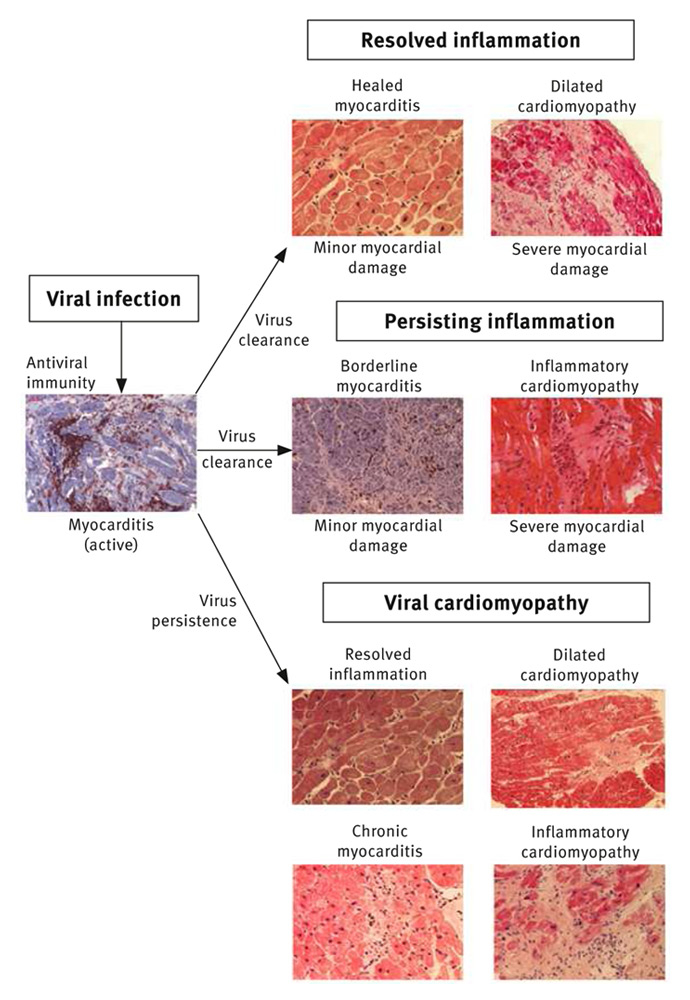
Figure 1
Histological, immunohistochemical and molecular biological findings at distinct phases of viral myocarditis.
DOI: https://doi.org/10.4414/smw.2014.14010
In acute disease, sudden onset of chest pain, dyspnoea, congestive heart failure with normal or enlarged ventricular chambers, ventricular arrhythmias and abnormal ST-T segments changes in the presence of elevated cardiac enzymes (CK/CKMB or TnT) are highly suspicious for an acute viral myocarditis, if other acute cardiac diseases with similar clinical presentation have been excluded. Without angiography accompanying acute viral myocarditis cannot be clinically distinguished from an acute coronary syndrome.
In the subacute and chronic phase after two to four weeks most clinical characteristics suggestive of acute myocarditis have been resolved. Patients present with uncharacteristic complaints such as persisting angina, dyspnoea, fatigue, reduced physical ability, or arrhythmias in the presence of a preserved or impaired systolic or diastolic ventricular function, or with idiopathic dilated cardiomyopathy. A virus-specific phenotype of myocarditis or inflammatory cardiomyopathy does not exist. The majority of non-acute viral infections are asymptomatic or oligosymptomatic and therefore, such infections are frequently not recognised prematurely as a possible cause of a delayed onset of heart disease. The clinical diagnostic challenge becomes even more complicated by the complex virus profiles of the myocardium with a number of distinct virus species and virus subtypes, different virus loads, or reactivated pathogens, all of which may be present in the presence or absence of inflammatory processes.
