Role of tumour angiogenesis in haematological malignancies
DOI: https://doi.org/10.4414/smw.2014.14050
Michael
Medinger, Jakob
Passweg
Summary
Tumour angiogenesis plays a key role in the pathogenesis and progression of haematological malignancies. Thereby, pro- and anti-angiogenic growth factors and cytokines regulate the angiogenic process. The most important growth factor, vascular endothelial growth factor (VEGF) and its signaling through its receptors 1 and 2, is not only involved in solid tumours, but there is also emerging evidence that tumour progression in haematological malignancies also depends on the induction of new blood vessel formation. The evidence supporting this theory includes the finding of increased bone marrow microvessel density and increased levels of plasma pro-angiogenic cytokines. Leukaemia cells interact with surrounding host cells and extracellular matrix, this crosstalk affecting the most important aspects of the malignant phenotype. The pathophysiology of leukaemia induced angiogenesis involves both direct production of angiogenic cytokines by leukaemia cells and their interaction with bone marrow microenvironment. The inhibition of VEGF signalling by monoclonal antibodies or small molecules (kinase inhibitors) has already been successfully used for the treatment of different cancer entities, and multiple new drugs are being tested. This review summarises recent advances in the basic understanding of the role of angiogenesis in haematological malignancies and the translation of such basic findings into clinical studies.
Abbreviations
ALL acute lymphoblastic leukaemia
AML acute myeloid leukaemia
Ang1 angiopoietin 1
ATRA all-trans-retinoic acid
bFGF basic fibroblast growth factor
CR complete remission
EPC endothelial progenitor cell
DII4 Delta-like ligand 4
ET essential thrombocythaemia
FLT3–ITD FMS-like tyrosine kinase 3 internal tandem duplications
HDAC – histone deacetylase
HGF hepatocyte growth factor
HIF-1 hypoxia-inducible factor 1
HSCs haematopoietic stem cells
IL-6 interleukin-6
IMiDs immunomodulatory drugs
KDR kinase domain receptor
MDS myelodysplastic syndrome
MGUS monoclonal gammopathy of undetermined significance
MM multiple myeloma
MPN myeloproliferative neoplasm
MVD – microvessel density
mTOR mammalian target of rapamycin
NOD/SCID non-obese diabetic severe combined immunodeficiency
PDGFR platelet-derived growth factor receptor
PIGF placental growth factor
PMF primary myelofibrosis
PV polycythaemia vera
SCFR stem cell factor receptor
TKI tyrosine kinase inhibitor
TNF – tumour necrosis factor
VEGF vascular endothelial growth factor
VEGFR vascular endothelial growth factor receptor
Introduction
The growth and metastatic potential of most tumours, both solid and haematological, are angiogenesis dependent [1, 2]. In adults, physiological angiogenesis is limited to a small number of specific processes, such as wound healing, tissue repair, and the female reproductive cycle [1, 3]. Most tumours can persist in situ for a considerable period of time ranging from months to years without neovascularisation. Folkman first described the phenomenon by which dormant tumours become vascularised when a subgroup of cells within the tumour acquire an angiogenic phenotype. According to Folkman tumours up to 2 to 3 mm3 can exist in a prevascular phase but in order to further increase in size they require neovascularisation [1, 3]. Tumour blood vessels are generated by various mechanisms, such as the co-option of the existing vascular network, the expansion of the host vascular network by the budding of endothelial sprouts (sprouting angiogenesis), the remodelling and expansion of vessels by the insertion of interstitial tissue columns into the lumen of pre-existing vessels (intussusceptive angiogenesis), and the homing of endothelial cell precursors (EPCs) from the bone marrow or peripheral blood into the endothelial lining of neovessels (vasculogenesis) [4]. Bone marrow-derived progenitor cells contribute significantly to neovascularisation in a variety of tumours [5, 6].
VEGF is the most well characterised proangiogenic factor and has at least six isoforms (VEGF, PlGF, VEGF-B, VEGF-C, VEGF-D and VEGF-E), all of which are secreted as dimeric glycoproteins [7, 8]. Tight control of angiogenesis is maintained by a balance of endogenous anti-angiogenic and pro-angiogenic factors. Three structurally similar receptor tyrosine kinases, designated VEGFR-1 (also known as Flt-1), VEGFR-2 (also known as KDR) and VEGFR-3 (also known as FLT4) are activated upon binding of VEGF ligands [9]. Leukaemia cells commonly express one or both of the major VEGF receptor tyrosine kinases, the c-fms-like tyrosine kinase (Flt-1) and the kinase domain receptor (KDR) and can produce and secrete VEGF [10]. As for other receptor tyrosine kinases, VEGF binding to the receptor leads to receptor homodimerisation or heterodimerisation and subsequent autophosphorylation on certain tyrosine residues, which in turn triggers intracellular signaling cascade mediated by several effectors, which are able to recognise and dock at phosphorylated tyrosine residues of the activated receptors.
VEGFR-1 and VEGFR-2 are predominantly expressed on vascular endothelial cells, and the activation of VEGFR-2 appears to be both necessary and sufficient to mediate VEGF-dependent angiogenesis and the induction of vascular permeability [9, 10]. VEGFR-1 is also expressed on haematopoietic stem cells (HSCs), vascular smooth muscle cells, monocytes, and leukaemic cells [11, 12], whereas VEGFR-2 is expressed on endothelial progenitor cells and megakaryocytes [13, 14]. VEGFR-3, which is largely restricted to lymphatic endothelial cells, binds the VEGF homologs VEGF-C and VEGF-D, and it may play an important role in the regulation of lymphangiogenesis. VEGF and VEGFR represent significant anticancer therapy targets that elegantly bypass potential tumour-related treatment barriers [9]. VEGF signalling inhibition has been shown to result in significant tumor growth delay in a wide range of animal models [15]. The clinical benefit of this approach has also been confirmed, and concerted efforts in recent years have resulted in a number of novel anti-angiogenic agents [16, 17].
This review summarises the role of pathological angiogenesis in haematological malignancies, with a focus on acute leukaemias, myelodysplastic syndromes (MDS), multiple myeloma (MM), and myeloproliferative neoplasms (MPNs), as well as therapeutic inhibition of pathological angiogenesis using novel agents (table 1).
|
Table 1:Selection of clinical trials and approved anti-angiogenic therapies in haematological malignancies. |
|
Drug
|
Target
|
Study entities
|
Approved for
|
|
Anti-VEGF strategies
|
|
|
|
| Bevacizumab (Avastin®) |
VEGF-A |
AML, MDS, CLL, CML, NHL, MM |
Colorectal cancer, NSCLC, breast cancer, RCC, ovarian cancer |
|
Receptor tyrosine kinase inhibitors
|
|
|
|
| PTK787/ZK 222584 (Vatalanib®) |
VEGFR1–3, PDGFRβ, c-Kit |
AML, PMF, MDS, CML, DLBCL, MM |
|
| SU5416
(Semaxinib) |
VEGFR1–2, c-kit, Flt3 |
AML, MDS, MM, MPN |
|
| Sorafenib (Nexavar®) |
VEGFR2–3, B-Raf, Faf-1, PDGFRβ |
AML, ALL, MDS, CML, CLL, NHL, MM |
RCC, HCC |
| Sunitinib (Sutent®) |
VEGFR1–3, PDGFRα+β, c-kit, Flt3 |
AML, MDS, CLL, MM, NHL |
RCC, GIST |
| PKC-412 (Midostaurin) |
VEGFR2, PKC, PDGFR, Flt3, c-Kit |
AML |
|
| Cediranib (Recentin®) |
VEGFR1–3, PDGFRβ, c-Kit |
AML, MDS, CLL |
|
|
Proteasome inhibitors
|
|
|
|
| Bortezomib (Velcade®) |
26S proteasome, NF-κB |
AML, ALL, MDS, CML, NHL, MCL |
MM, MCL |
|
Immunomodulatory drugs
|
|
|
|
| Thalidomide |
bFGF, VEGF, IL-6 |
AML, MDS, MPN, CLL, NHL, MM |
MM |
| Lenalidomide (Revlimid®) |
bFGF, VEGF, IL-6 |
AML, MDS, CLL, NHL |
MM, 5q- MDS |
| AML = acute myeloid leukaemia; bFGF = basic fibroblast growth factor; DLBCL = diffuse large B cell lymphoma; CLL = chronic lymphocytic leukaemia; CML = chronic myeloid leukaemia; GIST = gastrointestinal stromal tumours; HCC = hepatocellular carcinoma; IL-6 = Interleukin-6; NHL = non-Hodgkin lymphoma; NSCLC = non-small cell lung cancer; MCL = mantle cell lymphoma; MDS = myelodysplastic syndrome; MM = multiple myeloma; MPN = myeloproliferative neoplasm; PMF = primary myelofibrosis; RCC = renal cell carcinoma; VEGF = vascular endothelial growth factor. |
Pathophysiology of angiogenesis in haematological malignancies
Many studies have suggested a role for angiogenesis not only in the pathogenesis of solid tumours but also in haematological malignancies, such as acute and chronic leukaemia, lymphoma, myelodysplastic syndromes, myeloproliferative neoplasms, and multiple myeloma [16–23]. We and others have reported increased microvessel density (MVD) and VEGF expression in the bone marrow of patients with myeloproliferative neoplasms and lymphoma [19, 22].
