Toxic oligomer species of amyloid-β in Alzheimer's disease, a timing issue
DOI: https://doi.org/10.4414/smw.2014.14021
Summary
A decade following the paradigm-shifting concept that endogenous forms of soluble, non-fibrillar amyloid-β (Aβ) might constitute the major bioactive entity causing synaptic loss and cognitive decline in Alzheimer’s disease (AD), our understanding of these oligomeric species still remains conspicuously superficial. The current lack of direct evaluation tools for each endogenous Aβ oligomer hampers our ability to readily address crucial question such as: (i) where they form and accumulate?; (ii) when they first appear in human brains and body fluids?; (iii) what is the longitudinal expression of these putative toxins during the course of the disease?; (iv) and how do these soluble Aβ assemblies alter synaptic and neuronal function in the brain? Despite these limitations, indirect ex vivo measurement and isolation from biological specimens has been possible and have allowed parsing out intrinsic differences between putative endogenous Aβ oligomers. In this review, I integrated recent findings and extrapolated emerging hypotheses derived from these studies with the hope to provide a clarified view on the putative role of endogenous Aβ oligomers in AD, with a particular emphasis on the timing at which these soluble species might act in the aging and diseased brain.
Introduction
Many neurodegenerative diseases such as Alzheimer's disease (AD), Parkinson's disease or frontotemporal dementias are characterised by the presence of abnormal protein aggregates in the brain. In the case of AD, amyloid plaques (also called senile or neuritic) and neurofibrillary tangles are the two neuropathological lesions that characterise this neurological disorder in postmortem human brain tissue [1]. These lesions are respectively formed of insoluble fibrillar species of the amyloid-β peptide (Aβ) and the protein tau. While these deposited aggregates have been suspected to be the cause of AD for nearly a century, accumulating evidence over the past twelve years seems to suggest that soluble, non-fibrillar multimeric forms of these proteins might initiate the synaptic and neuronal dysfunction associated with the disease [2–11]. Generally, these molecular species of Aβ are referred to as Aβ oligomers (oAβ). Species composed of two (dimers), three (trimers) or four (tetramers) Aβ monomers are often placed under the umbrella term low molecular weight or low-n oAβ while larger assemblies are termed high molecular weight or high-n Aβ oligomers [12, 13].
Due to the dynamic nature of these oligomeric forms and the lack of specific molecular tools specific to each assembly, it has proven difficult to study oAβ present in human brain tissue, in brains of transgenic mice modeling AD and in cellular systems derived from these animal models. Despite this apparent obstacle and the accompanying ongoing debate surrounding the aforementioned points, several research groups have contributed to the reliable identification of several specific oAβ species by combining immunological and biochemical approaches developed for this purpose: dimers [5, 9, 10, 14], trimers [7, 9, 11, 14], Aβ*56 a possible dodecamer composed of twelve molecules Aβ [11, 14] and annular protofibrils [15, 16]. As a direct consequence of the identification of several oligomeric Aβ species, the hypothetical pathological cascade of events leading to the development of AD has exponentially increased in its complexity [12, 17]. In addition, multiple lines of evidence recently summarised [18–20] suggest that tau is mediating the deleterious effects of oAβ, and that abnormal changes in tau biology and subcellular localisation might be responsible for neuronal dysfunction and cognitive decline [21–23].
This paradigm shift also prompted the field to tackle questions that need to be answered in order to further improve our understanding of the series of events that might underlie AD pathogenesis. These questions are: [1] what is (are) the role(s) of Aβ oligomers in the pathogenesis of the disease?; [2] how do these molecular entities disrupt synaptic and neuronal homeostasis?; [3] when are Aβ oligomers active during aging, disease onset and progression? A recently published review from our group tried to summarise the existing knowledge on the production and toxicity of endogenous forms of Aβ oligomers [12]. This previous review is recommended for those interested in these aspects related to oAβ, as they will not be detailed in this review. Instead, the current article focuses on the third question aforementioned above, by integrating observations from experimental and human studies with a specific emphasis on the timing of the variation in oAβ brain levels and how they might provide clues about the biological cascade(s) involved in AD pathophysiology. In doing so, it is important to keep in mind, as a word of caution, that the comparisons made between mouse and human data in this review might be confounded by inherent differences in postmortem interval delays between animal and human studies (5.11 ± 2.26 hours, N = 88 in our ROS cohort [14]).
What are endogenous oligomeric forms of Aβ?
This term is used here to describe sodium dodecyl sulfate (SDS)-resistant multimeric Aβ assemblies that can be reliably detected in brain or cell protein extracts by combining size-exclusion chromatography coupled with gel electrophoresis and multiple antibodies detecting various domains of Aβ or various conformations of Aβ [11, 16, 24, 25]. The combined use of both approaches is thought to minimise the likelihood of mistakenly assigning the term oligomer to a molecule that might in fact correspond to a cleavage product of the amyloid protein precursor (APP) or to a heteromeric complex between Aβ and another protein. Additionally, concerns have emerged regarding the type of solvent used to extract proteins in studies of oAβ by independent groups. This review will not concentrate on how oAβ were obtained, as previous work has shown that both water- and detergent-soluble Aβ oligomers can be detected in mouse and human brain lysates [11, 14, 26, 27].