Since the pathological conditions in viral myocarditis take place at the cellular level, tissue analysis (endomyocardial biopsy) and not clinical tests are necessary to elucidate the true nature of the underlying acquired disease. Despite well-known limitations giving rise to false-negative results (sampling error) if only low numbers (<8–10 samples) are taken, endomyocardial biopsy is a safe diagnostic procedure and currently the gold standard for unequivocally establishing the diagnosis, particularly if histology is complemented by sensitive immunohistochemical methods which allow quantification and identification of inflammatory cell subtypes [1–4].
For the molecular biological virus analysis, at least three to four biopsy specimens should be analysed for DNA and RNA viruses, respectively, both in acute myocarditis and idiopathic dilated cardiomyopathy. With these biopsy numbers, the frequency of detectable viruses in right and left ventricular biopsies are not significantly different in chronic disease. In the acute stage of myocarditis with a history of less than two months, sampling error may become a concern due to focal infection but respective data for molecular analyses are still lacking.
Since immunosuppression may impair the outcome of virus-associated inflammatory cardiomyopathy, biopsies of all patients should undergo molecular analysis in addition to histological and immunohistochemical evaluations in order to allow optimal patient management [5, 6].
Early biopsy is recommended in clinically suspected acute disease in order to tailor personalised management of patients. Due to the lack of prognostic information, this also holds true for subacute myocarditis with its often uncharacteristic complaints and all patients with idiopathic dilated cardiomyopathy at first presentation or in cases of unexplained progression of heart failure. Since an incomplete diagnostic does not allow a safe specific treatment (see below), a complete diagnostic workup including molecular, histological and immunohistochemical analysis is mandatory.
Modern molecular virus diagnostics are not restricted to the solely PCR proof of viral RNA or DNA but also include quantification of the viral loads and of molecular markers of virus reactivation. Sequencing; furthermore confirms the involved virus subtypes (table 1) [7]. No other clinical diagnostic tool can recognise and quantify loads and types of different viruses or non-viral infectious pathogens, elucidate and quantify inflammatory cell subtypes, detect minor myocardial necroses, newly developing fibrosis, or circumscribed early scar formation characteristic of active infectious or postinfectious disease. Of note, neither a positive virus serology nor a positive virus-PCR in the peripheral blood can prove any organ involvement in acute or chronic disease and virus copy numbers in myocardial biopsies may be overestimated in the presence of high systemic virus loads by contamination with virus infected blood cells (e.g., in chronic HCV infection). Blood diagnostics may, however, allow the discrimination of an acute viral infection from endogeneous B19V or HHV6 reactivation, especially in cases with high virus loads as occasionally detected in patients with HHV6 and ciHHV6 reactivation.
The histological, immunohistological and molecular biological information are prerequisites to establish an accurate diagnosis of viral myocarditis and successful management of patients. They cannot be substituted by any non-invasive clinical analysis. Although imaging techniques including MRI can provide noninvasive tissue characterisation and may localise larger inflammatory infiltrates they are misleading if infectious agents are involved, since they neither detect nor quantify different virus types and loads or inflammatory cell numbers nor differentiate between cell subtypes of the immune response. On the other hand, MRI can provide prognostic information on outcome. If extended fibroses and scars have developed early, full recovery is less likely than in patients with an inconspicuous MRI.
| Table 1:Frequent viruses causing infectious and post-infectious myocarditis. |
| Viral (most common) |
| Picornavirus (coxsackie A/B, echo) |
| Adenovirus (A1, 2, 3, 5) |
| Erythrovirus (Parvovirus B19, B19V) |
| Herpes virus (HSV 1, 2 ,6A/B, ciHHV6 A,B, EBV) |
| Cytomegalovirus |
| Influenza (1, 2) |
| Human immunodeficiency virus |
| Mixed infections |
| Autoimmune activation |
| Post-infectious immunity/autoimmunity |
Infectious agents are the major causes of myocarditis and inflammatory cardiomyopathy (DCMI) in acquired “idiopathic” diseases of the heart muscle [4, 8, 9]. Although virtually any microbial agent can cause myocardial inflammation and dysfunction, non-viral infections are rare in these conditions, at least in western countries. Viral forms are considered the most common cause of acquired inflammatory cardiomyopathies nowadays [4, 10].