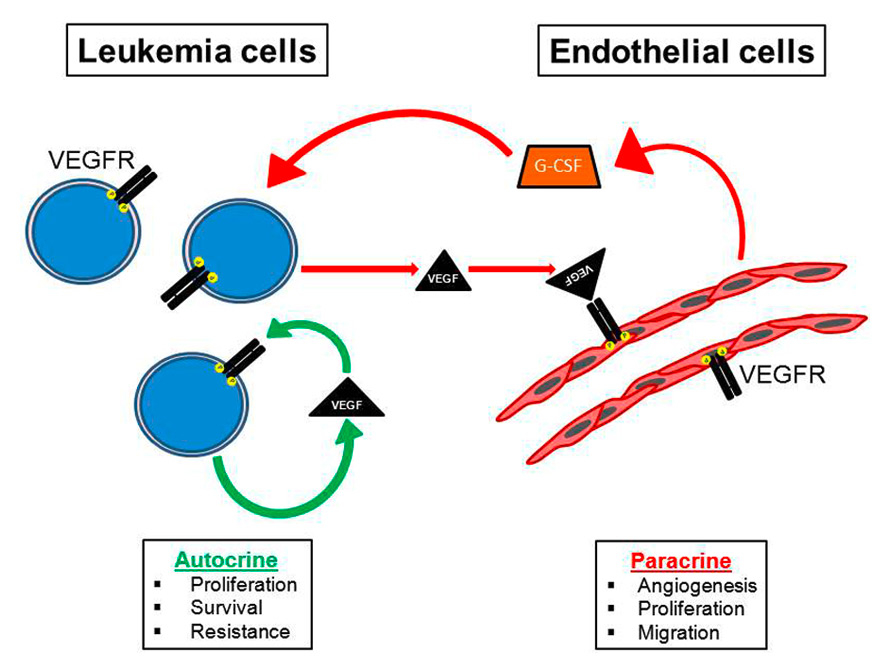
Figure 1
Cross-talk between leukaemic and endothelial cells. Autocrine and paracrine signalling via VEGF promotes the pathogenesis of leukaemia. Paracrine VEGF signalling influences angiogenesis and the bone marrow microenvironment.
G-CSF = granulocyte-colony stimulating factor; VEGF = vascular endothelial growth factor; VEGFR = vascular endothelial growth factor receptor
Thus, the extent of angiogenesis in the bone marrow often correlates with disease burden, prognosis, and treatment outcome [24, 25]. Cells, cytokines and growth factors that maintain physiological angiogenesis in normal bone marrow are unbalanced in the neoplastic marrow. Bone marrow tumour cells upregulate several factors, including interleukin-6, granulocyte-macrophage colony-stimulating factor and VEGF, producing autocrine and paracrine effects on multiple cell types and thereby stimulating angiogenesis and leading to increased vascularity (fig. 1) [12, 26]. Endothelial and haematopoietic cell lineages are therefore thought to be initiated from the same precursor cells [27, 28], and regulatory loops by which VEGF controls the survival of haematopoietic stem cells (HSCs) have been described [29]. It is now recognised that the haematopoietic and vascular systems are linked closely by virtue of the fact that both systems appear to be derived from a common precursor called the haemangioblast which originates from the mesoderm in the developing embryo [30]. Evidence supporting this hypothesis comes from studies showing that haematopoietic and endothelial cells share expression of a number of genes. CD34 enriched human peripheral blood cells were shown to have the ability to differentiate into endothelial cells in vitro [31].
The process of angiogenesis in haematological malignancies is similar to that seen in solid tumours. Endothelial cells from pre-existing venules within the bone marrow are activated, proliferate, migrate and form new blood vessels which in turn supports tumour proliferation [32]. Complex interactions take place between the neovasculature, tumour cells and the extracellular matrix mediated by growth factors, cytokines and by cell-cell contact in the bone marrow. Several primary cells and cell lines representing haematological neoplastic diseases express functional VEGFRs, and their signalling influences survival, proliferation, and migration [33, 34]. Thus, secreted VEGF contributes to disease progression by an autocrine or paracrine positive mechanism [32, 35].
Whether this observed increased neovascularisation in neoplastic bone marrow is instrumental in the neoplastic process or is an “innocent bystander” resulting from imbalances in growth factors and cytokines can only be determined by carefully designed experiments and clinical studies examining whether inhibiting the neovascular process inhibits the growth of the neoplasm.
Acute leukaemias
Numerous preclinical studies have also demonstrated the close relationship between angiogenesis and leukaemia cell survival. Human acute lymphoblastic leukaemia (ALL) cells injected into non-obese diabetic severe combined immunodeficiency (NOD/SCID) mice induce bone marrow neovascularisation [36]. Leukaemias have been associated with angiogenesis as the acute myeloid leukaemia (AML) cell line HL-60 was first used to clone the VEGFgene [37]. The first demonstration that leukaemia progression was accompanied by increased bone marrow vascularisation was provided by Judah Folkman’s group [2], who demonstrated that the bone marrow of ALL patients had increased blood vessel content compared with normal bone marrow. A detailed analysis of bone marrow sections from ALL patients showed irregular, albeit abundant, bone marrow vasculature. Moreover, urine and peripheral blood samples from ALL patients exhibited elevated levels of pro-angiogenic basic fibroblast growth factor (bFGF) and VEGF, which correlated with the increase in bone marrow angiogenesis [38]. The existence of an “angiogenesis switch”, first proposed for solid tumours [39], was therefore suggested for haematolymphoid malignancies as well. The presence of an “angiogenesis switch” in leukaemia has been demonstrated by findings of increased bone marrow MVD; increased expression of HIF-1, multiple pro-angiogenic factors (VEGF, bFGF, and angiopoietin-2), and soluble VEGFR; and decreased expression of endogenous angiogenesis inhibitors, such as thrombospondin-1 [40]. In patients with previously untreated AML, increased levels of plasma VEGF correlated with reduced rates of survival and remission [41]. Moreover, the level of plasma/serum VEGF correlated with the number of circulating blasts [42], indicating the probable cellular origin of this pro-angiogenic factor. In vitro studies have demonstrated the capacity of leukaemia cells to produce pro-angiogenic growth factors, such as VEGF and bFGF [32, 43, 44]. In patients with MDS, bone marrow VEGF expression was higher in the “very high risk” MDS than in the “very low risk” group according the World Health Organization classification-based prognostic scoring system. A high VEGF expression also predicted transfusion dependence [45]. A recent study examined the role of the Notch/Dll4 (Delta-like ligand 4) pathway in leukaemia-endothelium cross-talk and its functional link with VEGF [46]. The authors found that newly diagnosed AML patients had increased bone marrow vascularity, which was correlated with increases in VEGF and Dll4 expression. Furthermore, the upregulation of Dll4 expression in AML cells suppressed VEGF-induced endothelial cell proliferation and angiogenesis. The authors concluded that the modulation of the Notch/Dll4 pathway could be a novel anti-angiogenic strategy.