It is worth reiterating that the existence of these “endogenous” oAβ will only be validated when specific detection tools raised against each form of interest are generated and available to the community. Nonetheless, the generation of conformation-dependent antibodies pioneered by Charles Glabe and his group has led to the proposed classification of these oligomeric forms of Aβ into two large families [28]. For the purpose of this review, these families are referred to as pre-fibrillar and non-fibrillar assemblies (fig. 1). Pre-fibrillar oAβ are multimeric assemblies that lead to the formation of Aβ fibrils while non-fibrillar oAβ are not able to form fibrils. Of note, the exact structural motifs and quaternary structures of these Aβ oligomers are unknown and will require additional studies.
Aβ dimers as the molecular building block for pre-fibrillar assemblies?
With these caveats explicitly enunciated, numerous groups have focused their efforts on studying dimeric Aβ species. There are multiple reasons for this overall particular interest: [1] Aβ dimers appear to be stable in the presence of detergent such as SDS, [2] the abundance of Aβ dimers elevates with aging and amyloid burden in brains of transgenic mice used as models of AD and in patients with AD [3, 5, 6, 12] due to this remarkable increase (~20–fold in 26–month-old Tg2576 vs. 10–month-old animals [12]), it is fairly straight forward to detect and measure Aβ dimers in brain lysates from transgenic mouse and human brains [5, 12, 14, 29], and [4] Aβ dimers separated by size exclusion chromatography from AD brain lysates alter the cellular processes of long-term potentiation (LTP) and depression (LTD), and induce cognitive deficits in rodents when infused into the brain [2, 4, 5, 8].
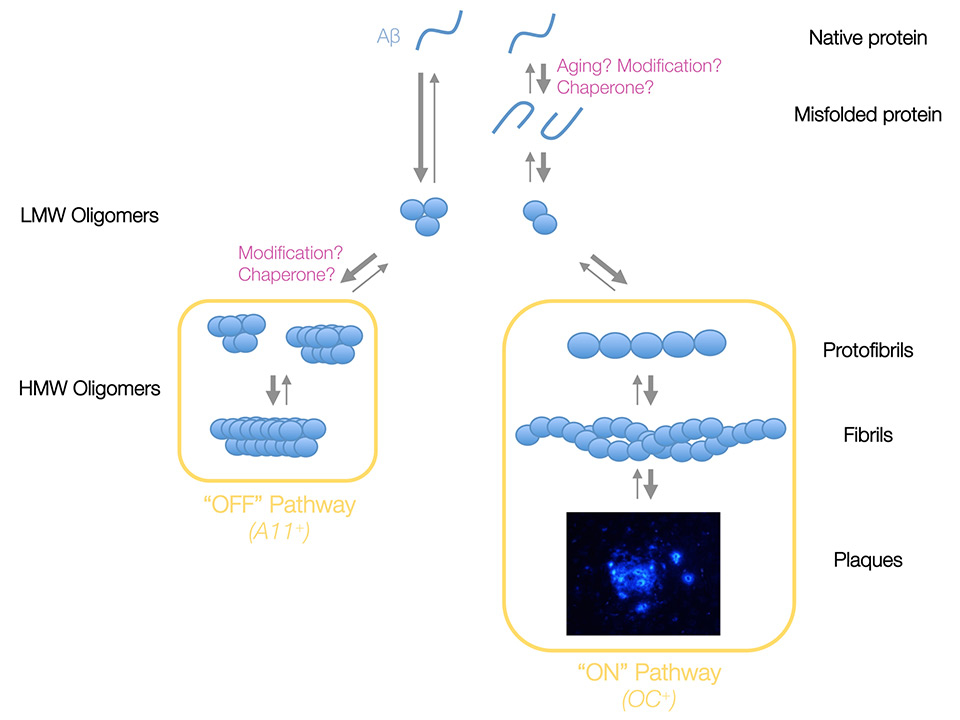
Figure 1
Proposed model of Aβ aggregation. In this model, natively folded Aβ monomers preferentially form Aβ trimers under physiological conditions, whereas dimers are formed following the misfolding of Aβ monomers induced by aging, post-translational modification of the primary sequence of Aβ or a chaperone protein. Both Aβ assemblies constitute the pool of low molecular weight (LMW) or low-n oAβ from which two pathways emerge. The “Off” pathway does not lead to the formation of insoluble fibrillar amyloid and is characterised by its immunoreactivity to the conformation-specific oligomer antibody A11. By contrast, the “On” pathway promotes the formation of fibrillar Aβ and molecules involved in this pathway are immunopositive to the conformation-specific fibril antibody OC. Once a species of Aβ has entered one of these paths, it cannot effectuate a horizontal transfer into the opposite pathway but instead it must de disassembled back into a monomeric state. The thickness of arrows illustrates the favoured direction of the putative equilibrium suggested by integrated experimental observations.