For decades coxsackieviruses [11, 12] and, to a lesser extent adenoviruses [12, 13], are well established in paediatric and adult myocarditis and chronic heart muscle disease (table 1). Furthermore, distinct genotypes of erythroviruses including parvovirus B19 (B19V), human herpesvirus type 6 (HHV6A/B and ciHHV6), human immune deficiency virus (HIV), cytomegalovirus (CMV), herpes simplex type 2 virus and hepatitis C virus, among many others have been identified with varying degrees of frequency in cardiac tissues.
In a comprehensive study by Bowles et al., nested PCR amplified a viral product in 40% of samples of 773 mostly younger American patients under 18 years of age with clinically suspected myocarditis (n = 624) or DCM (n = 149), with adenoviruses and enteroviruses predominating in the PCR analysis and only one percent was tested positive for parvovirus [13]. The frequency of myocardial virus species, however, changes with geographical differences and over time. In different European studies, viral genomes have been documented in 30% to 73% of EMB of patients with left ventricular dysfunction and a similar epidemiological shift with more frequent detection of B19V has recently been reported from the US [14–17].
Generally, erythrovirus and herpes virus genomes are detected much more frequently than the other viral species. Regarding such high numbers, it has to be kept in mind that in contrast to “classical” acquired enterovirus and adenovirus infections, erythroviruses and herpesviruses comprise life-long persistence after childhood infection [18, 19]. Particularly in adults, their detection in different tissues normally does not represent recently acquired but rather latent infections. Symptomatic diseases associated with such viruses are generally caused by reactivation of those lifelong persisting pathogens (see below) [4, 6].
The overall incidence of myocardial involvement in any viral infections is estimated at 3–6% [20]. The actual incidence of virus induced myocarditis or cardiomyopathy is less well established. The majority of viral infections is asymptomatic or oligosymptomatic and due to the infrequent use of biopsy based diagnoses, such infections are frequently not recognised as possible causes of acute or delayed onset heart failure. As far as human viral myocarditis and inflammatory cardiomyopathy are concerned, the underlying pathogenetic mechanisms are unknown for most of the infectious agents. A limited conception is available for enteroviruses and, to some extent, for erythroviruses and herpesviruses.
Figure 1
Histological, immunohistochemical and molecular biological findings at distinct phases of viral myocarditis.
Newly acquired viral myocarditis develops with three pathologically distinct phases (fig. 1 and 2) [9, 21]. The early phase of viral myocarditis is initiated by an infection of the cardiac myocytes, fibroblasts, or endothelial cells via receptor-mediated endocytosis [4, 22–24]. The resulting kind and extent of myocardial compromise and hence the prognosis of the disease depends on the nature of the offending infectious agent, the affected cardiac structures, and the degree of irreversible myocardial lesions caused by cytolytic viruses.
The activation of antigen-specific cell-mediated immunity initiates the second phase of virus clearance [25, 26]. Because virus-infected cells are destroyed by immune effector cells of the emerging cellular antiviral inflammatory response, virus clearance will occur at the expense of further loss of infected myocytes. The ensuing myocardial damage depends on the scale of the cellular virus infection and increases with growing virus dispersion which, in addition to the early virus- and immune-mediated injury of phase 1, contributes to tissue remodelling and progression of the disease. Hence, the resolving tissue infection occurs at the expense of a partial destruction of myocardial tissue that is not capable of regeneration.
In patients with a regularly controlled immune system the cellular inflammatory process fades away within the following weeks or months after successful elimination or substantial reduction of the infectious pathogen and thus prevents ongoing tissue damage by an extended immune response. Whether resolving inflammation contributes to myocardial injury and whether tissue alteration could be avoided by a more rapid decline of inflammation, e.g., supported by an early immunosuppressive treatment, is currently unknown. The mildly improved outcome reported for early immunosuppressive treatment in active myocarditis, however, would suppose such an assumption [27].
