Adopted orphans as regulators of inflammation, immunity and skeletal homeostasis
DOI: https://doi.org/10.4414/smw.2014.14055
Natacha
Ipseiz, Carina
Scholtysek, Stephan
Culemann, Gerhard
Krönke
Summary
Adopted orphan nuclear receptors, such as peroxisome proliferator-activated receptors (PPARs) and liver X receptors (LXRs), have emerged as key regulators of inflammation and immunity and likewise control skeletal homeostasis. These properties render them attractive targets for the therapy of various inflammatory and autoimmune diseases affecting the musculoskeletal system. This review summarises the current knowledge on the role of these families of receptors during innate and adaptive immunity as well as during the control of bone turnover and discuss the potential use of targeting these molecules during the treatment of chronic diseases such as osteoarthritis, rheumatoid arthritis and osteoporosis.
Introduction
The musculoskeletal system is a prevalent target of chronic inflammatory diseases such as rheumatoid arthritis (RA) and osteoarthritis. Typically, such disorders exhibit a non-resolving inflammatory response that does not only lead to the local destruction of bone and cartilage, but likewise results in the impairment of systemic homeostatic processes such as fat, glucose and bone metabolism. Current approaches for the treatment of chronic inflammatory diseases are still largely based on pharmacologic compounds that primarily interfere with pro-inflammatory signalling cascades and hence suppress the inflammatory response. The most widely used anti-inflammatory substances are still glucocorticoids, which have revolutionised the therapy of inflammatory diseases since its discovery in the 1940’s. The initial enthusiasm of clinicians using these compounds was soon compromised, when severe side effects of this therapeutic approach became evident. We now know that glucocorticoids exert their biological effects via the glucocorticoid receptor (GR), a member of the nuclear receptor (NR) super-family that shows a widespread expression throughout the body. In accordance with the ubiquitous expression profile of their receptor, glucocorticoids are not only involved in the modulation of inflammation, but likewise regulate diverse processes such as glucose metabolism and bone homeostasis. This fact, in turn, explains many side effects observed after glucocorticoid therapy, which range from iatrogenic diabetes mellitus to osteoporosis [1].
These side effects constrain the long-term use of glucocorticoids during chronic inflammatory diseases. However, scientific progress during the past decades has resulted in the identification and characterisation of a large panel of additional NRs, some of which share the anti-inflammatory potential of the GR, but exert differential effects on fat, glucose, cartilage and bone homeostasis. Notably, the exact effects individual NRs exert on inflammation vary considerably between the different receptors, which is linked to their individual properties and the panel of genes regulated. Many of these NRs were initially referred to as “orphan receptors” since their specific endogenous ligands and their functions were often unknown. Meanwhile, the identification of various ligands of such orphan receptors resulted in the emergence of “adopted” orphan receptors, which include NRs such as peroxisome proliferator-activated receptors (PPARs), liver X receptors (LXRs) and retinoic X receptors (RXRs) that bind fatty acids, oxysterols and retinoids, respectively [2].
Many of these NRs do not only exert potent anti-inflammatory effects, but likewise control homeostatic processes within the musculoskeletal system. However, their effects on bone, cartilage and muscle metabolism often oppose the effects exerted by glucocorticoids. Therefore, they have emerged as highly attractive targets for a therapeutic intervention during a variety of chronic inflammatory diseases. This review addresses the role of the PPAR, LXR and NR4a families of (adopted) orphan NRs during the regulation of inflammation and immunity and additionally highlights their impact on the musculoskeletal system in terms of bone, joint and cartilage metabolism.
Orphan NRs as regulators of inflammation and immunity
Most NRs act as ligand-dependent transcription factors that share a conserved structure consisting of a carboxy-terminal ligand-binding, a central DNA-binding and as well as an amino-terminal trans-activation domain. By recruitment of co-activator and co-repressor complexes, NRs can both positively and negatively affect gene expression [3]. Mechanisms underlying the NR-mediated transcriptional regulation are complex and often differ between individual NR subgroups. In general, NRs can form monomers, homodimers or heterodimers, and bind to hormone responsive elements at the promoters of their target genes. Upon binding of their ligand, NRs attach to specific responsive elements and subsequently promote transcription of target genes [4, 5]. Thus NRs induce expression of a large set of genes. As this includes certain anti-inflammatory genes such as heme-oxygenase-1 and can fundamentally change the phenotype of the respective cells, these positive transcriptional effects can indirectly contribute to the reported anti-inflammatory effects of NRs [6, 7]. Some NRs such as the members of the PPAR family are also able to constitutively bind to a distinct subset of promoter elements, where they attract co-repressor complexes and block transcription in the absence of a ligand [3, 8]. Upon ligand binding, these co-repressors are exchanged by co-activators and transcription is initiated. Furthermore, many NRs seem to be capable of actively blocking the transcription of individual genes in a ligand-dependent manner. The underlying molecular events are still incompletely understood and a matter of debate. A current working model suggests that during this process (referred to as “trans-repression”), the NR does not directly bind, but rather tethers to the repressed promoter, where it triggers recruitment or interferes with the dissociation of co-repressors such as NCoR or SMRT [9, 10]. A typical example is the trans-repression of distinct inflammatory genes by PPARγ and LXRs [11–13]. Many NRs do not only recruit co-activator and co-repressor complexes, but also directly interact with a variety of other proteins such as members of the NF-κB complex, different protein kinases and β-catenin [14–16]. Interactions can be direct or indirect sometimes using so-called co-regulators or even interfering with RNA elongation [17, 18]. Presumably larger complexes are involved. Thereby, NRs positively and negatively affect intracellular signalling and transcription at various levels. The effects distinct NR sub-families exert during inflammation and immunity are complex. Figure 1 summarises key findings on the role of the NR4a, LXR and PPAR families of NR during the innate and adaptive response.
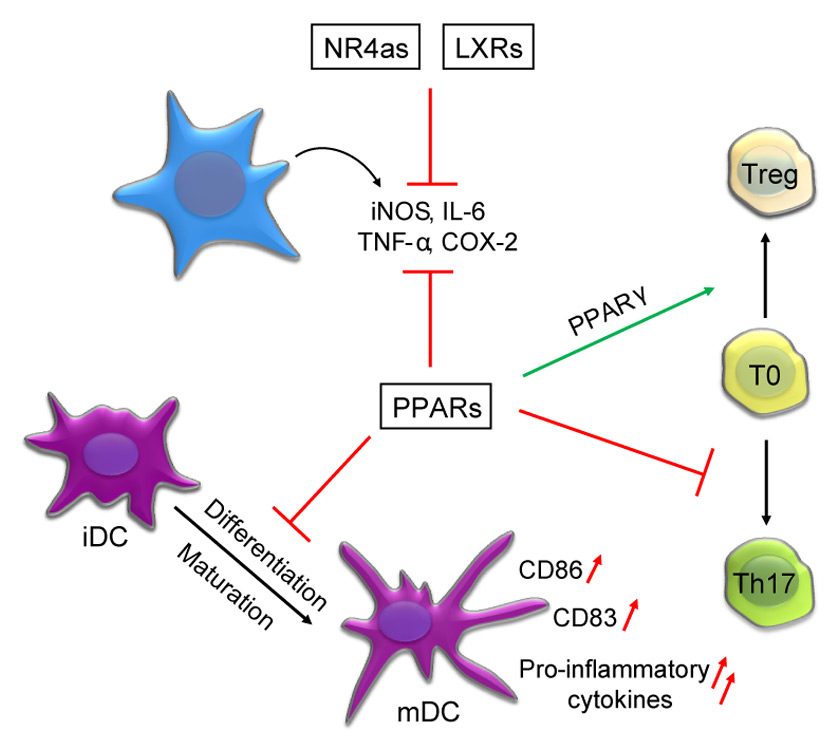
Figure 1
Summary of the key findings on the role of the PPAR, LXR and NR4a family of NRs during inflammation and immune homeostasis. A common feature of these families of NRs are their anti-inflammatory effects that result in the inhibition of the expression of multiple pro-inflammatory cytokines such as inducible NO-Synthase (iNOS), TNF-α, Interleukin-6 (IL-6) and cyclooxygenase-2 (COX-2). PPARs were additionally described to block the maturation of dendritic cells (DCs), which includes an attenuated expression of DC maturation markers such as CD86 and CD83 after activation of this NR sub-family. Also differentiation of T cells into distinct subsets was shown to be influenced by NRs, where PPARs interfere with the differentiation of Th17 cells and simultaneously promote the differentiation of regulatory T cells (Treg).