In transgenic mouse models of AD such as Tg2576 or J20 in which Aβ deposition is not very aggressive [30–32], Aβ dimers can be measured in soluble extracts of brain proteins around 10–14 months depending on gender and line. It is puzzling that these ages also correspond to the period of increased amyloid deposition in neocortical areas, perhaps hinting at a link between brain abundance of Aβ dimers and fibrillar forms of Aβ [12, 29, 33]. Consistent with this observation, Aβ dimers can be detected at earlier ages (~7–8 months) in APP transgenic mice displaying more aggressive amyloid deposition such as APP/PS1 or APP23 mice (Lesné et al., unpublished data). In another genetically engineered mouse line, Aβ dimers appear to be the major species formed in mice overexpressing the E693Δ mutant of APP [34]. Identified in 2008, this deletion in the middle region of Aβ preferentially engages the peptide into forming oligomers instead of fibrils [34, 35]. Given the panel of antibodies used to identify the putative oligomeric Aβ assemblies, apparent Aβ dimers were readily detected in TgAPPE693Δ brain tissue. Based on the biochemical segregation of the putative Aβ trimers detected in 1% Triton X-100 lysates and in 2% SDS lysates, the pair of antibodies used to detect Aβ peptides in this study (6E10 immunoprecipitation followed by western blotting using the rabbit polyclonal β001 antibody) and a slight upward shift in the electrophoretic migration compared to recombinant Aβ standards, it is very likely that the immunoreactive molecule labeled as Aβ trimers might in fact correspond to the carboxyl terminal fragment of APP generated by the β-secretase activity, CTF-β. If this assessment were correct, this would indicate that Aβ trimers are not readily detected in this mouse line. Furthermore, this would also suggest that the E693Δ mutation is somehow stabilising low-n prefibrillar Aβ oligomers such as Aβ dimers thereby preventing their aggregation into fibrils. This hypothesis is consistent with the exclusive segregation of AβE693Δ dimers in the insoluble protein fraction extracted with formic acid [34]. Interestingly, these oligomeric Aβ species were immunoreactive to the antibody NU-1 previously shown to detect diffuse and dense-core deposits in human brain tissue [36] labeled with the amyloid-binding dye thioflavin S, further suggesting that these oAβ are prefibrillar.
In studies using postmortem human brain specimens, soluble dimeric forms of Aβ seem to be detected in brains of subjects with abundant amyloid loads [5, 14]. This observation is further supported by the examination of the relative oAβ levels with respect to neuritic plaque pathology as assessed by the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) criteria. As shown in fig. 2A, CERAD scoring tracked well with amyloid loads detected in the brain region analysed (inferior temporal gyrus or ITG). Regarding oAβ, ITG Aβ dimers were most abundant in subjects with CERAD scores of 1 (Definite AD) or 2 (Probable AD) compared to those with a CERAD 3 (Possible AD) or 4 (No AD) (fig. 2C). It is worth noting that putative ITG Aβ trimers and Aβ*56 levels did not follow this pattern (fig. 2, D-F), suggesting that Aβ dimers and amyloid plaques could be interconnected in the brain.
Despite this noticeable relationship, the origin of dimeric Aβ species remains mysterious. Previous studies have revealed that Tg2576 cortical neurons maintained in culture produce mostly trimers and monomers while dimers of Aβ are produced and secreted in very small amounts (1:20 to 1:15 relative to Aβ monomers and trimers respectively) [11, 37]. These results contrast sharply with the relative abundance of putative Aβ dimers detected in the conditioned medium of 7PA2 cells, a stable line of Chinese hamster ovary cells overexpressing a mutant form of APP [9, 38]. Perhaps explaining this discrepancy, a recent study from the Blennow group indicated that part of the immunoreactivity of these apparent Aβ dimers in these cell lines might correspond instead to truncated Aβ peptides containing part of the APP molecule upstream of the β-secretase site [39]. This finding was recently confirmed and the authors additionally showed that these N-terminally extended (NTE) Aβ species can also impair in vivo LTP [40]. Although future work will evaluate the physiological relevance of these NTE Aβ molecules, this report also validated the presence of synaptotoxic Aβ homodimers in this cell line [40].
Consistent with recent work using artificially modified synthetic Aβ dimers [41, 42], these independent results support the notion that endogenous Aβ dimers could constitute the molecular building block for fibrillar forms of Aβ in vivo.
Aβ trimers as the molecular building block for non-fibrillar assemblies?
Although the detection of Aβ dimers in brain tissue appears to coincide with elevated amyloid loads in transgenic mouse and human brain [12, 14], this does not seem to be the case for endogenous Aβ trimers [14]. These putative assemblies are defined as such based on: (i) the ability to immunocapture these molecules with 6E10, 4G8, 82E1, 40/42–end specific Aβ antibodies from conditioned media of APP transgenic cultured neurons or brain lysates enriched in extracellular proteins and (ii) the ability to segregate them from other Aβ species by SEC [10, 11, 14, 37]. If this hypothesis were correct, Aβ trimers could therefore constitute the molecular building block for non-fibrillar Aβ oligomers. What then are the elements that support this argument?
