma have limited effectiveness [95].
Innate immunity is now emerging as an important factor that may explain childhood asthma. While innate immunity molecules protect the host from pathogens via recognition of pathogen associated patterns, it is now known that innate immunity also modulates a number of other processes such as allergic responses. At this point we would like to highlight the distinct role of AMs, the sentinel cells of lung innate immunity, that are involved in pathogen defence, recognition and removal of damaged tissue, neutralisation and generation of ROS [96]. These functions are directly linked to infection and OxS, both of which have been shown to trigger childhood asthma. In AMs from asthmatics, 38% of the differentially expressed genes were attributed to functions related to immune signaling response and stress, correlating thus the important role of AMs in childhood asthma pathogenesis [97]. In the course of breathing AMs are being exposed to allergens, microorganisms, and various other environmental insults. AMs interact with innate immunity molecules to bring about this first line of defence. These molecules include the surfactant protein A (SP-A) and SP-D [98, 99]. Many studies have shown that SP-A in addition to its role in surfactant-related (i.e., surfactant structure, function, and other) plays an important role in innate immune functions, including the enhancement of pathogen phagocytosis and cytokine production by AMs [66, 100], as well as the regulation of the AMs phenotype [69, 70, 101]. SP-A also has been shown to bind many allergens including dust mite, pollen, fungi, indicating that SP-A plays an important role in their clearance, and possibly in childhood asthma. SP-D is another important innate molecule that binds AMs and modulates the adaptive immune lymphocyte responses, playing a significant role in the immunologic environment of the lung and in allergic asthma [98].
The important roles of SP-A and SP-D in immunity have been further exemplified by their ability to mediate adaptive immune responses through interaction with dendritic cells (DCs) [98]. DCs the most potent antigen presenting cells, have the ability to activate naïve T-cells, and can initiate immune responses. DCs are distributed in lung parenchyma, airway epithelium, and alveolar space in order to capture inhaled antigens and present these antigens to T-cells. DCs mature in response to bacterial endotoxin, tissue damage, and pro-inflammatory cytokines. SP-D has been shown to enhance bacterial antigen uptake by DCs and to interact with DCs in order to present these antigens to T-cells. Specifically, SP-D binds immature DCs and enhances production of CD86, which is a T-cell activation ligand, and in this way augments antigen presentation [102]. On the other hand, SP-A has been shown to modulate lung inflammation by inhibiting DCs proliferation and preventing T cell activation. In specific, SP-A down-regulates the maturation of DCs by inhibiting the expression of co-stimulatory molecules CD86 and MHC-II, thus leading to inhibition of the activation of T-cells [103]. The above show that most probably SP-A and SP-D are the “yin” and “yang” of the lung’s DCs-mediated adaptive immunity.
SP-A is an important molecule in asthma and lung innate immunity. It has been shown to i) inhibit T-cell proliferation, ii) modulate phagocytosis of allergens and pathogens, iii) regulate cell surface proteins and metalloproteinases, and iv) modulate ROS production. SP-A and SP-D protect the host by recognising pathogen-associated molecular patterns and act as the bridging molecules in the clearance of apoptotic cells [104]. It is also known that SP-A and SP-D in asthmatic mouse models downregulate eosinophil inflammation, reduce production of IgE, and polarise T helper cell cytokines from Th2 towards Th1 phenotype. Moreover, these lung collectins (SP-A and SP-D) are implicated in Aspergillus fumigatus (Afu)-induced allergic asthma [105]. Specifically, these surfactant proteins have been shown to i) bind glycosylated antigens and allergens of Afu and to enhance their phagocytosis, ii) inhibit specific IgE binding to these allergens, iii) block histamine release from sensitised basophils, and when SP-A is administered exogenously to protect against Afu-induced pulmonary sensitivity.
Another plausible scenario for the involvement of SP-A in childhood asthma is that SP-A can alter the immune cells’ cytokine release. In vivo experimental models have demonstrated that in Afu-sensitised mice there was a transient decrease of SP-A levels while IL-4 and IL-5 mRNA and protein levels increased leading to an atopic Afu-induced T-cell proliferation [105, 106]. Moreover, SP-A has been shown not only to exert a protective effect in Afu-induced mouse asthma but also polymorphisms in the human SP-A2 gene have been associated with vulnerability to Afu-induced lung disease in humans [107].