Anti-angiogenic therapies
With significant evidence emerging to support a critical role for angiogenesis in the pathogenesis of haematological malignancies, there has been increasing interest in developing anti-angiogenic therapeutic strategies for these disorders. There are several theoretical reasons why targeting the neovasculature may be advantageous. Delivery of anti-cancer drugs to tumor cells is often impaired because of abnormalities in blood flow arising from altered tumour blood vessel architecture and function. In contrast to tumour cells, the endothelial cells lining the tumour blood vessels are more readily accessible to therapy. Furthermore endothelial cells unlike tumour cells are homogenous and genetically stable and less likely to develop mutations causing resistance to drugs [47]. Also the same drug can potentially be used to treat a range of malignancies when endothelial cells rather than the tumour cells are the target.
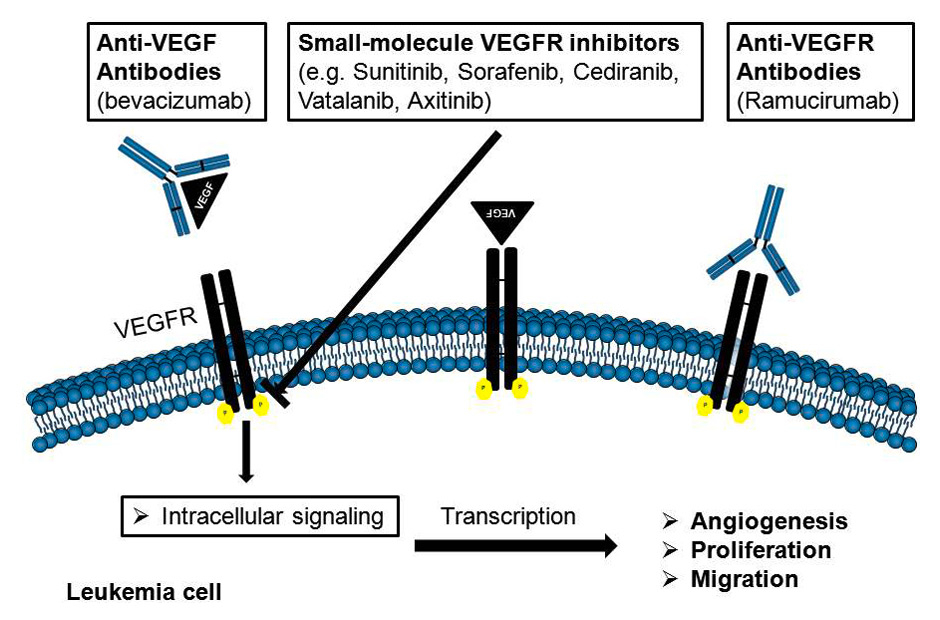
Figure 2
Mechanisms of action of angiogenic inhibitors in leukaemia.
VEGF = vascular endothelial growth factor VEGFR = vascular endothelial growth factor receptor
Most anti-angiogenic therapies are based on inhibiting the binding of VEGF to VEGFR using neutralising antibodies against the ligand or the receptor, soluble receptors, small molecule inhibitors or therapies directed against the tyrosine kinase activity of the VEGF receptors (fig. 2). A growing number of anti-angiogenics are now available, either in various stages of clinical development or as components of standard clinical regimens. The major classes of anti-angiogenic therapy include the following: (1) direct anti-VEGF-acting molecules (anti-VEGF antibodies and VEGF-antisense nucleotides); (2) receptor tyrosine kinase inhibitors that target VEGFR signalling and the receptors of other (pro-angiogenic) factors; (3) immunomodulatory drugs (IMiDs) with anti-angiogenic properties; (4) an anti-endothelial approach using metronomic therapy; and (5) other new compounds that target signals downstream of pro-angiogenic growth factors, such as mammalian target of rapamycin (mTOR) inhibitors, histone deacetylase (HDAC) inhibitors and proteasome inhibitors.
Anti-VEGF antibodies
The first anti-angiogenic agent to be approved for solid tumuors was bevacizumab (Avastin™), a humanised anti-VEGF monoclonal antibody that targets VEGF-A isoform and blocks its binding to the VEGF receptors [48]. The administration of bevacizumab, in combination with cytotoxic chemotherapy, conferred benefits to patients with several tumour entities, such as metastatic colorectal cancer, non-squamous, non-small cell lung cancer and metastatic breast cancer [48, 49]. In patients with refractory AML (n = 9), bevacizumab resulted in a reduction in VEGF expression in the bone marrow, but no clinical response [50]. Bevacizumab-associated side effects are generally mild to moderate in severity, although there are specific, uncommon events that are more severe and potentially life threatening. The most commonly observed adverse events are hypertension, proteinuria, bleeding and thrombosis, which are generally mild to moderate and manageable [50]. Bevacizumab was administered after induction chemotherapy (cytarabine and mitoxantrone) to adults with refractory or relapsed AML in a phase II clinical trial [51]. Induction therapy was received by forty-eight adults. The overall response was 23 of 48 (48%), with a complete response (CR) in 16 (33%) patients. Eighteen patients (14 CR and 4 partial responses) underwent one consolidation cycle, and 5 (3 CR and 2 partial responses) underwent allogeneic transplantation. The median overall and disease-free survivals for the CR patients were 16.2 months (64%, 1 year) and 7 months (35%, 1 year), respectively. Bone marrow samples demonstrated a marked MVD decrease after bevacizumab. VEGF was detected in pretreatment serum in 67% of the patients tested. The VEGF level increased by day 8 in 52% of the patients, and it decreased 2 h after bevacizumab in 93% of the patients (67% undetectable). In a further phase II study, elderly patients with AML (n = 171) were randomly assigned to receive standard chemotherapy (daunorubicin and cytarabine) with or without bevacizumab [52]. The complete remission rates in the 2 arms did not differ (65%). The event-free survival at 12 months was 33% for the standard arm versus 30% for the bevacizumab arm; at 24 months, it was 22% and 16%, respectively. The authors concluded that the addition of bevacizumab to standard chemotherapy did not improve the therapeutic outcome of older AML patients.
Receptor tyrosine kinase inhibitors
Small tyrosine kinase inhibitors that target VEGFR and other kinases are a further important class of anti-angiogenic drugs with applications in AML, although their efficacy in haematolymphoid neoplasias, especially AML, might be attributable to the inhibition of a variety of pathways, particularly those related to c-kit and FLT3 (FMS-like tyrosine kinase 3). Small-molecule inhibitors targeting VEGFRs and other kinases, e.g., sorafenib (Nexavar™) and sunitinib (Sutent™), have been approved based on their efficacy in treating renal cell carcinoma and hepatocellular carcinoma [53, 54].
Vatalanib (formerly PTK787/ZK 222584) is an oral multi-targeted inhibitor that was designed to target VEGFR-2 selectively, but was also demonstrated to inhibit VEGFR-1, VEGFR-3, and stem cell factor receptor (c-Kit) [55]. Vatalanib was well tolerated and showed clinical activity in a variety of solid tumours in a phase I trial [56]. In patients with MDS and AML, vatalanib was studied in a phase I clinical trial alone or in combination with cytosine-arabinoside and daunorubicin [57]. Sixty-three patients received vatalanib at doses of 500–1,000 mg/bid orally. At 1,000 mg/bid, dose-limiting toxicities resulting in lethargy, hypertension, nausea, emesis, and anorexia were observed. CR was observed in 5 of 17 evaluable AML patients treated with vatalanib combined with chemotherapy. The authors concluded that vatalanib is generally well tolerated and can be administered in combination with chemotherapy to patients with MDS and AML. In a recent phase II study, vatalanib was examined in patients with MDS. A total of 142 patients were evaluable for response. Only a small proportion of these MDS patients exhibited haematological improvement under vatalanib therapy [58].