Chronic immune stimulation or autoimmunity in chronic viral myocarditis results from incompletely cleared virus infection or in response to the preceded virus- or immune-mediated chronic tissue damage, respectively. Both the ongoing antigenic trigger from continuously synthesised viral proteins and the release of intracellular proteins from necrotic or apoptotic myocardial cells may stimulate chronic inflammation which initially damages some individual cells but ultimately can affect the whole myocardium [28–30].
In the third remodelling phase, the virus infection has been cleared completely and antiviral immune responses have been resolved. Nevertheless, the extent of the initially caused tissue damage determines the further clinical course of the disease. Biopsy-based diagnostics started so late can no longer elucidate the initial causes of the disease and will postulate an “idiopathic” disease. In those cases, a post-infectious or postmyocarditis disease can only be suspected but no longer proven by any diagnostic procedure. The clinical picture is consistent with an often irreversible dilated cardiomyopathy which develops in about 25% to 30% of concerned patients with biopsy proven myocarditis [31, 32].
| Table 2: Biopsy-based specific treatment options in patients with virus associated myocarditis. | |||||
| Clinical diagnosis | Virus PCR (myocardium) | Histology/IHS | Anti-viral treatment | Immuno-suppression | Comments |
| Acute myocarditis | Any virus | inflammation | No | No | Optimal heart failure medication (HFM) |
| Chronic inflammation | EV, ADV | IFN-β | No | HFM, 8x106 IU IFN-β sc for 6 months [45, 47] | |
| Latent B19V | No | Yes | HFM, no specific antiviral treatment Immunosuppression possible (see below), if acute infection has been excluded clinically and serologically | ||
| Reactivated B19V | IN-β | ± | HFM+ 8x106 IU IFN-β sc for 6 months [6, 48] | ||
| HHV6A/B | No | No | Immunosuppression possible (see below), if acute infection has been excluded clinically and serologically | ||
| ciHHV6A/B | Valganciclovir | No | HFM, 900–1800 mg valganciclovir [19] | ||
| Other viruses | No | No | HFM, no specific recommendations | ||
| Chronic virus persistence | EV, ADV | no inflammation | IFN-β | No | HFM, 8x106 IU IFN-β for 6 months [45, 47] |
| Latent B19V | No | No | HFM, no specific antiviral treatment | ||
| Reactivated B19V | IN-β | No | HFM+ 8x106 IU IFN-β sc for 6 months [6, 48] | ||
| HHV6A/B | No | No | Often spontaneous improvement | ||
| ciHHV6A/B | Valganciclovir | No | HFM, 900–1800 mg valganciclovir [6] | ||
| Other viruses | No | No | HFM, no specific recommendations | ||
| Post-infectious immunity | No virus | inflammation | No | Yes | Prednisolon 1 mg/kg bw + azathioprin 100 mg, daily. The steroid is tapered every 2 weeks by 10 mg. Maintainance dose 10 mg/day, treatment for 3–10 months [6] |
Enteroviruses and adenoviruses are established causes of acute myocarditis but are also detected in chronic heart failure presenting as DCM [12, 14, 33]. Both viruses infect cardiomyocytes in animal models and human disease after binding to the coxsackie-adenoviral receptor (CAR) and the decay accelerating factor (DAF, CD55) which serves as a co-receptor for enterovirus internalisation [23, 34]. After internalisation the enterovirus negative strand RNA is reversely transcribed into a positive strand for subsequent virus replication and spreading [12, 35] Myocardial injury is directly caused by the lytic viral infections (phase 1) or the antiviral immunity (phases 1 and 2, fig. 2).