SP-A genetic variants and childhood asthma: Human SP-A is encoded by two functional genes SP-A1 and SP-A2, and each gene has been identified with several polymorphic variants shown to associate with several lung diseases including asthma. The SP-A1 and SP-A2 variants have been identified with functional and regulatory differences [66, 100, 108, 109]. The function and the expression of some of these variants have been shown to change in response to OxS, indicating that these variants could contribute to lung disease via quantitative and/or qualitative derangement [63, 64, 68, 110, 111]. We have demonstrated that the ratio of SP-A1/SP-A differs among individuals as a function of age and health status (including asthma) [112, 113], and that SP-A from asthmatics does not abrogate inflammation as effectively as the SP-A from non-asthmatic subjects [113]. This dysfunction may be the result of an altered SP-A1/SP-A ratio. In fact this is a likely scenario because AMs from SP-A1 and SP-A2 humanised transgenic (hTG) mice (i.e. mice that lacked mouse SP-A but each carried a different human SP-A1 or SP-A2 variant) showed significant differences in the proteome profile derived from mice carrying the SP-A1 variant versus those carrying the SP-A2 variant [114]. Previous work from our laboratory has shown that a single exogenous treatment with SP-A of SP-A knockout mice is sufficient to nearly restore the AMs proteomic phenotype and make it similar to that of wild type [115]. In other words, when SP-A knockout mice (that lacked SP-A) were treated with SP-A, the AMs proteomic expression profile of the treated mice was closer to the wild type AM profile than the knockout AM profile indicating that SP-A has a major impact on the expression profile of the AMs. The fact that SP-A1 and SP-A2 differentially affect AMs indicates that an imbalance in the ratio (as it has been shown in asthmatics) may alter the phenotype of AMs and perhaps their function. This may in turn negatively affect the downstream processes and contribute to asthma and/or its exacerbation. The above data collectively provide evidence for a role of innate immunity and SP-A and SP-D in asthma. Moreover, the fairly large number of SP-A1 and SP-A2 variants, may have (via subtle quantitative and/or qualitative derangement) a further differential impact on asthma susceptibility among individuals. Future studies may help to understand SP-A variant-mediated susceptibilities and thus contribute to considerations in personalised medical therapies.
Breastfeeding and protection of childhood asthma risk
It is well established that infant breastfeeding is strongly associated with reduction of respiratory illnesses in later childhood and adolescence, although this association for childhood asthma is slightly different. It appears that there is a direct relation between the duration of breastfeeding and asthma protection when there is maternal history of asthma. On the other hand, no correlation has been found between lack of breastfeeding and infants who were atopic themselves or with no maternal asthma history [116]. Maternal milk apart from its nutritional components contains various molecules and cells shown to have a protective role in the newborn infant and also play an important role in the early acquired immune programming. For example one such molecule is IgA. This is produced by the secretory cells of the breast and protects the nursing infant from the microbial antigens that the mother has been exposed to [117].
Recently (2012) a report stated that breastfed infants are exposed to maternal microbiota. The authors suggested that exposure to mibrobiota determines the development of the infantile gut flora and possibly plays a protective role [118]. However, it is uncertain whether these bacteria originate from the maternal breast milk itself or the oral bacteria of the breast fed baby that enter breast milk and change its composition.
Therapeutic interventions
Childhood asthma is being treated by various medications either providing long term asthma control or quick-relief solutions. Long-term asthma medications include corticosteroids, long-acting beta agonists, leukotriene modifiers/inhibitors, theophylline, anticongestants and antihistamines. Quick-relief (rescue) medications include bronchodilators, sort-acting beta antagonists and oral or intravenous corticosteroids. It is important to highlight that the available medications merely target the symptoms and do not cure asthma.
Another option is subcutaneously administered immunotherapy (SCIT), which has shown significant improvement for control of allergic childhood asthma symptoms and medication needs. Although immunotherapy has proven to be efficacious for allergic asthma, it holds a major drawback, namely, the possibility of a major systemic allergic reaction in response to the treatment itself.
Omalizumab is a recombinant DNA-derived humanised IgG1k monoclonal antibody that selectively binds free IgE and is currently being used for Anti-IgE therapy in children over the age of 6 years. This therapy has been shown to reduce the free circulating IgE, the high affinity IgE receptors, and the mast cell and basophil activation [119].
Another treatment which is neither widely available nor appropriate for every asthmatic child is the bronchial thermoplasty (BT). BT was approved by the FDA in 2010 and is used primarily to treat severe persistent asthma that cannot be controlled with conventional therapies. BT reduces smooth muscle mass, via thermal energy, and thus attenuates the ability of smooth muscle cells to constrict. This can reduce the number of asthma attacks.
Since asthma appears to be a chronic inflammatory Th1/Th2–disease, a potential therapeutic intervention is to imitate the naturally occurring infections and/or to reduce regular early childhood vaccination regimens. As suggested by the authors of a multi-centre allergy study in 2002, one could stimulate Th1 immune system responses by exogenous administration of lactobacilli or endotoxin Gram-negative extracts early in infancy [79]. Although this was tested in a murine model in 2010 [120], so far it has not been tested on human subjects.