Cediranib (AZD2171, Recentin™) is a potent inhibitor of both VEGFR-1 and VEGFR-2; it also has activity against c-kit, PDGFR-β, and VEGFR-3 at nanomolar concentrations [59]. In a phase I trial in patients with a broad range of solid tumours, cediranib was well tolerated at doses up to 45 mg/d [60], with the most common adverse side effects being diarrhoea, dysphonia, and hypertension. In 35 patients with AML, the most common adverse events of cediranib were diarrhoea, hypertension, and fatigue in a phase I trial. An objective response was experienced by six patients (3 each at 20 and 30 mg). Dose- and time-dependent reductions in soluble VEGFR-2 were observed, and there was a correlation between cediranib exposure and plasma VEGF levels [61].
Sorafenib (Nexavar™; formerly BAY43–9006) is an orally available bisaryl urea derivative approved for clinical use in kidney cancer and hepatocellular carcinoma. It was originally developed as an inhibitor of the serine/ threonine kinase Raf. However, it also was found to effectively inhibit other kinases, including VEGFR, Kit, platelet-derived growth factor receptors, and fms-like tyrosine kinase receptor-3 (FLT3), all present on AML cells and in bone marrow stromal elements [62]. VEGF, Kit ligand, platelet-derived growth factor, and FLT3 ligand can have autocrine and/or paracrine functions supporting AML cell proliferation and survival [63]. The administration of sorafenib to treat AML patients who harboured an FLT3–ITD mutation was demonstrated effective before and after allogenic stem cell transplantation [62]. All the AML patients with an FLT3–ITD mutation (6/6) were shown to respond to sorafenib treatment. Because approximately 25% of the AML patients demonstrated an FLT3–ITD mutation, sorafenib appears to be a promising therapeutic for a subset of AML patients. Randomly assigned AML patients with an FLT3 mutation (74% ITD) demonstrated a higher partial response rate compared with AML patients with wild-type FLT3 (75 and 43%, respectively). None of the AML patients achieved complete remission. However, treatment duration was short and no durable responses were reported. Notably, a complete molecular remission has recently been reported in a patient relapsing after stem cell transplantation [64]. All these recent studies suggest that sorafenib deserves further evaluation in prospective clinical trials. A recent study examined the addition of sorafenib to standard induction and consolidation therapy in elderly patients with AML [65]. A total of 201 patients were randomly assigned to receive either sorafenib or placebo between the chemotherapy cycles and subsequently for up to 1 year after the beginning of therapy. Treatment with sorafenib did not result in significant improvements in event-free survival or overall survival. Similar results were revealed by subgroup analyses, including a subgroup positive for FLT3 internal tandem duplications. The authors concluded that the combination of sorafenib with standard induction and consolidation therapy in the investigated schedule is not beneficial for elderly patients with AML. In a further study, sorafenib was combined with all-trans retinoic acid (ATRA) in patients with refractory or relapsed FLT3–ITD-positive/ nucleophosmin1–positive AML. Three patients achieved significant response under this combination therapy [66]. In a phase II study of sorafenib, cytarabine and idarubicin for initial therapy in younger AML patients with a median follow-up of 18 months, the median event-free survival was 12.2 months in the placebo arm and was not reached in the sorafenib arm. Although there was an increased risk of hepatotoxicity and bleeding events in the sorafenib arm, the authors concluded that the addition of sorafenib to standard induction chemotherapy was associated with a prolongation of event-free survival, which was most marked in the patients with FLT3–ITD [67].
Immunomodulatory drugs (IMiDs)
As the first of the immune-modulatory drug (IMiD) class, thalidomide was introduced as a sedative used to prevent nausea during pregnancy in the late 1950s. In 1961, it was withdrawn due to teratogenicity and neuropathy [68]. Multiple myeloma is one of the first clinical entities for which this efficacy was demonstrated [68, 69]. The surprising effects of thalidomide have led to the development of a series of IMiDs with even higher anti-angiogenic potency. It was shown that thalidomide has important immunomodulatory effects by decreasing TNF-α synthesis and selectively modulating T cell subsets, shifting the T cell population toward T helper cells [70]. The interest in thalidomide as an anti-neoplastic agent rose after the demonstration of its anti-angiogenic activity in a rabbit model of corneal neovascularisation induced in response to bFGF [71]. The second generation of IMiDs include lenalidomide (CC-5013) and pomalidomide (CC4047), and both of them demonstrated more potent anti-cancer, anti-inflammatory and immunomodulatory activities than thalidomide [72, 73]. Although several mechanisms have been proposed to explain the activity of thalidomide, lenalidomide and pomalidomide, including demonstrable anti-angiogenic, anti-proliferative and immunomodulatory effects, the precise cellular targets and molecular mechanisms have not been understood in detail [72, 73].
Thalidomide therapy was assessed alone and in combination with other compounds in patients with AML. Thalidomide was administered in a phase II study to patients with relapsed or refractory AML who were previously treated with cytarabine-containing regimens [74]. A total of 16 patients were treated with 200 to 800 mg/d oral thalidomide. Overall, one patient (6%) achieved CR, which lasted for 36 months, and two patients had transient reductions in marrow blasts, from 8% and 7% to less than 5%. There was no correlation between reductions in angiogenesis marker levels and response. The safety and efficacy of thalidomide was tested in a phase I/II dose-escalating trial [75] in 20 AML patients. There were 13 patients who were assessable for both toxicity and response and were found to tolerate a maximum dose of 200 to 400 mg/d for at least one month. Overall, adverse events were fatigue, constipation, rash, and neuropathy (grades 1 to 2 in most patients). In four patients, a partial response was observed, defined as a reduction in the blast cell infiltration into the bone marrow by at least 50%, accompanied by increases in platelet counts and haemoglobin values. In parallel, MVD significantly decreased in 5 five patients during treatment with thalidomide.
A recent study of lenalidomide showed that two elderly patients with poor-risk cytogenetic AML achieved morphologic and cytogenetic complete remission [76]. In a phase II study, lenalidomide was administered to previously untreated older AML patients with del(5q) who declined standard chemotherapy [77]. Among 37 evaluable patients, the median age was 74 years. Five patients (14%) achieved a partial or complete response. The mean relapse-free survival was 5 months (range, 0–19). The median overall survival was two months for the entire population. In conclusion, lenalidomide as a single agent has modest activity in older del(5q) AML patients.
Proteasome inhibitors
Bortezomib (Velcade™) is a proteasome inhibitor known to induce apoptosis, reverse drug resistance of multiple myeloma cells, and block cytokine effects, cell adhesion, and angiogenesis in the myeloma cell microenvironment, all of which support the proliferation and migration of neoplastic plasma cells [78, 79]. It is approved for treating multiple myeloma and mantle cell lymphoma. In multiple myeloma, complete clinical responses have been obtained in patients with otherwise refractory or rapidly advancing disease.Beside its direct anti-tumour effects, anti-angiogenic actions of bortezomib have recently been described in vitro and in vivo [80]. In a study by Roccaro et al. the effect of bortezomib on the angiogenic phenotype of MM patient-derived endothelial cells was examined. Bortezomib inhibited the proliferation of endothelial cells and angiogeneis in a dose-dependent manner [80]. Bortezomib triggered a dose-dependent inhibition of VEGF and interleukin-6 (IL-6) secretion by the multiple myeloma patient-derived endothelial cells, and molecular testing confirmed drug-related down-regulation of VEGF, IL-6, insulin-like growth factor-I, Angiopoietin 1 (Ang1), and Ang2 transcription. In patients with untreated AML, bortezomib was combined with daunorubicin and cytarabine in induction and consolidation chemotherapy [81]. There were 95 patients (median age 67 years) included. A complete response was achieved by 65% (62 of 95 patients treated). Bortezomib in combination with daunorubicin and cytarabine showed a tolerable toxicity profile. Taken together, the combination of standard chemotherapy with bortezomib resulted in an encouraging remission rate.