PPARs
Among the different adopted orphan NRs, members of the PPAR sub-family of NRs were shown to exert various potent anti-inflammatory effects rendering them promising candidates for the therapy of chronic inflammatory diseases. This NR sub-family includes three members (PPARα, PPARβ/δ and PPARγ) that bind regulatory DNA elements as heterodimers together with the retinoic X receptor. Although all PPAR members act via conserved responsive elements, they differ in their ligand-specificity and are differentially expressed throughout the body. Endogenous ligands include various fatty acids as well as locally produced eicosanoids. Notably, different commonly used drugs exert their effects via PPARs. Prominent examples include the insulin-sensitising class of thiazolidinediones that act via PPARγ and lipid-lowering fibrates that act via PPARα. PPARs have been initially described as key regulators of fat and glucose metabolism, where the individual members of this NR family promote distinct metabolic programmes such as fatty acid oxidation (PPARα) and adipogenesis (PPARγ), respectively [19]. Furthermore, all three members were shown to exert potent regulatory functions during the innate and adaptive immune response [20].
The immune-modulatory role of PPARγ has been most intensively studied in monocytes and macrophages. Various ligands for PPARγ were shown to block the inflammatory response in these cell types, where PPARγ is the predominant PPAR isoform [21–23]. Although some of the anti-inflammatory effects observed after the use of high doses of PPARγ ligands were subsequently shown to be PPARγ-independent [24], the role of PPARγ as key regulator of macrophage activation has meanwhile been settled [14]. A set of carefully executed experiments using PPAR-deficient cells as controls, revealed that ligand-induced activation of PPARγ does not generally block inflammatory signalling, but represses a specific subset of toll-like receptor (TLR)-induced genes in macrophages [25]. Interestingly, most of the affected pro-inflammatory genes do not contain PPAR responsive elements in their promoters. Instead, PPARγ blocks their expression by trans-repressing NF-κB- and AP1–dependent transcriptional activation. Ligand binding was shown to induce allosteric changes of PPARγ that enable its SUMOylation by SUMO1. Subsequently, SUMOylated PPARγ interacts with the NCoR co-repressor in complex with Histone deacetylase 3. NCoR, in turn, is known to constitutively silence a distinct subset of inflammatory genes in the steady state and usually dissociates after arrival of an inflammatory signal. SUMOylated PPARγ, however, prevents the release of NCoR from these promoter regions thereby shutting down transcription of the respective genes [11]. This mechanism seems to mediate the ligand-induced repression of a number of inflammatory genes such as inducible NO-Synthase (iNOS) and TNF-α in macrophages [11, 13].
PPARα and PPARβ/δ are also able to attenuate pro-inflammatory gene expression. However underlying molecular mechanisms are less well investigated than the trans-repressive capacity of PPARγ [26]. PPARα is highly expressed in vascular smooth muscle and endothelial cells as well as in hepatocytes [27]. Here, this NR suppresses the expression of pro-inflammatory genes such as interleukin 6 (IL-6) and cyclooxygenase-2 (COX-2) by interference with p65 and c-Jun as well as by the transcriptional induction of the NF-κB Inhibitor IκB [28–31]. PPARβ/δ is the most widely expressed PPAR family member. Accordingly, this member of the PPAR family has been implicated in the regulation of inflammatory signalling in diverse cell types such as endothelial cells, smooth muscle cells and keratinocytes, where it negatively regulates the expression of a large panel of chemokines and cytokines [32, 33]. On a molecular level, PPARβ/δ seems to control the activation status of macrophages at least partially via sequestration of the transcriptional repressor BCL-6, which is released after the binding of a ligand to PPARβ/δ [34].
These potent anti-inflammatory effects of the PPAR family of NRs explain the fact that genetic deletion of PPARγ was shown to result in the exacerbation of chronic inflammatory diseases, where conditional deletion of PPAR family members in individual cell types revealed anti-inflammatory roles not only in monocytes and macrophages, but also in parenchymal cell types such as intestinal epithelial cells [35, 36]. Deletion of PPARα or PPARβ/δ also results in the exacerbation of the inflammatory response in murine models of contact dermatitis [37, 38], airway inflammation [39] or steatohepatitis [40]. Accordingly, treatment with PPARγ as well as with PPARα ligands attenuated disease severity in such models of inflammation. Amongst others, activation of these NRs were shown to exert protective effects during animal models of colitis [35, 36, 41–43] and allergic airway disease [44, 45].
Likewise, PPARs were shown to critically impact on the progression of atherosclerosis as one of the most frequent chronic inflammatory diseases and leading cause of death in modern societies. Within the vascular wall, PPARα and PPARβ/δ are mainly expressed in vascular smooth muscle cells and endothelial cells, whereas PPARγ is the predominant PPAR isoform in monocyte-derived macrophages and foam cells within the atherosclerotic plaque. Within cells of the vascular wall, ligand-induced activation of all three PPAR members was shown to exert anti-inflammatory effects thereby reducing the expression of adhesion molecules, chemokines and pro-inflammatory cytokines [28, 46–48]. In vivo studies determining the effects of PPAR ligands showed clear anti-atherogenic effect of PPARα and PPARγ ligands, which reduced the size of atherosclerotic lesions and foam cell formation within the plaque. In contrast, ligands for PPARβ/δ were not effective [49]. Studies using PPAR-deficient mice confirmed a protective role of PPARγ in murine models of atherosclerosis [49, 50], whereas studies using PPARα and PPARβ/δ- deficient mice revealed conflicting data on the exact role of these NR during atherogenesis [34, 51, 52]. In addition to the direct anti-inflammatory properties of PPAR agonists, the induction of anti-inflammatory and vascular-protective genes, such as heme oxygenase-1 [6], as well as changes in metabolic parameters [52] seems to contribute to their beneficial effects within the vascular wall.
In addition to their anti-inflammatory actions, PPARs were shown to directly and indirectly influence the adaptive immune response and to contribute to the maintenance of immunological tolerance. Notably, uptake of apoptotic cells by macrophages was shown to exert immune-modulatory effects via activation of PPARγ and PPARβ/δ within the phagocyte ensuring the non-inflammatory clearance of apoptotic cell-derived auto-antigens [53, 54]. On a molecular level, apoptotic cells seem to induce sumoylation of PPARγ and thereby prevent the dissociation of the NCoR co-repressor complex from promoters of inflammatory genes [53]. PPAR β/δ, in turn, promotes the expression of distinct opsonins such as complement component 1qb that facilitate the uptake of the apoptotic cell [54]. Furthermore, PPARs were shown to modulate the differentiation, maturation and function of professional antigen-presenting cells such as dendritic cells (DCs). Activation of all three PPAR subtypes results in an altered DC phenotype with an attenuated expression of co-stimulatory molecules and a reduced expression of pro-inflammatory cytokines as well as an impaired migratory capacity of the DC [55–63]. Accordingly, activation of PPARγ and PPARα were shown to reduce the antigen-presenting capacity of DCs [59, 62, 64]. Mechanistically, the effects PPARs exert on DCs seem to involve not only trans-repression mechanisms and interactions with other transcription factors such as STAT6 [58], but also a PPAR-induced shift in the metabolic programming of these cells [7, 65–67]. Apart from their regulatory role in antigen-presenting cells, PPARs also directly influence cells of the adaptive immune system. An example is the PPARγ- and PPARα-mediated blockade of the activation of T and B cells, where these NRs also exert pro-apoptotic effects in certain lymphocyte subsets [68–76]. PPARs also orchestrate the differentiation of naïve T cells into distinct T effector cell subsets such as Th1, Th17 or regulatory T cells. An example is the block of Th17 differentiation by PPARα, PPARβ/δ and PPARγ [77–79]. Th17 cells and regulatory T cells represent distinct T cell types that fulfil seemingly opposite tasks. Whereas Th17 cells have been both implicated in the defence against extracellular pathogens and the pathogenesis of distinct autoimmune diseases such as rheumatoid arthritis and multiple sclerosis [80], regulatory T cells provide protection against such autoimmune disorders [81]. Accordingly, T-cell-specific deletion of PPARγ results in an increased differentiation of Th17 cells as well as in an exacerbation of models for Th17–mediated autoimmune disease including experimental autoimmune encephalomyelitis [79], a murine model for multiple sclerosis. Mice that received PPARγ or PPARβ/δ ligands, in turn, showed an attenuated disease course [78, 79]. In addition to its negative effects on Th1 and Th17 differentiation, PPARγ seems to promote the accumulation of immune-modulatory regulatory T cells [82, 83].
Its multiple anti-inflammatory and immune-modulatory effects explain the reported beneficial effects that PPARs exert during chronic inflammatory diseases of the musculoskeletal system. In animal models of autoimmune arthritis, genetic deletion of PPARγ causes an exacerbation, whereas PPARγ ligand-treatment ameliorates disease severity [68, 84–86]. Furthermore, ligand-induced activation of PPARγ and PPARα was shown to beneficially influence inflammatory disease activity in patients suffering from rheumatoid arthritis [87–89] and psoriatic arthritis [90], respectively. Treatment with PPARγ and PPARα agonists, likewise, protects from degenerative joint disease in animal models of osteoarthritis, where activation of these NRs blocks expression of inflammatory cytokines and matrix metalloproteinases and thereby ameliorates proteoglycan degradation [91–95]. Cartilage-specific deletion of PPARγ, in turn, results in a spontaneous osteoarthritis phenotype [96, 97].