Conclusions
Childhood asthma is a complex multifactorial chronic bronchial inflammatory morbidity characterised by mast cell and mucosal eosinophil infiltration controlled by cytokines of Th2 lymphocytes. It appears that childhood asthma is the result of interplay between genetics, environmental insults, and epigenetic factors that favour the Th2 biased cell response and select against Th1 cell response. These events in turn trigger and exacerbate childhood asthma. Innate immunity is emerging as an important player in asthma, and the innate host defence molecules SP-A and SP-D appear to play a role as well. A diagrammatic summary of the susceptibility factors that may increase the risk of asthma or protect from asthma is shown in figure 1.
Comments
The available treatments seem to suppress rather than cure childhood asthma. The interplay of genetics with environmental insults through epigenetics may identify novel mechanisms of childhood asthma and unveil new potential points for therapeutic interventions. A concerted effort must be put forward to comprehend the underlying mechanisms as to why some children under certain circumstances develop asthma while others do not. We must understand the interplay between genetic variants of host defense molecules (such as SP-A and SP-D) and the environment and how these may differentially affect disease susceptibility and/or disease severity. This in turn may help explain the inter-individual vulnerability to asthma incidence and provide opportunities for therapeutic intervention and/or prevention of childhood asthma.
References
1 Barnett SB, Nurmagambetov TA. Costs of asthma in the United States: 2002–2007. J Allergy Clin Immunol. 2011;127(1):145–52.
2 Wenzel SE. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med. 2012;18(5):716–25.
3 Ricciardolo FL. Revisiting the role of exhaled nitric oxide in asthma. Curr Opin Pulm Med. 2014;20(1):53–9.
4 Lötvall J, Akdis CA, Bacharier LB, Bjermer L, Casale TB, Custovic A, et al. Asthma endotypes: a new approach to classification of disease entities within the asthma syndrome. J Allergy Clin Immunol. 2011;127(2):355–60.
5 Tantisira KG, Colvin R, Tonascia J, Strunk RC, Weiss ST, Fuhlbrigge AL, Group CAMPR. Airway responsiveness in mild to moderate childhood asthma: sex influences on the natural history. Am J Respir Crit Care Med. 2008;178(4):325–31.
6 Drevenstedt GL, Crimmins EM, Vasunilashorn S, Finch CE. The rise and fall of excess male infant mortality. Proc Natl Acad Sci U S A. 2008;105(13):5016–21.
7 Doctor TH, Trivedi SS, Chudasama RK. Pulmonary function test in healthy school children of 8 to 14 years age in south Gujarat region, India. Lung India. 2010;27(3):145–8.
8 Kirchengast S, Hartmannm B. The Male Disadvantage Hypothesis Reconsidered: Is There Really a Weaker Sex? An Analysis of Gender Differences in Newborn Somatometrics and Vital Parameters. In., vol. 1. J Life Sci; 2009:63–71.
9 Hussein MH, Daoud GA, Kakita H, Hattori A, Murai H, Yasuda M, et al. The sex differences of cerebrospinal fluid levels of interleukin 8 and antioxidants in asphyxiated newborns. Shock. 2007;28(2):154–9.
10 Etzel RA. How environmental exposures influence the development and exacerbation of asthma. Pediatrics. 2003;112(1 Pt 2):233–9.
11 Ley D, Wide-Swensson D, Lindroth M, Svenningsen N, Marsal K. Respiratory distress syndrome in infants with impaired intrauterine growth. Acta Paediatr. 1997;86(10):1090–6.
12 Collier CH, Risnes K, Norwitz ER, Bracken MB, Illuzzi JL. Maternal Infection in Pregnancy and Risk of Asthma in Offspring. Matern Child Health J. 2013.
13 Alshehri MA, Almegamesi TM, Alfrayh AS. Predictors of short-term hospital readmissions of asthmatic children. J Family Community Med. 2005;12(1):11–7.
14 Pido-Lopez J, Imami N, Aspinall R. Both age and gender affect thymic output: more recent thymic migrants in females than males as they age. Clin Exp Immunol. 2001;125(3):409–13.
15 Arshad SH, Karmaus W, Raza A, Kurukulaaratchy RJ, Matthews SM, Holloway JW, et al. The effect of parental allergy on childhood allergic diseases depends on the sex of the child. J Allergy Clin Immunol. 2012;130(2):427–34.e426.
16 Litonjua AA, Carey VJ, Burge HA, Weiss ST, Gold DR. Parental history and the risk for childhood asthma. Does mother confer more risk than father? Am J Respir Crit Care Med. 1998;158(1):176–81.
17 de Marco R, Pattaro C, Locatelli F, Svanes C, Group ES. Influence of early life exposures on incidence and remission of asthma throughout life. J Allergy Clin Immunol. 2004;113(5):845–52.