Multiple myeloma
Multiple myeloma was the first haematological malignancy in which an increased angiogenesis rate was detected [23]. As a progression from in situ to invasive and metastatic solid tumours is accompanied and enhanced by the switch from the perivascular to the vascular phase, these findings suggest that active MM may represent the ‘vascular phase’ of plasma cell tumours, and nonactive MM and MGUS their ‘prevascular phase’. Several studies show overexpression and secretion of VEGF by the clonal plasma cells [82]. VEGF stimulates proliferation and chemotaxis in both endothelial cells via VEGFR-2 and stromal cells via VEGFR-1. VEGFR-1, but not VEGFR-2, is commonly expressed on both MM cell lines and patient’s MM cells. Increased bone marrow MVD in patients with MM appears to also be an important prognostic factor [83]. As with other tumours induction of angiogenesis in myeloma is thought to be mediated by an alteration in the balance between pro- and anti-angiogenic cytokines. A number of pro-angiogenic cytokines are upregulated in patients with active myeloma. VEGF is one of the key pro-angiogenic cytokines thought to play a role in myeloma pathogenesis [84]. VEGF was shown to be overexpressed in plasma cells from myeloma patients with an increase in VEGF receptors in the surrounding stromal cells [85].
In a phase II trial, thalidomide monotherapy was assessed in 84 patients with relapsed and refractory MM who had received doses ranging from 200 to 800 mg/d. The overall response rate was 32%. The 2 year event-free and overall survival rates were 20% and 48%, respectively [68, 86]. In patients with newly diagnosed MM, the combination of thalidomide with dexamethasone led to a response rate of 63%, compared with 41% for dexamethasone alone [87]. Subsequent to these studies, thalidomide was approved for the treatment of newly diagnosed MM. Lenalidomide in combination with dexamethasone was tested in 2 phase III trials. The studies showed remarkable response rates and significantly less toxicity than thalidomide [88, 89]. The response rate increased from 22.5% to 59.2% compared with dexamethasone alone in patients with previously treated relapsed/refractory MM. Lenalidomide in combination with dexamethasone was approved as a second-line treatment for MM. In a recent study, the antiangiogenic effect of lenalidomide was studied in vitro in bone marrow endothelial cells from patients with multiple myeloma and in vivo using the chorioallantoic membrane assay. Lenalidomide exerted a relevant antiangiogenic effect in vivo at 1.75 μmol/l, a dose reached in the interstitial fluids of patients treated with 25 mg/d. In vitro, lenalidomide inhibited angiogenesis and the migration of multiple myeloma endothelial cells [90]. In a randomised phase II trial of bevacizumab versus bevacizumab and thalidomide for relapsed/refractory MM, 12 patients were evaluable. In the combination arm, two patients achieved a partial response and three achieved stable disease. Under bevacizumab monotherapy, one patient achieved stable disease [91].
Bortezomib (Velcade™) triggers its anti-MM activity at least in part through its anti-angiogenic effect and MM patients treated with bortezomib show a significant reduction in microvascular density. In a randomised phase II study, the combination of bortezomib with bevacizumab was tested in patients with relapsed/refractory MM [92]. The median PFS was 6.2 months for the bevacizumab-containing arm and 5.1 months for the bortezomib monotherapy arm, respectively; the overall response rates were 51% and 43.4% (p = 0.4029), respectively. Frequent adverse events occurred at similar rates across treatment arms, but hypertension, fatigue, and neuralgia occurred more frequently in the bevacizumab-containing arm. In conclusion, the addition of bevacizumab to bortezomib in unselected patients with pretreated MM did not result in significant improvements in efficacy outcomes.
Myeloproliferative neoplasms
Diseases commonly referred to as chronic BCR-ABL1–negative myeloproliferative neoplasms (MPNs) comprise essential thrombocythaemia (ET), polycythaemia vera (PV), and primary myelofibrosis (PMF), and are acquired, clonal disorders arising at the stem-cell level characterised by overproduction of terminally differentiated myeloid cells [93]. The identification of an acquired somatic mutation in the JAK2 gene, which results in a valine-to-phenylalanine substitution at position 617 (JAK2–V617F), has provided new insights into the pathogenesis of BCR-ABL1–negative MPNs, which are present in most patients with PV and in approximately 50% of patients with ET and PMF [93, 94]. Although the molecular diagnostic gap in JAK2V-617F-negative PV was adequately addressed by the discovery of other JAK2 mutations, this finding did not address the 35%–40% of patients with ET or PMF who do not have JAK2 mutations. In December 2013, two groups reported the occurrence of novel calreticulin (CALR) mutations in JAK2/MPL-unmutated PMF or ET [95, 96]. Both groups found mutual exclusivity between CALR, JAK2 and MPL mutations. In a study by Klampfl et al., CALR mutations were not observed in 382 cases of PV but were detected in 25% of patients with ET (n = 311) and in 35% of patients with PMF (n = 203).
Angiogenesis and the expression of VEGF and its receptors are increased in the bone marrow of patients with BCR-ABL1–negative MPNs, especially PMF, and increased angiogenesis might inversely correlate with survival [97, 98]. In a recent study, we found significant increases in MVD and VEGF expression in MPN patients compared with controls, especially in cases with high JAK2–V617Fmutant allele burdens [19]. Our results imply that elevated activity of Jak2–related pathways, as observed in cases with higher JAK2–V617F mutant allele burdens, may influence angiogenesis in MPN [19]. A very recent study identified the JAK2–V617Fmutation in microdissected endothelial cells from the liver veins of Budd-Chiari syndrome patients [99], raising the hypothesis that endothelial cells in PV are direct players in the neoplastic process.
Vatalanib, a VEGFR TKI, was administered to 29 PMF patients at doses of 500 or 750 mg bid in a phase I dose escalation study [100]. The response rate was modest in patients with PMF; one patient (3%) achieved CR, and five patients (17%) achieved clinical improvement. Transient, potentially vatalanib-related toxicities were observed, including mild nausea, vomiting, dizziness, fatigue, and thrombocytopenia. The efficacy of thalidomide monotherapy was assessed in a phase II study with 44 PMF patients [101]. There were 17 of 41 evaluable patients (41%) who received treatment for at least 15 days showed a response. CR (without reversal of bone marrow fibrosis) was achieved in 4 patients (10%), a partial response occurred in 4 patients (10%), and haematological improvements in anaemia, thrombopenia, and/or splenomegaly were observed in 9 patients (21%). In a placebo-controlled phase II study, the efficacy of thalidomide regarding the improvement anaemia in PMF patients was assessed [102]. The primary outcome was a 2 g/l increase in haemoglobin levels, resulting in a 20% reduction in transfusion needs. At 180 days, in an intention-to-treat analysis, no difference in haemoglobin levels was observed between the thalidomide and placebo groups. Lenalidomide monotherapy was tested in phase II studies with symptomatic PMF patients. The overall response rates were 22% for anaemia, 33% for splenomegaly, and 50% for thrombocytopenia [103]. In a combination study of lenalidomide with prednisone in 40 PMF patients [104], responses were recorded in 12 patients (30%) and are ongoing in 10 (25%). There was a median time to response of 12 weeks; 3 patients (7.5%) had partial responses, and 9 patients (22.5%) exhibited clinical improvement lasting a median of 18 months. The overall response rates were 30% for anaemia and 42% for splenomegaly. Interestingly, all eight JAK2–V617F-positive responders also experienced a reduction in the baseline mutant allele burden.