LXRs
In analogy to PPARs, the Liver X receptors (LXRα and LXRβ) are a second group of adopted orphan NRs that exert potent anti-inflammatory effects [98]. Like PPARs, they form heterodimers with RXR, whereas their natural ligands have been identified as oxysterols. While LXRs have been initially implicated in the regulation of cholesterol metabolism, these NRs have meanwhile emerged as crucial regulators of both innate and adaptive immunity. In response to the binding of a ligand, LXRs also become SUMOylated and prevent the dissociation of co-repressors from the promoter regions of inflammatory genes that partially overlap with genes that are suppressed by activation of the GR or PPARγ [13, 25, 98]. Thereby LXRs attenuate the expression of a distinct set of TLR-induced genes such as iNOS, COX-2 or IL-6 in macrophages [99]. Interestingly, there seems to be a reciprocal inhibition between LXR- and TLR-induced signalling, as pathogen-induced TLR activation blocks the LXR-mediated efflux of cholesterol in macrophages [100]. LXRαβ-/-mice show an impaired macrophage response to pathogens as well as an increased susceptibility to infection indicating that expression of LXRs is crucial for a proper function of macrophages [101, 102]. The uptake of apoptotic cells also results in the activation of LXRs ingesting macrophages, where these NRs contribute to the non-inflammatory removal of apoptotic cell-derived antigens and the maintenance of self-tolerance [103]. Furthermore, LXRs were shown to control the differentiation of distinct macrophage subsets such as marginal zone macrophages in the spleen and to contribute to neutrophil homeostasis in the steady state [104, 105]. Recent evidence also points towards a key role of LXR-mediated regulation of cholesterol homeostasis during the regulation of T cell proliferation implying a direct function of this NR during the adaptive immune response [106]. In accordance with an anti-inflammatory role of LXRs, synthetic LXR ligands were reported to exert anti-inflammatory effects in vivo and ameliorate the disease course in murine models of atherosclerosis and contact dermatitis [107–109]. The consequences of LXR activation during allergic airway disease and autoimmune arthritis are controversial, and both beneficial and detrimental effects of an LXR agonist treatment in murine asthma models and during collagen-induced arthritis have been described [110–114]. Reduced LXR signalling seems to contribute to catabolic metabolism in osteoarthritic cartilage [115], whereas LXR ligands block matrix degradation and alleviate pain during animal models of osteoarthritis [116].
NR4a1–3
Another group of NRs that were shown to crucially modulate the innate and adaptive immune response is the NR4a family of NRs [117, 118]. The three members (NR4a1–3 or alternatively termed as Nur77, Nurr1 and NOR-1) are differentially expressed throughout the body. Notably, they lack a classical ligand-binding domain and endogenous ligands for this NR subgroup have not been described so far. These findings render them “true orphans”, although recently a compound that was isolated from an endophytic fungus was identified to be able to act as an agonist for Nur77 [119]. Stimulation of cells with various pro-inflammatory, activation signals such as LPS or oxidised LDL results in the rapid transcriptional induction of NR4a expression within 1 hour [117]. By recruiting the CoREST co-repressor complex and blocking p65–mediated transcriptional activation of NF-κB target genes, Nurr1 was shown to prevent an overwhelming inflammatory response by microglia and astrocytes thereby providing protection from an inflammation-associated loss of dopaminergic neurons in the CNS [120]. Its family member, Nur77, was shown to interfere with the LPS-induced activation of macrophages by blocking the phosphorylation of the NF-κB subunit p65 [121]. In mouse models of atherosclerosis, deletion of Nur77 resulted in an increased expression of inflammatory genes in macrophages and an accelerated development of atherosclerotic plaques [121, 122]. Recent data from our lab suggests that the expression of Nur77 is rapidly induced in macrophages that recognise apoptotic cells. Subsequently this NR interferes with NF-κB-signalling and dampens pro-inflammatory signalling pathways within the phagocyte ensuring the non-inflammatory clearance of dying cells [123]. These findings suggest that the NR4a subgroup of NRs is not only involved in regulatory feedback loops, but also integrates pro- and anti-inflammatory signalling pathways to fine-tune the immune response. Furthermore, Nur77 was shown to act as an essential factor for the differentiation of Ly6Clowresident monocytes, a distinct subset of blood monocytes that, in contrast to Ly6Chigh monocytes, migrate into the tissue during the steady state and seem to fulfil homeostatic functions. Absence of Nur77 resulted in significantly reduced numbers of these “patrolling” monocytes in the bone marrow and blood of NR4a1–/– mice [124]. Notably, the three NR4a family members also control the development of thymic regulatory T cells and mice lacking all three NR4a members do not only lack this T cell subset, but also suffer from a fatal systemic autoimmune disorder [125]. As mice that are deficient for only one of the three NR4a members do not exhibit an obvious T cell phenotype, these findings point towards a certain redundancy in the function of members this NR sub-family. Little is known about a role of this NR sub-family during inflammatory joint disease. However, constitutive expression of Nur77 in T cells results in the amelioration of collagen-induced arthritis [126] and its family member Nurr1 was shown to repress the expression of matrix metalloproteinases in osteoarthritic cartilage [127].
Orphan nuclear receptors as regulators of skeletal homeostasis
Given the potential of NRs to serve as targets for the treatment of chronic inflammatory musculoskeletal diseases such as rheumatoid arthritis and osteoarthritis, it is important to consider their impact on skeletal homeostasis, where many of them control multiple aspects of bone biology including the differentiation and function of bone-forming osteoblasts and of bone-resorbing osteoclasts. Furthermore, NRs are involved in the coordination of systemic calcium and phosphate metabolism as well as in the differentiation of mesenchymal stem cells (MSCs). The role of classical steroid receptors such as oestrogen receptors, the glucocorticoid receptor as well as the vitamin D receptor during bone homeostasis is well studied and reviewed in detail elsewhere [128]. The beneficial effects of vitamin D on bone are exploited during the treatment of skeletal diseases such as osteoporosis, where the administration of vitamin D is a first line therapy [129]. The catabolic effects glucocorticoids exert on bone homeostasis, on the other hand, represent a major problem during the glucocorticoid-based treatment of chronic inflammatory diseases [130]. Recent data widened our understanding of the pivotal role adopted orphan receptors such as PPARs and LXRs play in bone biology. While the anti-inflammatory and immune modulatory effects of PPARs, LXRs and members of the NR4a family often overlap, these NRs differentially affect bone turnover. The various reported effects of PPARs and LXRs on bone homeostasis are summarised in figure 2.
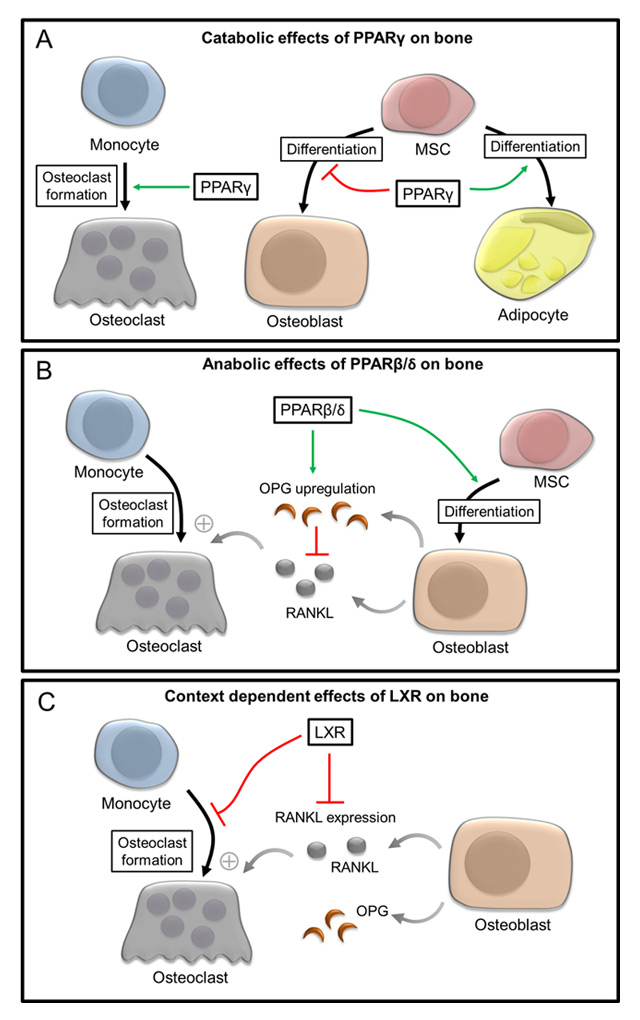
Figure 2
Role of PPARγ, PPARβ/δ and LXRs during the control of osteoblast and osteoclast differentiation. (A) Catabolic effects of PPARγ on bone homeostasis are attributed to the PPARγ-mediated promotion of osteoclast differentiation and its inhibitory role during the differentiation of mesenchymal stem cells (MSC) into osteoblasts. (B) PPARβ/δ exert its anabolic effects on bone homeostasis via promotion of the osteoblast differentiation and regulation of the RANKL/OPG ratio thereby indirectly influencing osteoclastogenesis. (C) During states of increased osteoclastogenesis and high bone turnover such as inflammation and osteoporosis, activation of LXRs is able to both directly and indirectly interfere with osteoclast differentiation via blockade of osteoclastogenesis and influencing the RANKL/OPG ratio.