18 Warner JO, Boner A. Paediatric allergy and asthma, vol. Chapter 18: Elsevier Ltd; 2012.
19 Klinnert MD, Nelson HS, Price MR, Adinoff AD, Leung DY, Mrazek DA. Onset and persistence of childhood asthma: predictors from infancy. Pediatrics. 2001;108(4):E69.
20 Metsälä J, Kilkkinen A, Kaila M, Tapanainen H, Klaukka T, Gissler M, Virtanen SM. Perinatal factors and the risk of asthma in childhood – a population-based register study in Finland. Am J Epidemiol. 2008;168(2):170–8.
21 Goodwin RD. Environmental tobacco smoke and the epidemic of asthma in children: the role of cigarette use. Ann Allergy Asthma Immunol. 2007;98(5):447–54.
22 Morgan WJ, Stern DA, Sherrill DL, Guerra S, Holberg CJ, Guilbert TW, Taussig LM, Wright AL, Martinez FD. Outcome of asthma and wheezing in the first 6 years of life: follow-up through adolescence. Am J Respir Crit Care Med. 2005;172(10):1253–8.
23 Traherne JA, Hill MR, Hysi P, D’Amato M, Broxholme J, et al. LD mapping of maternally and non-maternally derived alleles and atopy in FcepsilonRI-beta: Hum Mol Genet. 2003;12(20):2577–85.
24 Gilliland FD, Li YF, Dubeau L, Berhane K, Avol E, McConnell R, et al. Effects of glutathione S-transferase M1, maternal smoking during pregnancy, and environmental tobacco smoke on asthma and wheezing in children. Am J Respir Crit Care Med. 2002;166(4):457–63.
25 Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. 201;363(13):1211–21.
26 Torgerson DG, Ampleford EJ, Chiu GY, Gauderman WJ, Gignoux CR, Graves PE, et al. Meta-analysis of genome-wide association studies of asthma in ethnically diverse North American populations. Nat Genet. 2011;43(9):887–92.
27 Ober C, Yao TC. The genetics of asthma and allergic disease: a 21st century perspective. Immunol Rev. 2011;242(1):10–30.
28 Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747–53.
29 Parker CC, Palmer AA. Dark matter: are mice the solution to missing heritability? Front Genet. 2011;2:32.
30 Hansen G, Berry G, DeKruyff RH, Umetsu DT. Allergen-specific Th1 cells fail to counterbalance Th2 cell-induced airway hyperreactivity but cause severe airway inflammation. J Clin Invest. 1999;103(2):175–83.
31 Berger A. Th1 and Th2 responses: what are they? BMJ. 2000;321(7258):424.
32 Finn PW, Bigby TD: Innate immunity and asthma. Proc Am Thorac Soc. 2009;6(3):260–5.
33 Vock C, Hauber HP, Wegmann M. The other T helper cells in asthma pathogenesis. J Allergy (Cairo). 2010;2010:519298.
34 Kreymborg K, Etzensperger R, Dumoutier L, Haak S, Rebollo A, Buch T, Heppner FL, Renauld JC, Becher B. IL-22 is expressed by Th17 cells in an IL-23–dependent fashion, but not required for the development of autoimmune encephalomyelitis. J Immunol. 2007;179(12):8098–104.
35 Steinman L. A brief history of T(H)17, the first major revision in the T(H)1/T(H)2 hypothesis of T cell-mediated tissue damage. Nat Med. 2007;13(2):139–45.
36 Bullens DM, Truyen E, Coteur L, Dilissen E, Hellings PW, Dupont LJ, Ceuppens JL. IL-17 mRNA in sputum of asthmatic patients: linking T cell driven inflammation and granulocytic influx? Respir Res. 2006;7:135.
37 Duhen T, Geiger R, Jarrossay D, Lanzavecchia A, Sallusto F. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat Immunol. 2009;10(8):857–63.
38 Veldhoen M, Uyttenhove C, van Snick J, Helmby H, Westendorf A, Buer J, Martin B, Wilhelm C, Stockinger B. Transforming growth factor-beta “reprograms” the differentiation of T helper 2 cells and promotes an interleukin 9–producing subset. Nat Immunol. 2008;9(12):1341–6.
39 Presser K, Schwinge D, Wegmann M, Huber S, Schmitt S, Quaas A, Maxeiner JH, Finotto S, Lohse AW, Blessing M et al. Coexpression of TGF-beta1 and IL-10 enables regulatory T cells to completely suppress airway hyperreactivity. J Immunol. 2008;181(11):7751–8.
40 Akdis M, Verhagen J, Taylor A, Karamloo F, Karagiannidis C, Crameri R, et al. Immune responses in healthy and allergic individuals are characterized by a fine balance between allergen-specific T regulatory 1 and T helper 2 cells. J Exp Med. 2004;199(11):1567–75.