Conclusions
The autocrine regulators of angiogenesis are essential to the development of diseases. VEGF/VEGFR as key regulators of neoangiogenesis and vasculogenesis has been widely studied. Although the mechanism of the high expression of VEGF in leukaemia cells has not yet been identified, we are certain that VEGF/VEGFR interactions may stimulate proliferation, migration and survival of leukaemia/lymphoma cells by autocrine and paracrine loops. Novel agents targeting VEGF, its receptors, and other angiogenic pathways are in various stages of clinical development and investigation in haematological malignancies. The treatment of solid tumours has shown that combination therapies of different anti-angiogenic molecules with chemotherapy or irradiation increases treatment efficacy. Apart from anti-VEGF molecules, other anti-angiogenic therapies include IMiDs, receptor tyrosine kinase inhibitors, anti-endothelial approach to metronomic therapy, mTOR inhibitors, HDAC inhibitors and proteasome inhibitors. A better understanding of the role of VEGF in leukaemia and additional trials combining anti-angiogenic therapies will provide a greater insight to the mechanisms required for treatment. Blocking VEGF activity has been shown to be particularly effective at sensitising the vasculature and improving the delivery of cytotoxic drugs to tumour and endothelial cells. However, not all patients treated with anti-angiogenic therapies benefit from this type of therapy, and the effect is transient in most cases. Therefore, there is an urgent need for biomarkers to identify patients likely to benefit from anti-angiogenic treatments, to select the optimal dose to minimise side effects, and to understand the mechanisms of resistance. It must be kept in mind that most of these studies used anti-VEGF monotherapy to treat heavily pretreated, refractory, or relapsed patients. As multiple signal transduction pathways are activated in leukaemias, just a molecular targeted therapy is unlikely to be curative if used alone. Although, recently VEGF has brought much attention to the pathology of haematological malignancies, there is evidence that its only inhibition is not as effective as it was firstly thought. The responses are modest and transient. Thus, simultaneous inhibition of multiple targets is necessary in most patients.
References
1 Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27–31.
2 Perez-Atayde AR, Sallan SE, Tedrow U, Connors S, Allred E, Folkman J. Spectrum of tumor angiogenesis in the bone marrow of children with acute lymphoblastic leukemia. Am J Pathol. 1997;150:815–21.
3 Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–6.
4 Lyden D, Hattori K, Dias S, Costa C, Blaikie P, Butros L, et al. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med. 2001;7:1194–201.
5 Rafii S, Lyden D, Benezra R, Hattori K, Heissig B. Vascular and haematopoietic stem cells: novel targets for anti-angiogenesis therapy? Nat Rev Cancer. 2002;2:826–35.
6 Peters BA, Diaz LA, Polyak K, Meszler L, Romans K, Guinan EC, et al. Contribution of bone marrow-derived endothelial cells to human tumor vasculature. Nat Med. 2005;11:261–2.
7 Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–5.
8 Dor Y, Porat R, Keshet E. Vascular endothelial growth factor and vascular adjustments to perturbations in oxygen homeostasis. Am J Physiol Cell Physiol. 2001;280:C1367–74.
9 Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–76.
10 Gille H, Kowalski J, Li B, LeCouter J, Moffat B, Zioncheck TF, et al. Analysis of biological effects and signaling properties of Flt-1 (VEGFR-1) and KDR (VEGFR-2). A reassessment using novel receptor-specific vascular endothelial growth factor mutants. J Biol Chem. 2001;276:3222–30.
11 Hattori K, Dias S, Heissig B, Hackett NR, Lyden D, Tateno M, et al. Vascular endothelial growth factor and angiopoietin-1 stimulate postnatal hematopoiesis by recruitment of vasculogenic and hematopoietic stem cells. J Exp Med. 2001;193:1005–14.
12 Bellamy WT, Richter L, Frutiger Y, Grogan TM. Expression of vascular endothelial growth factor and its receptors in hematopoietic malignancies. Cancer Res. 1999;59:728–33.
13 Gill M, Dias S, Hattori K, Rivera ML, Hicklin D, Witte L, et al. Vascular trauma induces rapid but transient mobilization of VEGFR2(+)AC133(+) endothelial precursor cells. Circ Res. 2001;88:167–74.
14 Casella I, Feccia T, Chelucci C, Samoggia P, Castelli G, Guerriero R, et al. Autocrine-paracrine VEGF loops potentiate the maturation of megakaryocytic precursors through Flt1 receptor. Blood. 2003;101:1316–23.
15 Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, et al. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature. 1993;362:841–4.
16 Medinger M, Mross K. Clinical trials with anti-angiogenic agents in hematological malignancies. J Angiogenes Res. 2010;2:10.
17 Medinger M, Fischer N, Tzankov A. Vascular endothelial growth factor-related pathways in hemato-lymphoid malignancies. J Oncol. 2010;2010:729725.
18 Aguayo A. The role of angiogenesis in the biology and therapy of myelodysplastic syndromes. Curr Hematol Rep. 2004;3:184–91.
19 Medinger M, Skoda R, Gratwohl A, Theocharides A, Buser A, Heim D, et al. Angiogenesis and vascular endothelial growth factor-/receptor expression in myeloproliferative neoplasms: correlation with clinical parameters and JAK2–V617F mutational status. Br J Haematol. 2009;146:150–7.
20 Padro T, Ruiz S, Bieker R, Burger H, Steins M, Kienast J, et al. Increased angiogenesis in the bone marrow of patients with acute myeloid leukemia. Blood. 2000;95:2637–44.
21 Pruneri G, Bertolini F, Soligo D, Carboni N, Cortelezzi A, Ferrucci PF, et al. Angiogenesis in myelodysplastic syndromes. Br J Cancer. 1999;811398–1401.
22 Tzankov A, Heiss S, Ebner S, Sterlacci W, Schaefer G, Augustin F, et al. Angiogenesis in nodal B cell lymphomas: a high throughput study. J Clin Pathol. 2007;60:476–82.
23 Vacca A, Ribatti D, Roncali L, Ranieri G, Serio G, Silvestris F, et al. Bone marrow angiogenesis and progression in multiple myeloma. Br J Haematol. 1994;87:503–8.
24 Loges S, Heil G, Bruweleit M, Schoder V, Butzal M, Fischer U, et al. Analysis of concerted expression of angiogenic growth factors in acute myeloid leukemia: expression of angiopoietin-2 represents an independent prognostic factor for overall survival. J Clin Oncol. 2005;23:1109–17.
25 Vacca A, Ribatti D, Presta M, Minischetti M, Iurlaro M, Ria R, et al. Bone marrow neovascularization, plasma cell angiogenic potential, and matrix metalloproteinase2 secretion parallel progression of human multiple myeloma. Blood. 1999;93:3064–73.
26 Majka M, Janowska-Wieczorek A, Ratajczak J, Ehrenman K, Pietrzkowski Z, Kowalska MA, et al. Numerous growth factors, cytokines, and chemokines are secreted by human CD34(+) cells, myeloblasts, erythroblasts, and megakaryoblasts and regulate normal hematopoiesis in an autocrine/paracrine manner. Blood. 2001;97:3075–85.
27 Gordon-Keylock S, Medvinsky A. Endothelio–hematopoietic relationship: getting closer to the beginnings. BMC Biol. 2011;9:88.
28 Park C, Ma YD, Choi K. Evidence for the hemangioblast. Exp Hematol. 2005;33:965–70.
29 Gerber HP, Malik AK, Solar GP, Sherman D, Liang XH, Meng G, et al. VEGF regulates haematopoietic stem cell survival by an internal autocrine loop mechanism. Nature. 2002;417:954–8.
30 Choi K, Kennedy M, Kazarov A, Papadimitriou JC, Keller G. A common precursor for hematopoietic and endothelial cells. Development. 1998;125:725–32.
31 Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–7.
32 Dias S, Hattori K, Zhu Z, et al. Autocrine stimulation of VEGFR-2 activates human leukemic cell growth and migration. J Clin Invest. 2000;106:511–21.
33 Dias S, Shmelkov SV, Lam G, Rafii S. VEGF(165) promotes survival of leukemic cells by Hsp90–mediated induction of Bcl-2 expression and apoptosis inhibition. Blood. 2002;99:2532–40.
34 Fragoso R, Elias AP, Dias S. Autocrine VEGF loops, signaling pathways, and acute leukemia regulation. Leuk Lymphoma. 2007;48:481–8.
35 Santos SC, Dias S. Internal and external autocrine VEGF/ KDR loops regulate survival of subsets of acute leukemia through distinct signaling pathways. Blood. 2004;103:3883–9.
36 Veiga JP, Costa LF, Sallan SE, Nadler LM, Cardoso AA. Leukemia-stimulated bone marrow endothelium promotes leukemia cell survival. Exp Hematol. 2006;34:610–21.
37 Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246:1306–9.