PPARs
First clues for a key role of PPARγ during skeletal homeostasis came from the observation that patients suffering from type 2 diabetes, who received PPARγ-activating thiazolidinediones as insulin sensitisers, showed an increased risk of osteoporosis and osteoporosis-related fractures [131, 132]. This severe side effect of a therapy with these PPARγ-activating compounds is attributed to the fact that PPARγ serves as master regulator of adipogenesis and promotes the differentiation of mesenchymal stem cells (MSCs) into adipocytes resulting in a simultaneous PPARγ-mediated block of the differentiation of MSCs into chondrocytes, muscle cells and osteoblasts [133, 134] thereby explaining the detrimental effects this NR exerts on bone formation. Support for such a scenario comes from studies on mice carrying a haplo-insufficiency of PPARγ and that show an increased bone mass. Moreover, PPARγ+/- MSCs have an enhanced potential to differentiate into osteoblasts [135]. Treatment of mice with PPARγ agonists, on the other hand, resulted in a decreased bone mass, which was paralleled by an increased number of adipocytes in the bone marrow [136, 137]. On a molecular level, this PPARγ-mediated block of osteoblast differentiation seems to be based on a reciprocal inhibition between PPARγ and the Wnt-signalling pathway in MSCs and osteoblast precursors. Wnt signalling, in turn, acts as a key driver of osteoblast differentiation and bone formation [15, 138–141]. Whereas agonists of the Wnt pathway induce the expression of key transcription factors of osteoblastogenesis such as Runx2 and osterix, their expression is blocked by PPARγ [136]. Recent data suggest an additional role of PPARγ during the differentiation of bone-resorbing osteoclasts, which might contribute to the catabolic effects this NR exerts on bone homeostasis. Conditional deletion of PPARγ in osteoclast precursors resulted in increased bone mass and a reduced number of osteoclasts, whereas the PPARγ agonist rosiglitazone fostered the differentiation of osteoclasts, indicating an important pro-osteoclastogenic role of this NR [142].
In contrast to PPARγ, its family member PPARβ/δ exerts potent anabolic effects on bone turnover and homeostasis. Ligand-induced activation of this NR fosters the development of mature osteoblasts [16, 143, 144]. Mechanistically, activation of PPARβ/δ promotes the transcription of the Wnt co-receptor LRP5 and additionally interacts with β-catenin thereby amplifying Wnt signalling activity in osteoblast precursors [16]. Likewise, PPARβ/δ regulates the ratio between the pro-osteoclastogenic cytokine RANKL and its natural decoy receptor OPG. Both proteins are produced by osteoblasts and osteocytes and ligand-induced activation of PPARβ/δ results in a decreased RANKL/OPG ratio and an attenuation of osteoblast-mediated osteoclastogenesis [16]. Accordingly, PPARβ/δ-deficient mice exhibit a decreased bone mass, an increased RANKL/OPG ratio and a consecutive increase in the differentiation of osteoclasts. Pharmacological activation of PPARβ/δ, in turn, results in the protection from ovariectomy-induced bone loss highlighting the potential of PPARβ/δ as a novel target for the treatment of osteoporosis and related diseases in humans [16]. Importantly, the effects PPARβ/δ exerted on osteoclastogenesis were indirect and related to the regulation of the RANKL/OPG ratio provided by osteoblasts. Neither deletion of PPARβ/δ, nor treatment with specific concentrations of PPARβ/δ ligands altered the intrinsic potential of osteoclast precursors to form mature osteoclasts, although previous studies using high (and probably non-specific) concentrations of different PPAR agonists observed such inhibitory effects of various PPAR ligands on osteoclastogenesis [145]. Notably, treatment of ovariectomised rats with a PPARβ/δ-specific ligand resulted in a decreased bone density [146]. This, however, might be linked to the previously reported opposing consequences of a PPARβ/δ-activation on the metabolism of mice and humans on the one hand and of rats on the other hand [147].
In vitro data shows that the ligand-induced activation of PPARα mice also stimulates osteoblast differentiation [143]. Likewise, treatment of mice with the PPARα agonist bezafibrate resulted in increased periosteal bone formation [143]. However, PPARα-deficient mice do not exhibit any alterations in their bone structure [148] and patients receiving lipid-lowering fibrates did not show a reduced risk of bone fractures [149].
LXRs
The role of LXRs during bone metabolism is less clear. Activation of LXRs was shown to directly interfere with osteoclastogenesis and to additionally attenuate the RANKL/OPG ratio provided by osteoblasts [150, 151]. However, LXR-deficient mice show only mild alteration in their bone structure suggesting a rather minor role of LXRs during the regulation of physiological bone turnover [152]. Likewise, long-term treatment with LXR agonists did not significantly alter the structure and density of bones of non-challenged mice [153]. In contrast, treatment with LXR ligands efficiently blocked the increased osteoclastogenesis after ovariectomy or during inflammation, thereby protecting mice both from ovariectomy-induced osteoporosis and local inflammatory bone loss [151, 154]. These context-dependent effects LXR ligands exert on bone turnover render them highly attractive tools for future therapeutic approaches in the treatment of related human diseases.
NR4a1–3
Members of the NR4a family are also expressed in osteoblasts [155, 156], where they regulate genes involved in bone formation such as osteopontin, osteocalcin, alkaline phosphatase and collagen type I alpha I [155–157]. Interestingly, expression of NR4a receptors is rapidly induced following stimulation of osteoblasts or MSCs with parathyroid hormone. However the exact function of the NR4a sub-family of NRs during bone metabolism remains to be elucidated.
Implications and future therapeutic strategies
The identification of the PPAR, LXR and NR4a families of NRs as potent modulators of inflammation and immunity has rendered them attractive targets for the treatment of inflammatory diseases affecting the musculoskeletal system such as rheumatoid arthritis and osteoarthritis. Their role as master regulators of fat and glucose homeostasis as well as during skeletal development might be a simultaneous advantage and an obstacle for their clinical use. On the one hand, these pleiotropic functions increase the risk of side effects like osteoporosis as observed during a therapy with PPARγ-activating thiazolidinediones or glucocorticoids. On the other hand, agonists for PPARα or PPARβ/δ might evolve as anti-inflammatory compounds that allow the simultaneous treatment of alterations of fat, glucose and bone metabolism such as insulin resistance and osteoporosis as disorders that accompanied chronic inflammatory diseases. Another strategy would be the combination of different NR agonists that display a partial agonistic and antagonistic action profile such as ligands for PPARγ and PPARβ/δ in order to increase the anti-inflammatory potential of this treatment and minimise side effects (e.g. on bone). However, the prerequisite for a successful treatment strategy that targets NRs is a profound knowledge of their pleiotropic function and here we are just beginning to understand the complexity of the NR network that silently orchestrates our daily life in health and disease.
References
1 Baschant U, Lane NE, Tuckermann J. The multiple facets of glucocorticoid action in rheumatoid arthritis. Nat Rev Rheumatol. 2012;8:645–55.
2 Evans RM, Mangelsdorf DJ. Nuclear Receptors, RXR, and the Big Bang. Cell. 2014;157:255–66.
3 Sever R, Glass CK. Signaling by nuclear receptors. Cold Spring Harb Perspect Biol. 2013;5:a016709.
4 Kadmiel M, Cidlowski JA. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol Sci. 2013;34:518–30.
5 Baschant U, Culemann S, Tuckermann J. Molecular determinants of glucocorticoid actions in inflammatory joint diseases. Mol Cell Endocrinol. 2013;380:108–18.
6 Kronke G, Kadl A, Ikonomu E, Bluml S, Furnkranz A, Sarembock IJ, et al. Expression of heme oxygenase-1 in human vascular cells is regulated by peroxisome proliferator-activated receptors. Arterioscler Thromb Vasc Biol. 2007;27:1276–82.
7 Kiss M, Czimmerer Z, Nagy L. The role of lipid-activated nuclear receptors in shaping macrophage and dendritic cell function: From physiology to pathology. J Allergy Clin Immunol. 2013;132:264–86.