41 Tan Z, Randall G, Fan J, Camoretti-Mercado B, Brockman-Schneider R, Pan L, et al. Allele-specific targeting of microRNAs to HLA-G and risk of asthma. Am J Hum Genet. 2007;81(4):829–34.
42 Li YF, Langholz B, Salam MT, Gilliland FD. Maternal and grandmaternal smoking patterns are associated with early childhood asthma. Chest. 2005;127(4):1232–41.
43 Lima MR, Andrade MoA, Araújo AP, Figueroa JN, Andrade LB. Influence of maternal and neonatal factors on bronchopulmonary dysplasia development. Rev Assoc Med Bras. 2011;57(4):391–6.
44 Marra F, Lynd L, Coombes M, Richardson K, Legal M, Fitzgerald JM, Marra CA. Does antibiotic exposure during infancy lead to development of asthma?: a systematic review and metaanalysis. Chest. 2006;129(3):610–8.
45 Magnus MC, Håberg SE, Stigum H, Nafstad P, London SJ, Vangen S, Nystad W. Delivery by Cesarean section and early childhood respiratory symptoms and disorders: the Norwegian mother and child cohort study. Am J Epidemiol. 2011;174(11):1275–85.
46 Smith GC, Wood AM, White IR, Pell JP, Cameron AD, Dobbie R. Neonatal respiratory morbidity at term and the risk of childhood asthma. Arch Dis Child. 2004;89(10):956–60.
47 Sly PD, Kusel M, Holt PG. Do early-life viral infections cause asthma? J Allergy Clin Immunol. 2010;125(6):1202–5.
48 Whitrow MJ, Moore VM, Rumbold AR, Davies MJ. Effect of supplemental folic acid in pregnancy on childhood asthma: a prospective birth cohort study. Am J Epidemiol. 2009;170(12):1486–93.
49 Håberg SE, London SJ, Stigum H, Nafstad P, Nystad W. Folic acid supplements in pregnancy and early childhood respiratory health. Arch Dis Child. 2009;94(3):180–4.
50 Bekkers MB, Elstgeest LE, Scholtens S, Haveman-Nies A, de Jongste JC, Kerkhof M, et al. Maternal use of folic acid supplements during pregnancy, and childhood respiratory health and atopy. Eur Respir J. 2012;39(6):1468–74.
51 Dunstan JA, West C, McCarthy S, Metcalfe J, Meldrum S, Oddy WH, et al. The relationship between maternal folate status in pregnancy, cord blood folate levels, and allergic outcomes in early childhood. Allergy. 2012;67(1):50–7.
52 Miyake Y, Sasaki S, Tanaka K, Hirota Y. Maternal B vitamin intake during pregnancy and wheeze and eczema in Japanese infants aged 16–24 months: the Osaka Maternal and Child Health Study. Pediatr Allergy Immunol. 2011;22(1 Pt 1):69–74.
53 Magdelijns FJ, Mommers M, Penders J, Smits L, Thijs C. Folic acid use in pregnancy and the development of atopy, asthma, and lung function in childhood. Pediatrics. 2011;128(1):e135–144.
54 Kiefte-de Jong JC, Timmermans S, Jaddoe VW, Hofman A, Tiemeier H, Steegers EA, et al. High circulating folate and vitamin B-12 concentrations in women during pregnancy are associated with increased prevalence of atopic dermatitis in their offspring. J Nutr. 2012;142(4):731–8.
55 Martinussen MP, Risnes KR, Jacobsen GW, Bracken MB. Folic acid supplementation in early pregnancy and asthma in children aged 6 years. Am J Obstet Gynecol. 2012;206(1):72.e71–77.
56 Litonjua AA, Rifas-Shiman SL, Ly NP, Tantisira KG, Rich-Edwards JW, Camargo CA, et al. Maternal antioxidant intake in pregnancy and wheezing illnesses in children at 2 y of age. Am J Clin Nutr. 2006;84(4):903–11.
57 Granell R, Heron J, Lewis S, Davey Smith G, Smith GD, Sterne JA, et al. The association between mother and child MTHFR C677T polymorphisms, dietary folate intake and childhood atopy in a population-based, longitudinal birth cohort. Clin Exp Allergy. 2008;38(2):320–8.
58 Brown SB, Reeves KW, Bertone-Johnson ER. Maternal folate exposure in pregnancy and childhood asthma and allergy: a systematic review. Nutr Rev. 2014;72(1):55–64.
59 Tatum AJ, Shapiro GG. The effects of outdoor air pollution and tobacco smoke on asthma. Immunol Allergy Clin North Am. 2005;25(1):15–30.