38 Yetgin S, Yenicesu I, Cetin M, Tuncer M. Clinical importance of serum vascular endothelial and basic fibroblast growth factors in children with acute lymphoblastic leukemia. Leuk Lymphoma. 2001;42:83–8.
39 Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353–64.
40 Dong X, Han ZC, Yang R. Angiogenesis and antiangiogenic therapy in hematologic malignancies. Crit Rev Oncol Hematol. 2007;62:105–18.
41 Aguayo A, Kantarjian HM, Estey EH, Giles FJ, Verstovsek S, Manshouri T, et al. Plasma vascular endothelial growth factor levels have prognostic significance in patients with acute myeloid leukemia but not in patients with myelodysplastic syndromes. Cancer. 2002;95:1923–30.
42 Aguayo A, Estey E, Kantarjian H, Mansouri T, Gidel C, Keating M, et al. Cellular vascular endothelial growth factor is a predictor of outcome in patients with acute myeloid leukemia. Blood. 1999;94:3717–21.
43 Fiedler W, Graeven U, Ergün S, Verago S, Kilic N, Stockschläder M, et al. Vascular endothelial growth factor, a possible paracrine growth factor in human acute myeloid leukemia. Blood. 1997;89:1870–5.
44 Chen H, Treweeke AT, West DC, Till KJ, Cawley JC, Zuzel M, et al. In vitro and in vivo production of vascular endothelial growth factor by chronic lymphocytic leukemia cells. Blood. 2000;96:3181–7.
45 Gianelli U, Fracchiolla NS, Bucciarelli P, Ferla V, Boiocchi L, Savi F, et al. High levels of vascular endothelial growth factor protein expression are associated with an increased risk of transfusion dependence in myelodysplastic syndromes. Am J Clin Pathol. 2013;139:380–7.
46 Zhang J, Ye J, Ma D, Liu N, Wu H, Yu S, et al. Cross-talk between leukemic and endothelial cells promotes angiogenesis by VEGF activation of the Notch/Dll4 pathway. Carcinogenesis. 2013;34:667–77.
47 Kerbel RS. Inhibition of tumor angiogenesis as a strategy to circumvent acquired resistance to anti-cancer therapeutic agents. Bioessays. 1991;13:31–6.
48 Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–42.
49 Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, et al. Paclitaxel-carboplatin alone or with bevacizumab for nonsmall-cell lung cancer. N Engl J Med. 2006;355:2542–50.
50 Zahiragic L, Schliemann C, Bieker R, Thoennissen NH, Burow K, Kramer C, et al. Bevacizumab reduces VEGF expression in patients with relapsed and refractory acute myeloid leukemia without clinical antileukemic activity. Leukemia. 2007;21:1310–2.
51 Karp JE, Gojo I, Pili R, Gocke CD, Greer J, Guo C, et al. Targeting vascular endothelial growth factor for relapsed and refractory adult acute myelogenous leukemias: therapy with sequential 1–beta-d-arabinofuranosylcytosine, mitoxantrone, and bevacizumab. Clin Cancer Res. 2004;10:3577–85.
52 Ossenkoppele GJ, Stussi G, Maertens J, van Montfort K, Biemond BJ, Breems D, et al. Addition of bevacizumab to chemotherapy in acute myeloid leukemia at older age: a randomized phase 2 trial of the Dutch-Belgian Cooperative Trial Group for Hemato-Oncology (HOVON) and the Swiss Group for Clinical Cancer Research (SAKK). Blood. 2012;120:4706–11.
53 Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, et al., TARGET Study Group: Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356:125–34.
54 Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115–24.
55 Drevs J, Müller-Driver R, Wittig C, Fuxius S, Esser N, Hugenschmidt H, et al. PTK787/ZK 222584, a specific vascular endothelial growth factor-receptor tyrosine kinase inhibitor, affects the anatomy of the tumor vascular bed and the functional vascular properties as detected by dynamic enhanced magnetic resonance imaging. Cancer Res. 2002;62:4015–22.
56 Mross K, Drevs J, Müller M, Medinger M, Marmé D, Hennig J, et al. Phase I clinical and pharmacokinetic study of PTK/ZK, a multiple VEGF receptor inhibitor, in patients with liver metastases from solid tumours. Eur J Cancer. 2005;41:1291–9.
57 Roboz GJ, Giles FJ, List AF, Cortes JE, Carlin R, Kowalski M, et al. Phase 1 study of PTK787/ZK 222584, a small molecule tyrosine kinase receptor inhibitor, for the treatment of acute myeloid leukemia and myelodysplastic syndrome. Leukemia. 2006;20:952–7.
58 Gupta P, Mulkey F, Hasserjian RP, Sanford BL, Vij R, Hurd DD, et al.; Alliance for Clinical Trials in Oncology. A phase II study of the oral VEGF receptor tyrosine kinase inhibitor vatalanib (PTK787/ZK222584) in myelodysplastic syndrome: Cancer and Leukemia Group B study 10105 (Alliance). Invest New Drugs. 2013;31:1311–20.
59 Wedge SR, Kendrew J, Hennequin LF, Valentine PJ, Barry ST, Brave SR, et al. AZD2171: a highly potent, orally bioavailable, vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for the treatment of cancer. Cancer Res. 2005;65:4389–400.
60 Drevs J, Siegert P, Medinger M, Mross K, Strecker R, Zirrgiebel U, et al. Phase I clinical study of AZD2171, an oral vascular endothelial growth factor signaling inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2007;25:3045–54.
61 Fiedler W, Mesters R, Heuser M, Ehninger G, Berdel WE, Zirrgiebel U, et al. An open-label, Phase I study of cediranib (RECENTIN) in patients with acute myeloid leukemia. Leuk Res. 2010;34:196–202.
62 Metzelder S, Wang Y, Wollmer E, Wanzel M, Teichler S, Chaturvedi A, et al. Compassionate use of sorafenib in FLT3–ITD-positive acute myeloid leukemia: sustained regression before and after allogeneic stem cell transplantation. Blood. 2009;113:6567–71.
63 Zheng R, Levis M, Piloto O, Brown P, Baldwin BR, Gorin NC, et al. FLT3 ligand causes autocrine signaling in acute myeloid leukemia cells. Blood. 2004;103:267–74.
64 Safaian NN, Czibere A, Bruns I, Fenk R, Reinecke P, Dienst A, et al. Sorafenib (Nexavar) induces molecular remission and regression of extramedullary disease in a patient with FLT3–ITD+ acute myeloid leukemia. Leuk Res. 2009;33:348–50.
65 Serve H, Krug U, Wagner R, Sauerland MC, Heinecke A, Brunnberg U, et al. Sorafenib in combination with intensive chemotherapy in elderly patients with acute myeloid leukemia: results from a randomized, placebo-controlled trial. J Clin Oncol. 2013;31:3110–8.
66 Guenounou S, Delabesse E, Récher C. Sorafenib plus all-trans retinoic acid for AML patients with FLT3–ITD and NPM1 mutations. Eur J Haematol. 2014. doi: 10.1111/ejh.12334. [Epub ahead of print]
67 Ravandi F, Arana Yi C, Cortes JE, Levis M, Faderl S, Garcia-Manero G, et al. Final report of phase II study of sorafenib, cytarabine and idarubicin for initial therapy in younger patients with acute myeloid leukemia. Leukemia. 2014;28:1543–5.
68 Singhal S, Mehta J, Desikan R, Ayers D, Roberson P, et al. Antitumor activity of thalidomide in refractory multiple myeloma. N Engl J Med. 1999;341:1565–71.
69 Barlogie B, Desikan R, Eddlemon P, Spencer T, Zeldis J, Munshi N, et al. Extended survival in advanced and refractory multiple myeloma after single-agent thalidomide: identification of prognostic factors in a phase 2 study of 169 patients. Blood. 2001;98:492–4.
70 Moncada B, Baranda ML, Gonzalez-Amaro R, Urbina R, Loredo CE. Thalidomide-effect on T cell subsets as a possible mechanism of action. Int J Lepr Other Mycobact Dis. 1985; 53:201–5.