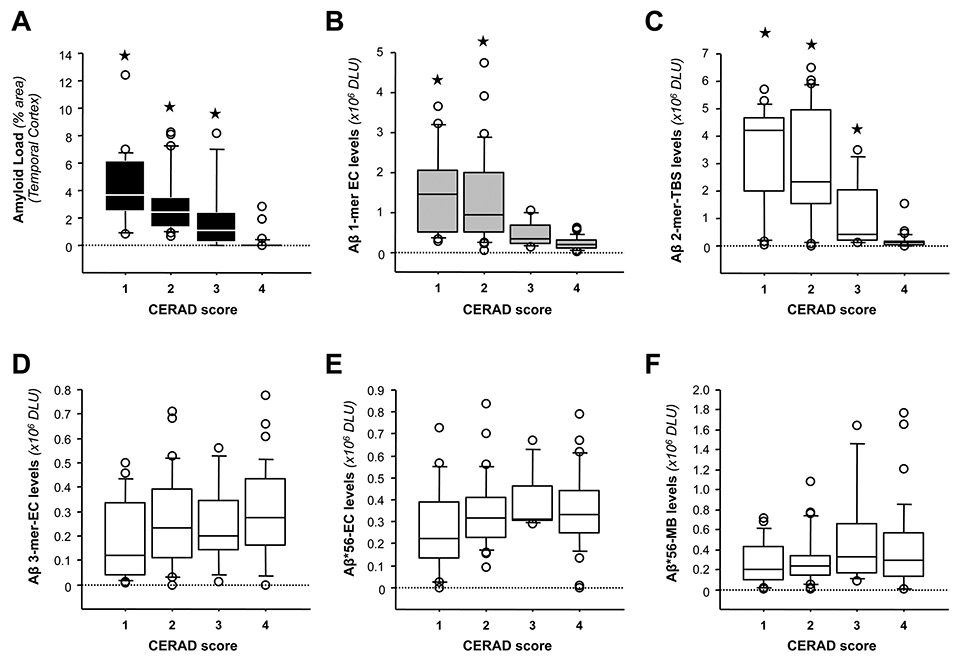
Figure 2
Relationships between specific Aβ oligomers in the brain and CERAD scores. (A-B) Relationships between CERAD scores and amyloid load in temporal cortex (A) and soluble Aβ monomers detected in EC fractions (B). CERAD scores: 1, Definite AD; 2, Probable AD; 3, Possible AD and 4, No AD. (C–F) Relationships between CERAD scores and the levels of various oligomeric Aβ species, including Aβ dimers in TBS extracts (C), Aβ trimers in EC fractions (D), and Aβ*56 in EC (E) and MB (F) lysates. (★
p <0.05 vs. the CERAD score 4 group using Kruskal-Wallis test followed by Mann-Whitney U test with Bonferroni corrections). Abbreviations: EC: extracellular-enriched, MB: membrane-enriched; TBS: Tris Buffered Saline; DLU = densitometry light units.)
Firstly, Aβ trimers appear to be by far the most abundant oligomeric Aβ species produced and secreted by primary cultured neurons [11, 37]. Interestingly, we did not observe the presence of Aβ assemblies larger than a putative tetramer (~18kDa) in near-pure or mixed cultures of mouse cortical neurons.
Secondly, Aβ trimers are readily detected at low levels as early as 14 days of gestation in brain tissue of Tg2576 mice [11] and at 1 year of age in post-mortem human brain tissue [14]. Both in mouse and human brain, the abundance of Aβ trimers increased very slowly but steadily with aging [11]. Obviously, the presence of these putative Aβ trimers was detected months or decades prior to Aβ deposition in APP transgenic mouse and human brain tissue respectively [12]. This apparent dissociation in time between Aβ trimers and amyloid plaque formation proves difficult to reconcile with the hypothesis that Aβ trimers are pre-fibrillar Aβ oligomers.
Thirdly, brain levels of Aβ trimers did not correlate with amyloid burden (Spearman rho = -0.2682, p >0.05, N = 85) in our human studies [14]. By contrast, brain levels of Aβ dimers strongly correlated with amyloid loads (Spearman rho = 0.68, p <0.0001, N = 85).
Fourth, the disassembly of Aβ oligomers present in 13–month-old Tg2576 mice induced by the solvent hexafluoroisopropanol (HFIP) led to a decrease in 6E10–immnoreactive bands consistent with putative hexamers, nonamers and dodecamers and a parallel elevation of Aβ trimers by more than 3–fold [11]. These results suggest that the 28 kDa, 42 kDa and 56 kDa Aβ species might contain or be composed of Aβ trimers.
Fifth, the detection of extracellular Aβ trimers seems to depend on Aβ production in vivo. Although Tg2576 and Tg5469 mice express comparable protein levels of transgene-derived APP, soluble SDS-Aβ levels are ~10–fold higher in Tg2576 (~20–25 pmol/g) than in Tg5469 (~2 pmol/g) mice [43, 44]. Upon analysing extracellular-enriched protein fractions from 12–month-old Tg5469 animals, no Aβ oligomers were detected while Aβ trimers were readily observed in lysates of age-matched Tg2576 animals [43].
Overall, it is clear that additional research is needed to determine whether Aβ trimers truly exist in situ, what governs the formation of Aβ trimers, where trimers are located and how trimers might alter neuronal function.
Very few studies have been performed to examine the role of Aβ trimers in AD pathogenesis [2, 7, 8, 37]. Preparations of conditioned media from 7PA2 cells enriched for putative Aβ trimers were reported to inhibit LTP in mouse hippocampal slices [7]. Intracerebroventricular injections of either 7PA2– or Tg2576–derived Aβ trimers in healthy rats were however unable to cause cognitive impairments during the alternating lever cyclic ratio (ALCR) assay despite a noticeable trend [2]. This finding might be inherent to the concentrations used in the study or to the stability of the species tested. In fact, when Aβ trimers purified from AD brain tissue were applied to cortical primary neurons at low nanomolar concentrations (1–5 nM), these assemblies were bioactive by inducing the phosphorylation of the Src kinase Fyn and its downstream target tau [37]. Overall, these observations suggest: [1] the formation of Aβ trimers in vivo appears to be dependent on Aβ production, [2] Aβ trimers are the first detectable endogenous oligomeric species produced and secreted by neurons.