60 David GL, Romieu I, Sienra-Monge JJ, Collins WJ, Ramirez-Aguilar M, del Rio-Navarro BE, et al. Nicotinamide adenine dinucleotide (phosphate) reduced:quinone oxidoreductase and glutathione S-transferase M1 polymorphisms and childhood asthma. Am J Respir Crit Care Med. 2003;168(10):1199–204.
61 Careau E, Proulx LI, Pouliot P, Spahr A, Turmel V, Bissonnette EY. Antigen sensitization modulates alveolar macrophage functions in an asthma model. Am J Physiol Lung Cell Mol Physiol. 2006;290(5):L871–879.
62 Dalle-Donne I, Aldini G, Carini M, Colombo R, Rossi R, Milzani A. Protein carbonylation, cellular dysfunction, and disease progression. J Cell Mol Med. 2006;10(2):389–406.
63 Haque R, Umstead TM, Ponnuru P, Guo X, Hawgood S, Phelps DS, et al. Role of surfactant protein-A (SP-A) in lung injury in response to acute ozone exposure of SP-A deficient mice. Toxicol Appl Pharmacol. 2007;220(1):72–82.
64 Mikerov AN, Umstead TM, Gan X, Huang W, Guo X, Wang G, et al. Impact of ozone exposure on the phagocytic activity of human surfactant protein A (SP-A) and SP-A variants. Am J Physiol Lung Cell Mol Physiol. 2008;294(1):L121–130.
65 Wang G, Umstead T, Phelps D, Al-Mondhiry H, Floros J. The effect of ozone exposure on the ability of human surfactant protein a variants to stimulate cytokine production. Environ Health Perspect. 2002;110(1):79–84.
66 Mikerov AN, Umstead TM, Huang W, Liu W, Phelps D, Floros J. SP-A1 and SP-A2 variants differentially enhance association of Pseudomonas aeruginosa with rat alveolar macrophages. Am J Physiol Lung Cell Mol Physiol. 2005;288:L150–L158.
67 Mikerov AN, Gan X, Umstead TM, Miller L, Chinchilli VM, Phelps DS, et al. Sex differences in the impact of ozone on survival and alveolar macrophage function of mice after Klebsiella pneumoniae infection. Respir Res. 2008;9:24.
68 Haque R, Umstead TM, Freeman WM, Floros J, Phelps DS. The impact of surfactant protein-A on ozone-induced changes in the mouse bronchoalveolar lavage proteome. Proteome Sci. 2009;7:12.
69 Floros J, Phelps DS. Pulmonary surfactant protein A; structure, expression, and its role in innate host defense. In: Surfactant-Update of Intensive Care Medicine. Volume 6, edn. Edited by Nakos G, Lekka ME. Ioannina, Greece: University of Ioannina; 2002;87–102.
70 Floros J, Wang G, Mikerov AN. Genetic complexity of the human innate host defense molecules, surfactant protein A1 (SP-A1) and SP-A2–-impact on function. Crit Rev Eukaryot Gene Expr. 2009;19(2):125–37.
71 Kann S, Huang MY, Estes C, Reichard JF, Sartor MA, Xia Y, et al. Arsenite-induced aryl hydrocarbon receptor nuclear translocation results in additive induction of phase I genes and synergistic induction of phase II genes. Mol Pharmacol. 2005;68(2):336–46.
72 Fahy O, Sénéchal S, Pène J, Scherpereel A, Lassalle P, Tonnel AB, et al. Diesel exposure favors Th2 cell recruitment by mononuclear cells and alveolar macrophages from allergic patients by differentially regulating macrophage-derived chemokine and IFN-gamma-induced protein-10 production. J Immunol. 2002;168(11):5912–9.
73 Kim H, Herbert R, Landrigan P, Markowitz SB, Moline JM, Savitz DA, et al. Increased rates of asthma among World Trade Center disaster responders. Am J Ind Med. 2012;55(1):44–53.
74 Colilla S, Nicolae D, Pluzhnikov A, Blumenthal MN, Beaty TH, Bleecker ER, Lange EM, Rich SS, Meyers DA, Ober C et al. Evidence for gene-environment interactions in a linkage study of asthma and smoking exposure. J Allergy Clin Immunol. 2003;111(4):840–6.
75 Larsson ML, Frisk M, Hallström J, Kiviloog J, Lundbäck B. Environmental tobacco smoke exposure during childhood is associated with increased prevalence of asthma in adults. Chest. 2001;120(3):711–7.
76 Cross CE, Traber M, Eiserich J, van der Vliet A. Micronutrient antioxidants and smoking. Br Med Bull. 1999;55(3):691–704.
77 Rahman I, MacNee W. Role of transcription factors in inflammatory lung diseases. Thorax. 1998;53(7):601–12.
78 Francus T, Klein RF, Staiano-Coico L, Becker CG, Siskind GW. Effects of tobacco glycoprotein (TGP) on the immune system. II. TGP stimulates the proliferation of human T cells and the differentiation of human B cells into Ig secreting cells. J Immunol. 1988;140(6):1823–9.