71 D’Amato RJ, Loughnan MS, Flynn E, Folkman J. Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci U S A. 1994;91:4082–5.
72 Teo SK. Properties of thalidomide and its analogues: implications for anticancer therapy. AAPS J. 2005, 7:E14–19.
73 Dredge K, Marriott JB, Macdonald CD, Man HW, Chen R, Muller GW, et al. Novel thalidomide analogues display antiangiogenic activity independently of immunomodulatory effects. Br J Cancer. 2002;87:1166–72.
74 Thomas DA, Estey E, Giles FJ, Faderl S, Cortes J, Keating M, et al. Single agent thalidomide in patients with relapsed or refractory acute myeloid leukaemia. Br J Haematol. 2003;123:436–41.
75 Steins MB, Padró T, Bieker R, Ruiz S, Kropff M, Kienast J, et al. Efficacy and safety of thalidomide in patients with acute myeloid leukemia. Blood. 2002;99:834–9.
76 Fehniger TA, Byrd JC, Marcucci G, Abboud CN, Kefauver C, Payton JE, et al. Single-agent lenalidomide induces complete remission of acute myeloid leukemia in patients with isolated trisomy 13. Blood. 2009;113:1002–5.
77 Sekeres MA, Gundacker H, Lancet J, Advani A, Petersdorf S, Liesveld J, et al. A phase 2 study of lenalidomide monotherapy in patients with deletion 5q acute myeloid leukemia: Southwest Oncology Group Study S0605. Blood. 2011;118:523–8.
78 Shin DH, Chun YS, Lee DS, Huang LE, Park JW. Bortezomib inhibits tumor adaptation to hypoxia by stimulating the FIH-mediated repression of hypoxia-inducible factor-1. Blood. 2008;111:3131–6.
79 Williams S, Pettaway C, Song R, Papandreou C, Logothetis C, McConkey DJ. Differential effects of the proteasome inhibitor bortezomib on apoptosis and angiogenesis in human prostate tumor xenografts. Mol Cancer Ther. 2003;2:835–43.
80 Roccaro AM, Hideshima T, Raje N, Kumar S, Ishitsuka K, Yasui H, et al. Bortezomib mediates antiangiogenesis in multiple myeloma via direct and indirect effects on endothelial cells. Cancer Res. 2006;66:184–91.
81 Attar EC, Johnson JL, Amrein PC, Lozanski G, Wadleigh M, DeAngelo DJ, et al. Bortezomib added to daunorubicin and cytarabine during induction therapy and to intermediate-dose cytarabine for consolidation in patients with previously untreated acute myeloid leukemia age 60 to 75 years: CALGB (Alliance) study 10502. J Clin Oncol. 2013;31:923–9.
82 Kumar S, Witzig TE, Timm M, Haug J, Wellik L, Fonseca R, et al. Expression of VEGF and its receptors by myeloma cells. Leukemia. 2003;17:2025–31.
83 Munshi NC, Wilson C. Increased bone marrow microvessel density in newly diagnosed multiple myeloma carries a poor prognosis. Semin Oncol. 2001;28:565–9.
84 Podar K, Tai YT, Davies FE, Lentzsch S, Sattler M, Hideshima T, et al. Vascular endothelial growth factor triggers signaling cascades mediating multiple myeloma cell growth and migration. Blood. 2001;98:428–35.
85 Podar K, Anderson KC. The pathophysiological role of VEGF in hematological malignancies: Therapeutic implications. Blood. 2005;105:1383–95.
86 Barlogie B, Desikan R, Eddlemon P, Spencer T, Zeldis J, Munshi N, et al. Extended survival in advanced and refractory multiple myeloma after single-agent thalidomide: identification of prognostic factors in a phase 2 study of 169 patients. Blood. 2001;98:492–4.
87 Rajkumar SV, Blood E, Vesole D, Fonseca R, Greipp PR; Eastern Cooperative Oncology Group. Phase III clinical trial of thalidomide plus dexamethasone compared with dexamethasone alone in newly diagnosed multiple myeloma: a clinical trial coordinated by the Eastern Cooperative Oncology Group. J Clin Oncol. 2006;24:431 6.
88 Dimopoulos M, Spencer A, Attal M, Prince HM, Harousseau JL, Dmoszynska A, et al.; Multiple Myeloma (010) Study Investigators. Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma. N Engl J Med. 2007;357:2123–32.
89 Weber DM, Chen C, Niesvizky R, Wang M, Belch A, Stadtmauer EA, et al.; Multiple Myeloma (009) Study Investigators. Lenalidomide plus dexamethasone for relapsed multiple myeloma in North America. N Engl J Med. 2007;357:2133–42.
90 De Luisi A, Ferrucci A, Coluccia AM, et al. Lenalidomide restrains motility and overangiogenic potential of bone marrow endothelial cells in patients with active multiple myeloma. Clin Cancer Res. 2011;17:1935–46.
91 Somlo G, Lashkari A, Bellamy W, Zimmerman TM, Tuscano JM, O’Donnell MR, et al. Phase II randomized trial of bevacizumab versus bevacizumab and thalidomide for relapsed/refractory multiple myeloma: a California Cancer Consortium trial. Br J Haematol. 2011;154:533–5.
92 White D, Kassim A, Bhaskar B, Yi J, Wamstad K, Paton VE. Results from AMBER, a randomized phase 2 study of bevacizumab and bortezomib versus bortezomib in relapsed or refractory multiple myeloma. Cancer. 2013;119:339–47.
93 Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, et al. A gain-of-function mutation of JAK2 in myeloproliferative isorders. N Engl J Med. 2005;352:1779–90.
94 Kralovics R, Teo SS, Li S, Theocharides A, Buser AS, Tichelli A, et al. Acquisition of the V617F mutation of JAK2 is a late genetic event in a subset of patients with myeloproliferative disorders. Blood. 2006;108:1377–80.
95 Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369:2379–90.
96 Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369:2391–405.
97 Lundberg LG, Lerner R, Sundelin P, Rogers R, Folkman J, Palmblad J. Bone marrow in polycythemia vera, chronic myelocytic leukemia, and myelofibrosis has an increased vascularity. Am J Pathol. 2000;157:15–19.
98 Gianelli U, Vener C, Raviele PR, Savi F, Somalvico F, Calori R, et al. VEGF expression correlates with microvessel density in Philadelphia chromosomenegative chronic myeloproliferative disorders. Am J Clin Pathol. 2007;128:966–73.
99 Sozer S, Fiel MI, Schiano T, Xu M, Mascarenhas J, Hoffman R. The presence of JAK2V617F mutation in the liver endothelial cells of patients with Budd-Chiari syndrome. Blood. 2009;113:5246–9.
100 Giles FJ, List AF, Carroll M, Cortes JE, Valickas J, Chen BL, et al. PTK787/ZK 222584, a small molecule tyrosine kinase receptor inhibitor of vascular endothelial growth factor (VEGF), has modest activity in myelofibrosis with myeloid metaplasia. Leuk Res. 2007;31:891–7.
101 Thomas DA, Giles FJ, Albitar M, Cortes JE, Verstovsek S, Faderl S, et al. Thalidomide therapy for myelofibrosis with myeloid metaplasia. Cancer. 2006;106:1974–84.
102 Abgrall JF, Guibaud I, Bastie JN, Flesch M, Rossi JF, Lacotte-Thierry L, et al.; Groupe Ouest-Est Leucémies et Maladies du Sang (GOELAMS). Thalidomide versus placebo in myeloid metaplasia with myelofibrosis: a prospective, randomized, double-blind, multicenter study. Haematologica. 2006;91:1027–32.
103 Tefferi A, Cortes J, Verstovsek S, Mesa RA, Thomas D, Lasho TL, et al. Lenalidomide therapy in myelofibrosis with myeloid metaplasia. Blood. 2006;108:1158–64.
104 Quintás-Cardama A, Kantarjian HM, Manshouri T, Thomas D, Cortes J, Ravandi F, et al. Lenalidomide plus prednisone results in durable clinical, histopathologic, and molecular responses in patients with myelofibrosis. J Clin Oncol. 2009;27:4760–6.