Importance of trimer-based Aβ assemblies exemplified by the oligomer Aβ*56
While searching to identify the soluble molecules of Aβ associated with the onset of spatial reference memory deficit observed in young Tg2576 mice prior to the formation of amyloid plaques, the examination of brain lysates from these animals by western blotting revealed apparent trimers (molecular weight of about 13 kDa), hexamers (~27 kDa), nonamers (~40 kDa) and dodecamers (~56 kDa) using antibodies targeting the amino terminus and the central area of the Aβ peptide such as 6E10 and using the A11 antibody that detects non-fibrillar oligomeric forms of amyloid proteins [11, 25]. Of note, the OC antibody, which recognises pre-fibrillar oligomers [24], does not detect Aβ*56 (data not shown). This combined epitope specificity therefore categorises Aβ*56 as a non-fibrillar Aβ oligomer. As summarised elsewhere [12], Aβ*56 was also reported in other APP transgenic lines [33, 45, 46], in APP23 (Lesné & Ashe, unpublished data) and APPPS1 mice (Lesné, unpublished data), and in 7-month-old regulatable tTA-APP mice [47] confirming that its formation was not restricted to the Tg2576 mouse line. In these animal models, Aβ*56 was found in two soluble protein fractions, extracellular and membrane extracts [11, 45, 47, 48], suggesting that this oligomer was formed extracellularly and that it may interact with a putative surface receptor at the neuronal plasma membrane. A similar profile has been observed in recently published studies with two human cohorts (n = 135), in which Aβ*56 increased with aging starting in midlife years [14].
Contrasting with these in vivo observations, we have not been able to detect Aβ*56 in cellular protein lysates of APP transgenic primary neurons or in the conditioned media of these cells (Lesné et al., unpublished data). Based on these results, we believe that Aβ*56 is not produced and secreted by primary neurons [11, 37]. Considering that Tg2576 primary neurons generate and secrete relatively large quantities of apparent Aβ trimers [11, 37], one likely explanation as to why Aβ*56 is not formed in vitro might be due to the possibility that this multimeric form Aβ requires an unidentified cofactor or chaperone "X" in brain tissue in order to promote its formation. The obvious alternative is that the abundance of Aβ*56 in vitro lies below our detection limit (~1–2 pg of recombinant Aβ). If this cofactor X is indeed required to generate Aβ*56, aging should regulate the expression of this cofactor to explain the sudden accumulation of Aβ*56 at the age of 6 months in Tg2576 brain tissue [11] and the rise of Aβ*56 abundance in brain tissue of subjects in their mid-forties [14]. Furthermore, the relative levels of extracellular Aβ*56 in human brains showed no relation to Aβ dimers or Aβ monomers [14]. By contrast, the brain abundance of Aβ*56 was positively correlated with that of trimeric Aβ. This observation is consistent with the hypothesis that Aβ trimers are the molecular building block for larger oligomers of Aβ in the non-fibrillar pathway (fig. 1).
What is the temporal profile of these oligomeric forms of Aβ in the human brain during aging and AD?
To answer this question we analysed the expression of Aβ oligomers, which we can measure reliably and reproducibly (i.e., Aβ dimers, trimers and Aβ*56), in postmortem brain tissue of 135 subjects aged between 1 and 97 years [14]. Of these, 75 individuals had no cognitive impairment (NCI), including young children to the elderly, and 58 subjects were clinically diagnosed with mild memory impairment (MCI) or probable AD. It is important to reiterate here that it is currently believed that AD begins about two to three decades before the onset of symptoms and neuronal loss [49].
Similarly to APP transgenic mouse models of AD in which Aβ trimers were present in young mice before any memory decline [11], Aβ trimers were detected in postmortem human brain tissue of children and adolescents [14]. In combination with the observation that cultured primary neurons secrete putative Aβ trimers, the findings in our human studies suggest that Aβ trimers might be physiologically produced and secreted in the infant and adult brain. If true, trimeric Aβ could be regarded as the first step in the oligomerisation of natively-folded Aβ monomers since Aβ trimers are not immunoreactive to either A11 or OC antibodies. With aging, their relative expression gradually increased and became elevated above baseline levels in subjects over 70 years of age [14]. In the brain of subjects with mild cognitive impairment (MCI), trimeric Aβ levels seemed particularly high compared to those measured in age-matched control individuals [14]. Unexpectedly, the relative abundance of Aβ trimers diminished in postmortem AD brain tissue below the levels detected in the NCI group. Although longitudinal studies are needed to confirm these variations, we propose that these changes are suggestive of a transient, perhaps stage-specific, elevation of this Aβ oligomer [14]. We also suggest that the apparent decrease in the brain abundance of Aβ trimers in AD compared to NCI might indicate a switch in the On/Off pathway balance (fig. 1) in favour of prefibrillar Aβ species, including Aβ dimers (see below).
Contrasting with Aβ trimers, brain levels of Aβ*56 initially appeared below the range of detection in our assay (~1–2 pg of Aβ) in children and young adults but, unlike Aβ trimers, abnormally increased in subjects in their forties to culminate in subjects over 80 years of age [14]. This elevation coincided with the onset of subtle cognitive decline linked to aging [50, 51]. It is also interesting to note that this age-dependent elevation of Aβ*56 levels exceeded the respective abundance of Aβ trimers, suggesting that the formation of Aβ*56 is favoured to the detriment of trimeric Aβ species (pending that Aβ*56 is a trimer-based oAβ). However, to our initial surprise, soluble or membrane-bound Aβ*56 levels decreased in what seemed a step-wise fashion in MCI and AD [14]. Since the levels of Aβ*56 decreased while Aβ trimers increased in MCI brain tissue, we propose that this change reflects a possible reversal of the trimer-Aβ*56 equilibrium. With disease progression, the relative brain abundance of both Aβ*56 and Aβ trimers returned to baseline or lowered below baseline suggesting a possible collapse of the “Off pathway” or trimer-based oligomerisation pathway (fig. 1). If this scenario truly exists, it could allow for a greater pool of soluble Aβ potentially available for sustaining or exacerbating the “On pathway” or dimer-based oligomerisation pathway.