8 Adhikary T, Kaddatz K, Finkernagel F, Schonbauer A, Meissner W, Scharfe M, et al. Genomewide analyses define different modes of transcriptional regulation by peroxisome proliferator-activated receptor-beta/delta (PPARbeta/delta). PLoS One 2011;6:e16344.
9 Glass CK, Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat Rev Immunol. 2010;10:365–76.
10 Perissi V, Jepsen K, Glass CK, Rosenfeld MG. Deconstructing repression: evolving models of co-repressor action. Nat Rev Genet. 2010;11:109–23.
11 Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, et al. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437:759–63.
12 Ghisletti S, Huang W, Jepsen K, Benner C, Hardiman G, Rosenfeld MG, et al. Cooperative NCoR/SMRT interactions establish a corepressor-based strategy for integration of inflammatory and anti-inflammatory signaling pathways. Genes Dev. 2009;23:681–93.
13 Ghisletti S, Huang W, Ogawa S, Pascual G, Lin ME, Willson TM, et al. Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARgamma. Mol Cell. 2007;25:57–70.
14 Straus DS, Glass CK. Anti-inflammatory actions of PPAR ligands: new insights on cellular and molecular mechanisms. Trends Immunol. 2007;28:551–8.
15 Mulholland DJ, Dedhar S, Coetzee GA, Nelson CC. Interaction of nuclear receptors with the Wnt/beta-catenin/Tcf signaling axis: Wnt you like to know? Endocr Rev. 2005;26:898–915.
16 Scholtysek C, Katzenbeisser J, Fu H, Uderhardt S, Ipseiz N, Stoll C, et al. PPARbeta/delta governs Wnt signaling and bone turnover. Nature medicine. 2013;19:608–13.
17 Flammer JR, Rogatsky I. Minireview: Glucocorticoids in autoimmunity: unexpected targets and mechanisms. Mol Endocrinol. 2011;25:1075–86.
18 Taubert S, Ward JD, Yamamoto KR. Nuclear hormone receptors in nematodes: evolution and function. Mol Cell Endocrinol. 2011;334:49–55.
19 Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nat Med. 2004;10:355–61.
20 Glass CK, Ogawa S. Combinatorial roles of nuclear receptors in inflammation and immunity. Nat Rev Immunol. 2006;6:44–55.
21 Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–6.
22 Straus DS, Pascual G, Li M, Welch JS, Ricote M, Hsiang CH, et al. 15–deoxy-delta 12,14–prostaglandin J2 inhibits multiple steps in the NF-kappa B signaling pathway. Proc Natl Acad Sci U S A. 2000;97:4844–9.
23 Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82.
24 Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RMPPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med. 2001;7:48–52.
25 Ogawa S, Lozach J, Benner C, Pascual G, Tangirala RK, Westin S, et al. Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell. 2005;122:707–21.
26 Ricote M, Glass CK. PPARs and molecular mechanisms of transrepression. Biochim Biophys Acta. 2007;1771:926–35.
27 Pyper SR, Viswakarma N, Yu S, Reddy JK. PPARalpha: energy combustion, hypolipidemia, inflammation and cancer. Nuclear receptor signaling. 2010;8:e002.
28 Staels B, Koenig W, Habib A, Merval R, Lebret M, Torra IP, et al. Activation of human aortic smooth-muscle cells is inhibited by PPARalpha but not by PPARgamma activators. Nature. 1998;393:790–3.
29 Delerive P, De Bosscher K, Besnard S, Vanden Berghe W, Peters JM, Gonzalez FJ, et al. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J Biol Chem. 1999;274:32048–54.
30 Delerive P, Gervois P, Fruchart JC, Staels B. Induction of IkappaBalpha expression as a mechanism contributing to the anti-inflammatory activities of peroxisome proliferator-activated receptor-alpha activators. J Biol Chem. 2000;275:36703–7.
31 Xu X, Otsuki M, Saito H, Sumitani S, Yamamoto H, Asanuma N, et al. PPARalpha and GR differentially down-regulate the expression of nuclear factor-kappaB-responsive genes in vascular endothelial cells. Endocrinology. 2001;142:3332–9.
32 Barish GD, Atkins AR, Downes M, Olson P, Chong LW, Nelson M, et al. PPARdelta regulates multiple proinflammatory pathways to suppress atherosclerosis. Proc Natl Acad Sci U S A. 2008;105:4271–6.
33 Bishop-Bailey D, Bystrom J. Emerging roles of peroxisome proliferator-activated receptor-beta/delta in inflammation. Pharmacol Ther. 2009;124:141–50.
34 Lee CH, Chawla A, Urbiztondo N, Liao D, Boisvert WA, Evans RM, et al. Transcriptional repression of atherogenic inflammation: modulation by PPARdelta. Science. 2003;302:453–7.
35 Shah YM, Morimura K, Gonzalez FJ. Expression of peroxisome proliferator-activated receptor-gamma in macrophage suppresses experimentally induced colitis. Am J Physiol Gastrointest Liver Physiol. 2007;292:G657–666.
36 Adachi M, Kurotani R, Morimura K, Shah Y, Sanford M, Madison BB, et al. Peroxisome proliferator activated receptor gamma in colonic epithelial cells protects against experimental inflammatory bowel disease. Gut. 2006;55:1104–13.
37 Dubrac S, Elentner A, Schoonjans K, Auwerx J, Schmuth M. Lack of IL-2 in PPAR-alpha-deficient mice triggers allergic contact dermatitis by affecting regulatory T cells. Eur J Immunol. 2011;41:1980–91.
38 Man MQ, Barish GD, Schmuth M, Crumrine D, Barak Y, Chang S, et al. Deficiency of PPARbeta/delta in the epidermis results in defective cutaneous permeability barrier homeostasis and increased inflammation. J Invest Dermatol. 2008;128:370–7.
39 Delayre-Orthez C, Becker J, Guenon I, Lagente V, Auwerx J, Frossard N, et al. PPARalpha downregulates airway inflammation induced by lipopolysaccharide in the mouse. Respir Res. 2005;6:91.
40 Stienstra R, Mandard S, Patsouris D, Maass C, Kersten S, Muller M. Peroxisome proliferator-activated receptor alpha protects against obesity-induced hepatic inflammation. Endocrinology. 2007;148:2753–63.
41 Tanaka T, Kohno H, Yoshitani S, Takashima S, Okumura A, Murakami A, et al. Ligands for peroxisome proliferator-activated receptors alpha and gamma inhibit chemically induced colitis and formation of aberrant crypt foci in rats. Cancer Res. 2001;61:2424–8.
42 Esposito E, Mazzon E, Paterniti I, Dal Toso R, Pressi G, Caminiti R, Cuzzocrea S. PPAR-alpha Contributes to the Anti-Inflammatory Activity of Verbascoside in a Model of Inflammatory Bowel Disease in Mice. PPAR Res. 2010: 917312.
43 Su CG, Wen X, Bailey ST, Jiang W, Rangwala SM, Keilbaugh SA, et al. A novel therapy for colitis utilizing PPAR-gamma ligands to inhibit the epithelial inflammatory response. J Clin Invest. 1999;104:383–9.
44 Woerly G, Honda K, Loyens M, Papin JP, Auwerx J, Staels B, et al. Peroxisome proliferator-activated receptors alpha and gamma down-regulate allergic inflammation and eosinophil activation. J Exp Med. 2003;198:411–21.
45 Trifilieff A, Bench A, Hanley M, Bayley D, Campbell E, Whittaker P. PPAR-alpha and -gamma but not -delta agonists inhibit airway inflammation in a murine model of asthma: in vitro evidence for an NF-kappaB-independent effect. Br J Pharmacol. 2003;139:163–71.
46 Marx N, Sukhova GK, Collins T, Libby P, Plutzky J. PPARalpha activators inhibit cytokine-induced vascular cell adhesion molecule-1 expression in human endothelial cells. Circulation. 1999;99:3125–31.
47 Israelian-Konaraki Z, Reaven PD. Peroxisome proliferator-activated receptor-alpha and atherosclerosis: from basic mechanisms to clinical implications. Cardiology. 2005;103:1–9.
48 Li AC, Glass CK. PPAR- and LXR-dependent pathways controlling lipid metabolism and the development of atherosclerosis. J Lipid Res. 2004;45:2161–73.
49 Li AC, Binder CJ, Gutierrez A, Brown KK, Plotkin CR, Pattison JW, et al. Differential inhibition of macrophage foam-cell formation and atherosclerosis in mice by PPARalpha, beta/delta, and gamma. J Clin Invest. 2004;114:1564–76.
50 Chawla A, Boisvert WA, Lee CH, Laffitte BA, Barak Y, Joseph SB, et al. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Molecular Cell. 2001;7:161–71.
51 Tordjman K, Bernal-Mizrachi C, Zemany L, Weng S, Feng C, Zhang F, et al. PPARalpha deficiency reduces insulin resistance and atherosclerosis in apoE-null mice. J Clin Invest. 2001;107:1025–34.