79 Umetsu DT, McIntire JJ, Akbari O, Macaubas C, DeKruyff RH. Asthma: an epidemic of dysregulated immunity. Nat Immunol. 2002;3(8):715–20.
80 Murk W, Risnes KR, Bracken MB. Prenatal or early-life exposure to antibiotics and risk of childhood asthma: a systematic review. Pediatrics. 2011;127(6):1125–38.
81 McRae WM, Wong CS. Asthma, allergy and the hygiene hypothesis. In., vol. 29. Continuing Medical Education; 2002:31–7.
82 Gern JE, Busse WW. Association of rhinovirus infections with asthma. Clin Microbiol Rev. 1999;12(1):9–18.
83 McIntire JJ, Umetsu DT, DeKruyff RH. TIM-1, a novel allergy and asthma susceptibility gene. Springer Semin Immunopathol. 2004;25(3–4):335–48.
84 Tripathi P, Awasthi S, Husain N, Prasad R, Mishra V. Increased expression of ADAM33 protein in asthmatic patients as compared to non-asthmatic controls. Indian J Med Res. 2013;137(3):507–14.
85 Reijmerink NE, Bottema RW, Kerkhof M, Gerritsen J, Stelma FF, Thijs C, et al. TLR-related pathway analysis: novel gene-gene interactions in the development of asthma and atopy. Allergy. 2010;65(2):199–207.
86 Papoutsakis C, Priftis KN, Drakouli M, Prifti S, Konstantaki E, Chondronikola M, Antonogeorgos G, Matziou V. Childhood overweight/obesity and asthma: is there a link? A systematic review of recent epidemiologic evidence. J Acad Nutr Diet. 2013;113(1):77–105.
87 Dietert RR. Maternal and childhood asthma: risk factors, interactions, and ramifications. Reprod Toxicol. 2011;32(2):198–204.
88 Miller RL, Ho SM. Environmental epigenetics and asthma: current concepts and call for studies. Am J Respir Crit Care Med. 2008;177(6):567–73.
89 Tagaya E, Tamaoki J. Mechanisms of airway remodeling in asthma. Allergol Int. 2007;56(4):331–40.
90 Holgate ST. Pathogenesis of asthma. Clin Exp Allergy. 2008;38(6):872–97.
91 Yoshimura A, Wakabayashi Y, Mori T. Cellular and molecular basis for the regulation of inflammation by TGF-beta. J Biochem. 2010;147(6):781–92.
92 Costa R, Marques C, Rios R, Silva H, Carneiro N, Cana Brasil T, et al. Are TGF-β gene polymorphisms associated with asthma risk? In., vol. 1. OA Immunology; 2013: 5.
93 Tran DQ. TGF-β: the sword, the wand, and the shield of FOXP3(+) regulatory T cells. J Mol Cell Biol. 2012;4(1):29–37.
94 Prakash YS, Martin RJ. Brain-derived neurotrophic factor in the airways. Pharmacol Ther. 2014;143(1):74–86.
95 Murdoch JR, Lloyd CM. Chronic inflammation and asthma. Mutat Res. 2010;690(1–2):24–39.
96 Balhara J, Gounni AS. The alveolar macrophages in asthma: a double-edged sword. Mucosal Immunol. 2012;5(6):605–9.
97 Madore AM, Perron S, Turmel V, Laviolette M, Bissonnette EY, Laprise C. Alveolar macrophages in allergic asthma: an expression signature characterized by heat shock protein pathways. Hum Immunol. 2010;71(2):144–50.
98 Hohlfeld JM, Erpenbeck VJ, Krug N. Surfactant proteins SP-A and SP-D as modulators of the allergic inflammation in asthma. Pathobiology. 2002;70(5):287–92.
99 Silveyra P, Floros J. Genetic variant associations of human SP-A and SP-D with acute and chronic lung injury. Frontiers in Bioscience. 2012;17:407–29.
100 Wang G, Phelps DS, Umstead TM, Floros J. Human SP-A protein variants derived from one or both genes stimulate TNF-alpha production in the THP-1 cell line. Am J Physiol Lung Cell Mol Physiol. 2000;278(5):L946–54.
101 Phelps DS. Surfactant regulation of host defense function in the lung: a question of balance. Pediatr Pathol Mol Med. 2001;20(4):269–92.
102 Brinker KG, Martin E, Borron P, Mostaghel E, Doyle C, Harding CV, et al. Surfactant protein D enhances bacterial antigen presentation by bone marrow-derived dendritic cells. Am J Physiol Lung Cell Mol Physiol. 2001;281(6):L1453–63.