Conversely, Aβ dimers were not detected in brain tissue of individuals younger than 60 years of age. At older ages, their abundance increased strongly in parallel with the accumulation of amyloid plaques [14]. Although the magnitude of the change was lessened compared to the effect observed across aging, we found that Aβ dimers increased in AD compared to MCI and NCI groups [14] consistent with previous observations [5].
Toxicity of discrete brain Aβ oligomers
To my knowledge, very little is known about the toxicity of Aβ oligomers purified from brain tissue. While numerous reports have observed the presence of putative oAβ in brain lysates [12], only two studies have attempted to determine the effects on signaling of discrete oAβ species immunopurified from AD brain tissue [3, 37]. In the first study, Jin and colleagues demonstrated that application of apparent Aβ dimers at ~0.5 nM could induce tau hyperphosphorylation and neurotoxicity in primary hippocampal neurons after 3 days [3]. Similarly, our own group demonstrated that apparent Aβ dimers selectively induced the hyperphosphorylation of tau on tyrosine 18 following the formation of a signaling complex containing the cellular prion protein PrPC and the protein kinase Fyn [37] previously associated to neuronal death induced by Aβ [52]. Neither Aβ*56 nor protofibrillar Aβ species of molecular weight approaching 150 kDa were able to activate Fyn in primary neurons [37] suggesting that each form of Aβ oligomers might induce very specific effects in brain cells.
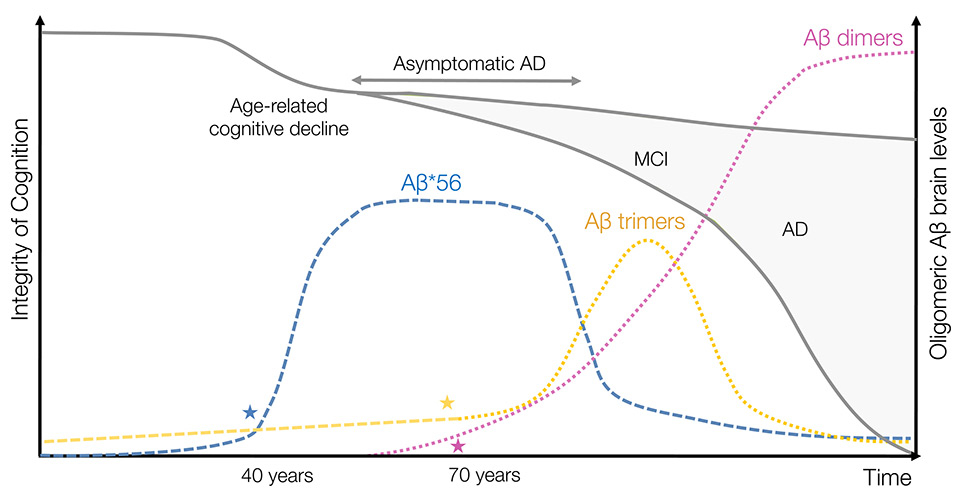
Figure 3
Revised model of endogenous oligomeric Aβ brain levels in aging and Alzheimer’s disease. In this model limited to three readily detectable oAβ in human brain tissues (i.e., Aβ dimers, trimers and Aβ*56), Aβ*56 is the first assembly to rise abnormally in midlife coinciding with subtle age-related cognitive decline although Aβ trimers are the first soluble species expressed by neurons. This scenario implies that energetic requirements for forming Aβ*56 are relatively low. The high expression levels of Aβ*56 in the brain are sustained over decades possibly contributing to early disruption in neuronal physiology and to the long silent asymptomatic phase of AD. Concomitant to a lowering of brain levels of Aβ*56, Aβ trimers accumulate transiently during the initial stage of the symptomatic phase of AD, MCI. This sequence implies that Aβ trimers are indeed the molecular building block of Aβ*56 and that the putative cofactor responsible for sustaining Aβ*56 production declines with disease progression/aging. With the emerging accumulation of prefibrillar and fibrillar Aβ assemblies, brain levels of “trimer-based” oligomers (i.e., Aβ*56 and Aβ trimers) decline with disease progression. In this setting, Aβ dimers contribute to the local toxicity in the vicinity of amyloid plaques, finishing off neurons that might have resisted the toxic waves induced by Aβ*56 and Aβ trimers. Stars (★) indicate the ages at which statistically significant changes in the respective levels of oAβ were observed.
Consistent with this hypothesis, we found strong positive correlations between Aβ*56 and two pathological forms of tau protein (hyperphosphorylated tau at serine 202 and tau conformers immunoreactive with Alz50), and negative correlations between Aβ*56 and two postsynaptic proteins (drebrin and fyn) in individuals without apparent memory problems for their age category [53]. By contrast, no association was found between Aβ dimers or Aβ trimers and these same tau changes. These observations suggest that Aβ*56 could induce abnormal changes of tau and neuronal connections early in the pathogenesis of AD. Studies are ongoing to identify the molecular mechanism induced by Aβ*56 and to confirm whether these effects on tau are also observed in mice and in primary neurons.
This obvious scarcity in available data surrounding this topic is extremely problematic and additional research studies are needed to decipher the role of oAβ endogenous to brain tissues.