52 Brown JD, Plutzky J. Peroxisome proliferator-activated receptors as transcriptional nodal points and therapeutic targets. Circulation. 2007;115:518–33.
53 Jennewein C, Kuhn AM, Schmidt MV, Meilladec-Jullig V, von Knethen A, Gonzalez FJ, et al. Sumoylation of peroxisome proliferator-activated receptor gamma by apoptotic cells prevents lipopolysaccharide-induced NCoR removal from kappaB binding sites mediating transrepression of proinflammatory cytokines. J Immunol. 2008;181:5646–52.
54 Mukundan L, Odegaard JI, Morel CR, JHeredia E, Mwangi JW, Ricardo-Gonzalez RR, et al. PPAR-delta senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat Med. 2009;15:1266–72.
55 Gosset P, Charbonnier AS, Delerive P, Fontaine J, Staels B, Pestel J, et al. Peroxisome proliferator-activated receptor gamma activators affect the maturation of human monocyte-derived dendritic cells. Eur J Immunol. 2001;31:2857–65.
56 Faveeuw C, Fougeray S, Angeli V, Fontaine J, Chinetti G, Gosset P, et al. Peroxisome proliferator-activated receptor gamma activators inhibit interleukin-12 production in murine dendritic cells. FEBS Lett. 2000;486:261–6.
57 Szatmari I, Gogolak P, Im JS, Dezso B, Rajnavolgyi E, Nagy L. Activation of PPARgamma specifies a dendritic cell subtype capable of enhanced induction of iNKT cell expansion. Immunity. 2004;21:95–106.
58 Szanto A, Balint BL, Nagy ZS, Barta E, Dezso B, Pap A, et al. STAT6 transcription factor is a facilitator of the nuclear receptor PPARgamma-regulated gene expression in macrophages and dendritic cells. Immunity. 2010;33:699–712.
59 Nencioni A, Grunebach F, Zobywlaski A, Denzlinger C, Brugger W, Brossart P. Dendritic cell immunogenicity is regulated by peroxisome proliferator-activated receptor gamma. J Immunol. 2002;169:1228–35.
60 Appel S, Mirakaj V, Bringmann A, Weck MM, Grunebach F, Brossart P. PPAR-gamma agonists inhibit toll-like receptor-mediated activation of dendritic cells via the MAP kinase and NF-kappaB pathways. Blood. 2005;106:3888–94.
61 Angeli V, Hammad H, Staels B, Capron M, Lambrecht BN, Trottein F. Peroxisome proliferator-activated receptor gamma inhibits the migration of dendritic cells: consequences for the immune response. J Immunol. 2003;170:5295–301.
62 Klotz L, Dani I, Edenhofer F, Nolden L, Evert B, Paul B, et al. Peroxisome proliferator-activated receptor gamma control of dendritic cell function contributes to development of CD4+ T cell anergy. J Immunol. 2007;178:2122–31.
63 Jakobsen MA, Petersen RK, Kristiansen K, Lange M, Lillevang ST. Peroxisome proliferator-activated receptor alpha, delta, gamma1 and gamma2 expressions are present in human monocyte-derived dendritic cells and modulate dendritic cell maturation by addition of subtype-specific ligands. Scand J Immunol. 2006;63:330–7.
64 Dubrac S, Stoitzner P, Pirkebner D, Elentner A, Schoonjans K, Auwerx J, et al. Peroxisome proliferator-activated receptor-alpha activation inhibits Langerhans cell function. J Immunol. 2007;178:4362–72.
65 Szatmari I, Torocsik D, Agostini M, Nagy T, Gurnell M, Barta E, et al. PPARgamma regulates the function of human dendritic cells primarily by altering lipid metabolism. Blood. 2007;110:3271–80.
66 Szatmari I, Pap A, Ruhl R, Ma JX, Illarionov PA, Besra GS, et al. PPARgamma controls CD1d expression by turning on retinoic acid synthesis in developing human dendritic cells. J Exp Med. 2006;203:2351–62.
67 Gogolak P, Rethi B, Szatmari I, Lanyi A, Dezso B, Nagy L, Rajnavolgyi E. Differentiation of CD1a- and CD1a+ monocyte-derived dendritic cells is biased by lipid environment and PPARgamma. Blood. 2007;109:643–52.
68 Setoguchi K, Misaki Y, Terauchi Y, Yamauchi T, Kawahata K, Kadowaki T, et al. Peroxisome proliferator-activated receptor-gamma haploinsufficiency enhances B cell proliferative responses and exacerbates experimentally induced arthritis. J Clin Invest. 2001;108:1667–75.
69 Clark RB, Bishop-Bailey D, Estrada-Hernandez T, Hla T, Puddington L, Padula SJ. The nuclear receptor PPAR gamma and immunoregulation: PPAR gamma mediates inhibition of helper T cell responses. J Immunol. 2000;164:1364–71.
70 Cunard R, DiCampli D, Archer DC, Stevenson JL, Ricote M, Glass CK, et al. WY14,643, a PPAR alpha ligand, has profound effects on immune responses in vivo. J Immunol. 2002;169:6806–12.
71 Schlezinger JJ, Jensen BA, Mann KK, Ryu HY, Sherr DH. Peroxisome proliferator-activated receptor gamma-mediated NF-kappa B activation and apoptosis in pre-B cells. J Immunol. 2002;169:6831–41.
72 Padilla J, Leung E, Phipps RP. Human B lymphocytes and B lymphomas express PPAR-gamma and are killed by PPAR-gamma agonists. Clin Immunol. 2002;103:22–33.
73 Padilla J, Kaur K, Cao HJ, Smith TJ, Phipps RP. Peroxisome proliferator activator receptor-gamma agonists and 15–deoxy-Delta(12,14)(12,14)-PGJ(2) induce apoptosis in normal and malignant B-lineage cells. J Immunol. 2000;165:6941–8.
74 Ray DM, Akbiyik F, Bernstein SH, Phipps RP. CD40 engagement prevents peroxisome proliferator-activated receptor gamma agonist-induced apoptosis of B lymphocytes and B lymphoma cells by an NF-kappaB-dependent mechanism. J Immunol. 2005;174:4060–9.
75 Ramon S, Bancos S, Thatcher TH, Murant TI, Moshkani S, Sahler JM, et al. Peroxisome proliferator-activated receptor gamma B cell-specific-deficient mice have an impaired antibody response. J Immunol. 2012;189:4740–7.
76 Schmidt S, Moric E, Schmidt M, Sastre M, Feinstein DL, Heneka MT. Anti-inflammatory and antiproliferative actions of PPAR-gamma agonists on T lymphocytes derived from MS patients. J Leukoc Biol. 2004;75:478–85.
77 Zhang MA, Rego D, Moshkova M, Kebir H, Chruscinski A, Nguyen H, et al. Peroxisome proliferator-activated receptor (PPAR)alpha and -gamma regulate IFNgamma and IL-17A production by human T cells in a sex-specific way. Proc Natl Acad Sci U S A. 2012;109:9505–10.
78 Dunn SE, Bhat R, Straus DS, Sobe RA l, Axtell R, Johnson A, et al. Peroxisome proliferator-activated receptor delta limits the expansion of pathogenic Th cells during central nervous system autoimmunity. J Exp Med. 2010;207:1599–608.
79 Klotz L, Burgdorf S, Dani I, Saijo K, Flossdorf J, Hucke S, et al. The nuclear receptor PPAR gamma selectively inhibits Th17 differentiation in a T cell-intrinsic fashion and suppresses CNS autoimmunity. J Exp Med. 2009;206:2079–89.
80 Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23–IL-17 immune axis: from mechanisms to therapeutic testing. Nature reviews. Immunology. 2014;14:585–600.
81 Ohkura N, Kitagawa Y, Sakaguchi S. Development and maintenance of regulatory T cells. Immunity. 2013;38:414–23.
82 Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, et al. PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature. 2012;486:549–53.
83 Housley WJ, O’Conor CA, Nichols F, Puddington L, Lingenheld EG, Zhu L, et al. PPARgamma regulates retinoic acid-mediated DC induction of Tregs. J Leukoc Biol. 2009;86:293–301.
84 Kawahito Y, Kondo M, Tsubouchi Y, Hashiramoto A, Bishop-Bailey D, Inoue K, et al. 15–deoxy-delta(12,14)-PGJ(2) induces synoviocyte apoptosis and suppresses adjuvant-induced arthritis in rats. J Clin Invest. 2000;106:189–97.
85 Cuzzocrea S, Mazzon E, Dugo L, Patel NS, Serraino I, Di Paola R, et al. Reduction in the evolution of murine type II collagen-induced arthritis by treatment with rosiglitazone, a ligand of the peroxisome proliferator-activated receptor gamma. Arthritis Rheum. 2003;48:3544–56.
86 Tomita T, Kakiuchi Y, Tsao PS. THR0921, a novel peroxisome proliferator-activated receptor gamma agonist, reduces the severity of collagen-induced arthritis. Arthritis Res Ther. 2006;8:R7.