103 Brinker KG, Garner H, Wright JR. Surfactant protein A modulates the differentiation of murine bone marrow-derived dendritic cells. Am J Physiol Lung Cell Mol Physiol. 2003;284(1):L232–41.
104 Jäkel A, Clark H, Reid KB, Sim RB. The human lung surfactant proteins A (SP-A) and D (SP-D) interact with apoptotic target cells by different binding mechanisms. Immunobiology. 2010;215(7):551–8.
105 Madan T, Kishore U, Singh M, Strong P, Clark H, Hussain EM, et al. Surfactant proteins A and D protect mice against pulmonary hypersensitivity induced by Aspergillus fumigatus antigens and allergens. J Clin Invest. 2001;107(4):467–75.
106 Madan T, Reid KB, Clark H, Singh M, Nayak A, Sarma PU, Hawgood S, Kishore U. Susceptibility of mice genetically deficient in SP-A or SP-D gene to invasive pulmonary aspergillosis. Mol Immunol. 2010;47(10):1923–30.
107 Saxena S, Madan T, Shah A, Muralidhar K, Sarma PU. Association of polymorphisms in the collagen region of SP-A2 with increased levels of total IgE antibodies and eosinophilia in patients with allergic bronchopulmonary aspergillosis. J Allergy Clin Immunol. 2003;111(5):1001–7.
108 Wang G, Guo X, Diangelo S, Thomas NJ, Floros J. Humanized SFTPA1 and SFTPA2 transgenic mice reveal functional divergence of SP-A1 and SP-A2: formation of tubular myelin in vivo requires both gene products. J Biol Chem. 2010;285(16):11998–2010.
109 Wang G, Myers C, Mikerov A, Floros J. Effect of cysteine 85 on biochemical properties and biological function of human surfactant protein A variants. Biochemistry. 2007;46(28):8425–35.
110 Wang G, Bates-Kenney SR, Tao JQ, Phelps DS, Floros J. Differences in biochemical properties and in biological function between human SP-A1 and SP-A2 variants, and the impact of ozone-induced oxidation. Biochemistry. 2004;43(14):4227–39.
111 Huang W, Wang G, Phelps DS, Al-Mondhiry H, Floros J. Human SP-A genetic variants and bleomycin-induced cytokine production by THP-1 cells: effect of ozone-induced SP-A oxidation. Am J Physiol Lung Cell Mol Physiol. 2004;286(3):L546–53.
112 Tagaram HR, Wang G, Umstead TM, Mikerov AN, Thomas NJ, Graff GR, et al. Characterization of a human surfactant protein A1 (SP-A1) gene-specific antibody; SP-A1 content variation among individuals of varying age and pulmonary health. Am J Physiol Lung Cell Mol Physiol. 2007;292(5):L1052–63.
113 Wang Y, Voelker DR, Lugogo NL, Wang G, Floros J, Ingram JL, et al. Surfactant protein A is defective in abrogating inflammation in asthma. Am J Physiol Lung Cell Mol Physiol. 2011;301(4):L598–606.
114 Phelps D, Umstead T, Silveyra P, Hu S, Wang G, Floros J. Differences in the alveolar macrophage proteome in transgenic mice expressing human SP-A1 and SP-A2. Journal of Proteomics and Genomics Research; 2013;1:2–26.
115 Phelps DS, Umstead TM, Quintero OA, Yengo CM, Floros J. In vivo rescue of alveolar macrophages from SP-A knockout mice with exogenous SP-A nearly restores a wild type intracellular proteome; actin involvement. Proteome Sci. 2011;9:67.
116 Wright AL. Epidemiology of asthma and recurrent wheeze in childhood. Clin Rev Allergy Immunol. 2002;22(1):33–44.
117 Hanson LA, Adlerberth I, Carlsson B, Zaman S, Hahn-Zoric M, Jalil F. Antibody-mediated immunity in the neonate. Padiatr Padol. 1990;25(5):371–6.
118 Cabrera-Rubio R, Collado MC, Laitinen K, Salminen S, Isolauri E, Mira A. The human milk microbiome changes over lactation and is shaped by maternal weight and mode of delivery. Am J Clin Nutr. 2012;96(3):544–51.
119 Schulman ES. Development of a monoclonal anti-immunoglobulin E antibody (omalizumab) for the treatment of allergic respiratory disorders. Am J Respir Crit Care Med. 2001;164(8 Pt 2):S6–11.
120 Schwarzer M, Repa A, Daniel C, Schabussova I, Hrncir T, Pot B, et al. Neonatal colonization of mice with Lactobacillus plantarum producing the aeroallergen Bet v 1 biases towards Th1 and T-regulatory responses upon systemic sensitization. Allergy. 2011;66(3):368–75.