Hypothetical model of the timing of oligomeric forms of Aβ
Altogether, these observations posit that the human brain might sustain the attack of (at least) three successive waves of exposure to Aβ oligomers (i.e., Aβ*56, Aβ trimers and Aβ dimers) during aging and AD (fig. 3). In this model, Aβ*56 would be the main initiator toxin of the first wave during midlife, leading to abnormal changes in tau protein but without causing neuronal death, possibly through compensatory cellular mechanisms. The role of Aβ*56 could be envisioned as priming the cells for a secondary insult, rendering them more sensitive to the exposure of additional toxins. This model could explain a long silent asymptomatic phase of the disease. The second toxic wave would be mediated by exposure to a rapid accumulation of Aβ trimers around neurons already stressed by Aβ*56, triggering additional pathological changes of tau molecules. In this scenario, neurons exposed to the first two waves could no longer avoid cellular death, likely due to the depletion in resources required to maintain compensatory mechanisms. I propose that this phase would coincide with mild cognitive impairment, the preclinical phase of AD, and a nascent neuronal atrophy in sensitive brain regions. The third and final wave of exposure to Aβ oligomers would be induced by Aβ dimers, which would act as executioner species in this model. Due to the apparent relationship between Aβ dimers and dense-core amyloid plaques, these assemblies of Aβ would induce additional and specific changes of the tau protein and lead to neuronal death in the vicinity of amyloid deposits.
To conclude, if these soluble Aβ species prove to be the major bioactive toxins initiating the synaptic and neuronal loss associated with Alzheimer's disease, I believe that decoding the precise molecular role of each oAβ must constitute a priority. Armed with this new knowledge, we as a field should be in a better position to conceive and develop novel therapeutic strategies aimed at countering cellular processes activated by these Aβ oligomers. In this context, taking one or multiple drugs at specific ages/clinical phases of the disease might prevent, limit or stop its progression.
Acknowledgement: We thank Dr. David Bennett and the participants of the Religious Orders Study.
References
1 Serrano-Pozo A, Frosch MP, Masliah E & Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harbor perspectives in medicine. 2011;1:a006189, doi:10.1101/cshperspect.a006189.
2 Reed MN, et al. Cognitive effects of cell-derived and synthetically derived Abeta oligomers. Neurobiol Aging. 2011;32:1784–94, doi:10.1016/j.neurobiolaging.2009.11.007.
3 Jin M, et al. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci U S A. 2011;108:5819–24, doi:10.1073/pnas.1017033108.
4 Li S, et al. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62:788–801, doi:10.1016/j.neuron.2009.05.012.
5 Shankar GM, et al. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med 2008;14:837–42, doi:10.1038/nm1782.
6 Shankar GM, et al. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–75, doi:10.1523/JNEUROSCI.4970–06.2007.
7 Townsend M, Shankar GM, Mehta T, Walsh DM & Selkoe DJ. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol. 2006;572:477–92, doi:10.1113/jphysiol.2005.103754.
8 Cleary JP, et al. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84, doi:10.1038/nn1372.
9 Walsh DM, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–39, doi:10.1038/416535a.
10 Walsh DM, Tseng BP, Rydel RE, Podlisny MB & Selkoe DJ. The oligomerization of amyloid beta-protein begins intracellularly in cells derived from human brain. Biochemistry. 2000;39:10831–9.
11 Lesne S, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–7, doi:10.1038/nature04533.
12 Larson ME & Lesne SE. Soluble Abeta oligomer production and toxicity. J Neurochem. 2012;120:Suppl 1, 125–39, doi:10.1111/j.1471–4159.2011.07478.x.
13 Walsh DM & Selkoe DJ. A beta oligomers – a decade of discovery. J Neurochem. 2007 ;101:1172–84, doi:10.1111/j.1471–4159.2006.04426.x.
14 Lesne SE, et al. Brain amyloid-beta oligomers in ageing and Alzheimer's disease. Brain. 2013;136:1383–98, doi:10.1093/brain/awt062.
15 Lasagna-Reeves CA, Glabe CG & Kayed R. Amyloid-{beta} Annular Protofibrils Evade Fibrillar Fate in Alzheimer Disease Brain. J Biol Chem. 2011;286:22122–30, doi:10.1074/jbc.M111.236257.
16 Kayed R, et al. Annular protofibrils are a structurally and functionally distinct type of amyloid oligomer. J Biol Chem. 2009;284:4230–7, doi:10.1074/jbc.M808591200.
17 Benilova I, Karran E & De Strooper B. The toxic Abeta oligomer and Alzheimer's disease: an emperor in need of clothes. Nat Neurosci. 2012;15:349–57, doi:10.1038/nn.3028.
18 Ittner LM & Gotz. J. Amyloid-beta and tau--a toxic pas de deux in Alzheimer's disease. Nat Rev Neurosci. 2011;12:65–72, doi:10.1038/nrn2967.
19 Huang Y & Mucke, L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148:1204–22, doi:10.1016/j.cell.2012.02.040.
20 Morris M, Maeda S, Vossel K & Mucke L. The many faces of tau. Neuron. 2011;70:410–26, doi:10.1016/j.neuron.2011.04.009.
21 Ittner LM, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–97, doi:10.1016/j.cell.2010.06.036.
22 Roberson ED, et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–4, doi:10.1126/science.1141736.
23 Hoover BR, et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68:1067–81, doi:10.1016/j.neuron.2010.11.030.
24 Glabe CG & Kayed R. Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology. 2006;66:S74–8, doi:10.1212/01.wnl.0000192103.24796.42.