87 Shirinsky I, Polovnikova O, Kalinovskaya N, Shirinsky V. The effects of fenofibrate on inflammation and cardiovascular markers in patients with active rheumatoid arthritis: a pilot study. Rheumatol Int. 2013;33:3045–8.
88 Shahin D, Toraby EE, Abdel-Malek H, Boshra V, Elsamanoudy AZ, Shaheen D. Effect of peroxisome proliferator-activated receptor gamma agonist (pioglitazone) and methotrexate on disease activity in rheumatoid arthritis (experimental and clinical study). Clin Med Insights Arthritis Musculoskelet Disord. 2011;4:1–10.
89 Okamoto H, Kamatani N. Successful treatment with fenofibrate, a peroxisome proliferator activated receptor alpha ligand, for a patient with rheumatoid arthritis. Ann Rheum Dis. 2004;63:1002–3.
90 Bongartz T, Coras B, Vogt T, Scholmerich J, Muller-Ladner U. Treatment of active psoriatic arthritis with the PPARgamma ligand pioglitazone: an open-label pilot study. Rheumatology (Oxford). 2005;44:126–9.
91 Sabatini M, Bardiot A, Lesur C, Moulharat N, Thomas M, Richard, I et al. Effects of agonists of peroxisome proliferator-activated receptor gamma on proteoglycan degradation and matrix metalloproteinase production in rat cartilage in vitro. Osteoarthritis and cartilage / OARS, Osteoarthritis Research Society. 2002;10:673–9.
92 Kobayashi T, Notoya K, Naito T, Unno S, Nakamura A, Martel-Pelletier J, et al. Pioglitazone, a peroxisome proliferator-activated receptor gamma agonist, reduces the progression of experimental osteoarthritis in guinea pigs. Arthritis Rheum. 2005;52:479–87.
93 Boileau C, Martel-Pelletier J, Fahmi H, Mineau F, Boily M, Pelletier JP. The peroxisome proliferator-activated receptor gamma agonist pioglitazone reduces the development of cartilage lesions in an experimental dog model of osteoarthritis: in vivo protective effects mediated through the inhibition of key signaling and catabolic pathways. Arthritis Rheum. 2007;56:2288–98.
94 Fahmi H, Martel-Pelletier J, Pelletier JP, Kapoor M. Peroxisome proliferator-activated receptor gamma in osteoarthritis. Mod Rheumatol. 2011;21:1–9.
95 Clockaerts S, Bastiaansen-Jenniskens YM, Feijt C, JVerhaar A, Somville J, De Clerck LS, et al. Peroxisome proliferator activated receptor alpha activation decreases inflammatory and destructive responses in osteoarthritic cartilage. Osteoarthritis and cartilage / OARS, Osteoarthritis Research Society. 2011;19:895–902.
96 Vasheghani F, Monemdjou R, Fahmi H, Zhang Y, Perez G, Blati M, et al. Adult cartilage-specific peroxisome proliferator-activated receptor gamma knockout mice exhibit the spontaneous osteoarthritis phenotype. Am J Pathol. 2013;182:1099–106.
97 Monemdjou R, Vasheghani F, Fahmi H, Perez G, Blati M, Taniguchi N, et al. Association of cartilage-specific deletion of peroxisome proliferator-activated receptor gamma with abnormal endochondral ossification and impaired cartilage growth and development in a murine model. Arthritis Rheum. 2012;64:1551–61.
98 Hong C, Tontonoz P. Coordination of inflammation and metabolism by PPAR and LXR nuclear receptors. Current opinion in genetics & development. 2008;18:461–7.
99 Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat Med. 2003;213–9.
100 Castrillo A, Joseph SB, Vaidya SA, Haberland M, Fogelman AM, Cheng G, et al. Crosstalk between LXR and toll-like receptor signaling mediates bacterial and viral antagonism of cholesterol metabolism. Molecular cell. 2003;12:805–16.
101 Joseph SB, Bradley MN, Castrillo A, Bruhn KW, Mak PA, Pei L, et al. LXR-dependent gene expression is important for macrophage survival and the innate immune response. Cell. 2004;119:299–309.
102 Valledor AF, Hsu LC, Ogawa S, Sawka-Verhelle D, Karin M, Glass CK. Activation of liver X receptors and retinoid X receptors prevents bacterial-induced macrophage apoptosis. Proc Natl Acad Sci U S A. 2004;101:17813–8.
103 N, A. G., Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity. 2009;31:245–58.
104 Hong C, Kidani Y, A. G. N, Phung T, Ito A, Rong X, Ericson K, et al. Coordinate regulation of neutrophil homeostasis by liver X receptors in mice. J Clin Invest. 2012;122:337–47.
105 N, A. G., Guillen JA, Gallardo G, Diaz M, de la Rosa JV, Hernandez IH, et al. The nuclear receptor LXRalpha controls the functional specialization of splenic macrophages. Nat Immunol. 2013;14:831–9.
106 Bensinger SJ, Bradley MN, Joseph SB, Zelcer N, Janssen EM, Hausner MA, et al. LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell. 2008;134:97–111.
107 Joseph SB, McKilligin E, Pei L, Watson MA, Collins AR, Laffitte BA, et al. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc Natl Acad Sci U S A. 2002;99:7604–9.
108 Fowler AJ, Sheu MY, Schmuth M, Kao J, Fluhr JW, Rhein L, et al. Liver X receptor activators display anti-inflammatory activity in irritant and allergic contact dermatitis models: liver-X-receptor-specific inhibition of inflammation and primary cytokine production. J Invest Dermatol. 2003;120:246–55.
109 Sheu MY, Fowler AJ, Kao J, Schmuth M, Schoonjans K, Auwerx J, et al. Topical peroxisome proliferator activated receptor-alpha activators reduce inflammation in irritant and allergic contact dermatitis models. J Invest Dermatol. 2002;118:94–101.
110 Asquith DL, Miller AM, Hueber AJ, McKinnon HJ, Sattar N, Graham GJ, et al. Liver X receptor agonism promotes articular inflammation in murine collagen-induced arthritis. Arthritis Rheum. 2009;60:2655–65.
111 Park MC, Kwon YJ, Chung SJ, Park YB, Lee SK. Liver X receptor agonist prevents the evolution of collagen-induced arthritis in mice. Rheumatology (Oxford). 2010;49:882–90.
112 Asquith DL, Miller AM, Reilly J, Kerr S, Welsh P, Sattar N, et al. Simultaneous activation of the liver X receptors (LXRalpha and LXRbeta) drives murine collagen-induced arthritis disease pathology. Ann Rheum Dis. 2011;70:2225–8.
113 Birrell MA, De Alba J, Catley MC, Hardaker E, Wong S, Collins M, et al. Liver X receptor agonists increase airway reactivity in a model of asthma via increasing airway smooth muscle growth. J Immunol. 2008;181:4265–71.
114 Shi Y, Xu X, Tan Y, Mao S, Fang S, Gu W. A Liver-X-Receptor Ligand, T0901317, Attenuates IgE Production and Airway Remodeling in Chronic Asthma Model of Mice. PLoS One 2014;9:e92668.
115 Collins-Racie LA, Yang Z, Arai M, Li N, Majumdar MK, Nagpal S, et al. Global analysis of nuclear receptor expression and dysregulation in human osteoarthritic articular cartilage: reduced LXR signaling contributes to catabolic metabolism typical of osteoarthritis. Osteoarthritis and cartilage / OARS, Osteoarthritis Research Society. 2009;17:832–42.
116 Li N, Rivera-Bermudez MA, Zhang M, Tejada J, Glasson SS, Collins-Racie LA, et al. LXR modulation blocks prostaglandin E2 production and matrix degradation in cartilage and alleviates pain in a rat osteoarthritis model. Proc Natl Acad Sci U S A. 2010;107:3734–9.
117 McMorrow JP, Murphy EP. Inflammation: a role for NR4A orphan nuclear receptors? Biochem Soc Trans. 2011;39:688–93.
118 Hamers AA, Hanna RN, Nowyhed H, Hedrick CC, de Vries CJ. NR4A nuclear receptors in immunity and atherosclerosis. Curr Opin Lipidol. 2013;24:381–5.
119 Zhan Y, Du X, Chen H, Liu J, Zhao B, Huang D, et al. Cytosporone B is an agonist for nuclear orphan receptor Nur77. Nat Chem Biol. 2008;4:548–56.
120 Saijo K, Winner B, Carson CT, Collier JG, Boyer L, Rosenfeld MG, et al. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell. 2009;137:47–59.
121 Hanna RN, Shaked I, Hubbeling HG, Punt JA, Wu R, Herrley E, et al. NR4A1 (Nur77) deletion polarizes macrophages toward an inflammatory phenotype and increases atherosclerosis. Circulation research. 2012;110:416–27.
122 Hamers AA, M Vos, F Rassam, G Marinkovic, K Kurakula, PJ van Gorp, et al. Bone marrow-specific deficiency of nuclear receptor Nur77 enhances atherosclerosis. Circ Res. 2012;110:428–38.
123 Ipseiz N, Uderhardt S, Scholtysek C, Steffen M, Schabbauer G, Bozec A, et al. The nuclear receptor Nr4a1 mediates anti-inflammatory effects of apoptotic cells. J Immunol. 2014;192:4852–8.
124 Hanna RN, Carlin LM, Hubbeling HG, Nackiewicz D, Green AM, Punt JA, et al. The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C- monocytes. Nat Immunol. 2011;12:778–85.
125 Sekiya T, Kashiwagi I, Yoshida R, Fukaya T, Morita R, Kimura A, et al. Nr4a receptors are essential for thymic regulatory T cell development and immune homeostasis. Nat Immunol. 2013;14:230–7.
126 De Silva S, Han S, Zhang X, Huston DP, Winoto A, Zheng B. Reduction of the incidence and severity of collagen-induced arthritis by constitutive Nur77 expression in the T cell lineage. Arthritis Rheum. 2005;52:333–8.
127 Mix KS, Attur MG, Al-Mussawir H, Abramson SB, Brinckerhoff CE, Murphy EP. Transcriptional repression of matrix metalloproteinase gene expression by the orphan nuclear receptor NURR1 in cartilage. J Biol Chem. 2007;282:9492–504.
128 Imai Y, Youn MY, Inoue K, Takada I, Kouzmenko A, Kato S. Nuclear receptors in bone physiology and diseases. Physiol Rev. 2013;93:481–523.
129 Warriner AH, Saag KG. Osteoporosis diagnosis and medical treatment. Orthop Clin North Am. 2013;44:125–35.
130 Seibel MJ, Cooper MS, Zhou H. Glucocorticoid-induced osteoporosis: mechanisms, management, and future perspectives. The lancet. Diabetes & endocrinology. 2013;1:59–70.
131 Schwartz AV, Sellmeyer DE, Vittinghoff E, Palermo L, Lecka-Czernik B, Feingold KR, et al. Thiazolidinedione use and bone loss in older diabetic adults. J Clin Endocrinol Metab. 2006;91:3349–54.
132 Kahn SE, Zinman B, Lachin JM, Haffner SM, Herman WH, Holman RR, et al. Rosiglitazone-associated fractures in type 2 diabetes: an Analysis from A Diabetes Outcome Progression Trial (ADOPT). Diabetes care. 2008;31:845–51.
133 Hu E, Tontonoz P, Spiegelman BM. Transdifferentiation of myoblasts by the adipogenic transcription factors PPAR gamma and C/EBP alpha. Proc Natl Acad Sci U S A. 1995;92:9856–60.
134 Lecka-Czernik B, Moerman EJ, Grant DF, Lehmann JM, Manolagas SC, Jilka RL. Divergent effects of selective peroxisome proliferator-activated receptor-gamma 2 ligands on adipocyte versus osteoblast differentiation. Endocrinology. 2002;143:2376–84.
135 Akune T, Ohba S, Kamekura S, Yamaguchi M, Chung UI, Kubota N, et al. PPARgamma insufficiency enhances osteogenesis through osteoblast formation from bone marrow progenitors. J Clin Invest. 2004;113:846–55.
136 Ali AA, Weinstein RS, Stewart SA, Parfitt AM, Manolagas SC, Jilka RL. Rosiglitazone causes bone loss in mice by suppressing osteoblast differentiation and bone formation. Endocrinology. 2005;146:1226–35.
137 Rzonca SO, Suva LJ, Gaddy D, Montague DC, Lecka-Czernik B. Bone is a target for the antidiabetic compound rosiglitazone. Endocrinology. 2004;145:401–6.
138 Takada I, Kouzmenko AP, Kato S. Wnt and PPARgamma signaling in osteoblastogenesis and adipogenesis. Nat Rev Rheumatol. 2009;5:442–7.
139 Takada I, Suzawa M, Matsumoto K, Kato S. Suppression of PPAR transactivation switches cell fate of bone marrow stem cells from adipocytes into osteoblasts. Ann N Y Acad Sci. 2007;1116:182–95.
140 Okamura M, Kudo H, Wakabayashi K, Tanaka T, Nonaka A, Uchida A, et al. COUP-TFII acts downstream of Wnt/beta-catenin signal to silence PPARgamma gene expression and repress adipogenesis. Proc Natl Acad Sci U S A. 2009;106:5819–24.
141 Kawai M, Rosen CJ. PPARgamma: a circadian transcription factor in adipogenesis and osteogenesis. Nat Rev Endocrinol. 6:629–36.
142 Wan Y, Chong LW, Evans RM. PPAR-gamma regulates osteoclastogenesis in mice. Nat Med. 2007;13:1496–503.
143 Still K, Grabowski P, Mackie I, Perry M, Bishop N. The peroxisome proliferator activator receptor alpha/delta agonists linoleic acid and bezafibrate upregulate osteoblast differentiation and induce periosteal bone formation in vivo. Calcif Tissue Int. 2008;83:285–92.
144 Kim DH, Liu J, Bhat S, Benedict G, Lecka-Czernik B, Peterson SJ, et al. Peroxisome proliferator-activated receptor delta agonist attenuates nicotine suppression effect on human mesenchymal stem cell-derived osteogenesis and involves increased expression of heme oxygenase-1. J Bone Miner Metab. 2013;31:44–52.
145 Chan BY, Gartland A, Wilson PJ, Buckley KA, Dillon JP, Fraser WD, et al. PPAR agonists modulate human osteoclast formation and activity in vitro. Bone. 2007;40:149–59.
146 Mosti M, Stunes A, Ericsson M, Pullisaar H, Reseland J, Shabestari M, et al. Effects of the peroxisome proliferator-activated receptor (PPAR) delta agonist GW501516 on bone and muscle in ovariectomized rats. Endocrinology: 2014. en20131166.
147 Ye JM, Tid-Ang J, Turner N, Zeng XY, Li HY, Cooney GJ, et al. PPARdelta agonists have opposing effects on insulin resistance in high fat-fed rats and mice due to different metabolic responses in muscle. Br J Pharmacol. 2011;163:556–66.
148 Wu X, Peters JM, Gonzalez FJ, Prasad HS, Rohrer MD, Gimble JM. Frequency of stromal lineage colony forming units in bone marrow of peroxisome proliferator-activated receptor-alpha-null mice. Bone. 2000;6:21–6.
149 Meier CR, Schlienger RG, Kraenzlin ME, Schlegel B, Jick H. HMG-CoA reductase inhibitors and the risk of fractures. JAMA. 2000;283:3205–10.
150 Remen KM, Henning P, Lerner UH, Gustafsson JA, Andersson G. Activation of liver X receptor (LXR) inhibits receptor activator of nuclear factor kappaB ligand (RANKL)-induced osteoclast differentiation in an LXRbeta-dependent mechanism. J Biol Chem. 2011;286:33084–94.
151 Kleyer A, Scholtysek C, Bottesch E, Hillienhof U, Beyer C, Distler JH, et al. Liver X receptors orchestrate osteoblast/osteoclast crosstalk and counteract pathologic bone loss. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2012;27:2442–51.
152 Robertson KM, Norgard M, Windahl SH, Hultenby K, Ohlsson C, Andersson G, et al. Cholesterol-sensing receptors, liver X receptor alpha and beta, have novel and distinct roles in osteoclast differentiation and activation. J Bone Miner Res. 2006;21:1276–87.
153 Prawitt J, Beil FT, Marshall RP, Bartelt A, Ruether W, Heeren J, et al. Short-term activation of liver X receptors inhibits osteoblasts but long-term activation does not have an impact on murine bone in vivo. Bone. 2011;48:339–46.
154 Kim HJ, Yoon KA, Yoon HJ, Hong JM, Lee MJ, Lee IK, et al. Liver X receptor activation inhibits osteoclastogenesis by suppressing NF-kappaB activity and c-Fos induction and prevents inflammatory bone loss in mice. J Leukoc Biol. 2013;94:99–107.
155 Lammi J, Huppunen J, Aarnisalo P. Regulation of the osteopontin gene by the orphan nuclear receptor NURR1 in osteoblasts. Mol Endocrinol. 2004;18:1546–57.
156 Lee MK, Choi H, Gil M, Nikodem VM. Regulation of osteoblast differentiation by Nurr1 in MC3T3–E1 cell line and mouse calvarial osteoblasts. J Cell Biochem. 2006;99:986–94.
157 Pirih FQ, Tang A, Ozkurt IC, Nervina JM, Tetradis S. Nuclear orphan receptor Nurr1 directly transactivates the osteocalcin gene in osteoblasts. J Biol Chem. 2004;279:53167–74.