25 Kayed R, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–9, doi:10.1126/science.1079469.
26 McDonald JM, Cairns NJ, Taylor-Reinwald L, Holtzman D & Walsh DM. The levels of water-soluble and triton-soluble Abeta are increased in Alzheimer's disease brain. Brain research. 2012;1450:138–47, doi:10.1016/j.brainres.2012.02.041.
27 Shankar GM, Welzel AT, McDonald JM, Selkoe DJ & Walsh DM. Isolation of low-n amyloid beta-protein oligomers from cultured cells, CSF, and brain. Methods Mol Biol. 2011;670:33–44, doi:10.1007/978–1–60761–744–0_3.
28 Glabe CG. Structural classification of toxic amyloid oligomers. J Biol Chem. 2008;283:29639–43, doi:10.1074/jbc.R800016200.
29 Shankar GM, et al. Biochemical and immunohistochemical analysis of an Alzheimer's disease mouse model reveals the presence of multiple cerebral Abeta assembly forms throughout life. Neurobiol Dis. 2009;36:293–302, doi:10.1016/j.nbd.2009.07.021.
30 Mucke L, et al. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–8.
31 Hsiao K, et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102.
32 Kawarabayashi T, et al. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci. 2001;21:372–81.
33 Meilandt WJ, et al. Neprilysin overexpression inhibits plaque formation but fails to reduce pathogenic Abeta oligomers and associated cognitive deficits in human amyloid precursor protein transgenic mice. J Neurosci. 2009;29:1977–86, doi:10.1523/JNEUROSCI.2984–08.2009.
34 Tomiyama T, et al. A mouse model of amyloid beta oligomers: their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. J Neurosci. 2010;30:4845–56, doi:10.1523/JNEUROSCI.5825–09.2010.
35 Tomiyama T, et al. A new amyloid beta variant favoring oligomerization in Alzheimer's-type dementia. Ann Neurol. 2008;63:377–87, doi:10.1002/ana.21321.
36 Lambert MP, et al. Monoclonal antibodies that target pathological assemblies of Abeta. J Neurochem. 2007;100:23–35, doi:10.1111/j.1471–4159.2006.04157.x.
37 Larson M, et al. The complex PrP(c)-Fyn couples human oligomeric Abeta with pathological tau changes in Alzheimer's disease. J Neurosci. 2012;32:16857–71a, doi:10.1523/JNEUROSCI.1858–12.2012.
38 Podlisny MB, et al. Oligomerization of endogenous and synthetic amyloid beta-protein at nanomolar levels in cell culture and stabilization of monomer by Congo red. Biochemistry. 1998;37:3602–11, doi:10.1021/bi972029u.
39 Portelius E, et al. Mass spectrometric characterization of amyloid-beta species in the 7PA2 cell model of Alzheimer's disease. J Alzheimers Dis. 2013 ;33:85–93, doi:10.3233/JAD-2012–120994.
40 Welzel AT, et al. Secreted Amyloid beta-Proteins in a Cell Culture Model Include N-Terminally Extended Peptides That Impair Synaptic Plasticity. Biochemistry. 2014;doi:10.1021/bi5003053.
41 O'Nuallain B, et al. Amyloid beta-protein dimers rapidly form stable synaptotoxic protofibrils. J Neurosci. 2010;30:14411–9, doi:10.1523/JNEUROSCI.3537–10.2010.
42 Tsigelny IF, et al. Structural diversity of Alzheimer's disease amyloid-beta dimers and their role in oligomerization and fibril formation. J Alzheimers Dis. 2014;39:583–600, doi:10.3233/JAD-131589.
43 Ma H, et al. Involvement of beta-site APP cleaving enzyme 1 (BACE1) in amyloid precursor protein-mediated enhancement of memory and activity-dependent synaptic plasticity. Proc Natl Acad Sci U S A. 2007;104:8167–72, doi:10.1073/pnas.0609521104.
44 Kawarabayashi T et al. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci. 2001;21:372–81.
45 Cheng IH, et al. Accelerating amyloid-beta fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J Biol Chem. 2007;282:23818–28, doi:10.1074/jbc.M701078200.
46 Oddo S, et al. Reduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem. 2006;281:39413–23, doi:10.1074/jbc.M608485200.
47 Fowler SW, et al. Genetic modulation of soluble abeta rescues cognitive and synaptic impairment in a mouse model of Alzheimer's disease. J Neurosci. 2014;34:7871–85, doi:10.1523/JNEUROSCI.0572–14.2014.
48 Sherman MA & Lesne SE. Detecting abeta*56 oligomers in brain tissues. Methods Mol Biol. 2011;670:45–56, doi:10.1007/978–1–60761–744–0_4.
49 Jack CR, Jr. et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet neurology. 2013;12:207–16, doi:10.1016/S1474–4422(12)70291–0.
50 Salthouse TA. When does age-related cognitive decline begin? Neurobiology of aging. 2009;30:507–14, doi:10.1016/j.neurobiolaging.2008.09.023.
51 Singh-Manoux A, et al. Timing of onset of cognitive decline: results from Whitehall II prospective cohort study. Bmj. 2012;344 :d7622, doi:10.1136/bmj.d7622.
52 Lambert MP, et al. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–53.
53 Lesne SE. Breaking the Code of Amyloid- Oligomers. Int J Cell Biol. 2013;2013:950783, doi:10.1155/2013/950783.