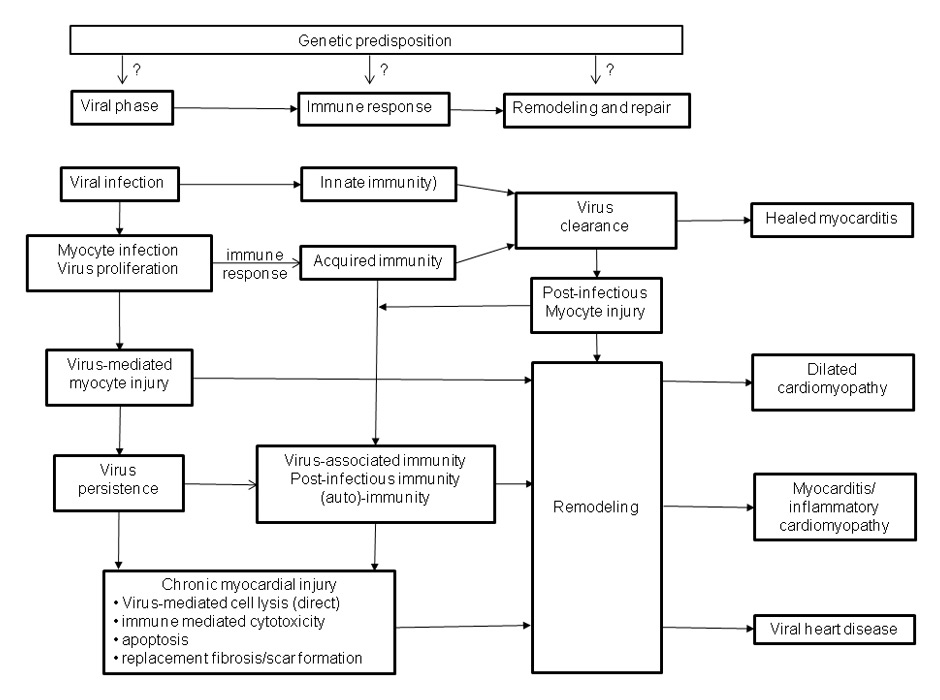
Figure 2
Three phase model of cardiovascular infection: A genetic predisposition is supposed (?) for some viruses but currently not proven. During the viral phase, infected cardiomyocytes become injured and the virus may persist due to an inadequate immune response. A regularly mounted immune response (phase 2) eliminates the viral infection and may further damage the myocardium if it declines improperly. Post-infectious, post-inflammatory immunity and preceding myocardial damage may stimulate ongoing tissue remodelling (phase 3) which affects cardiac function in the long run despite clearance of the initial causes.
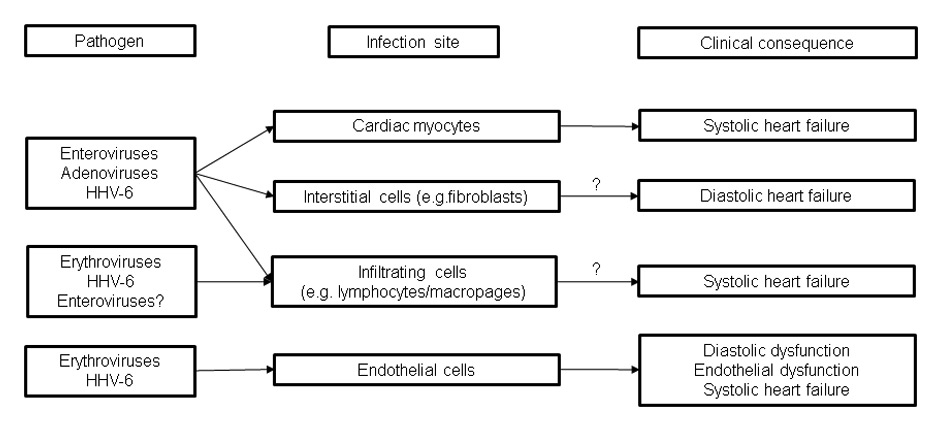
Figure 3
Clinical impact of viruses on the myocardium. Distinct cardiac dysfunctions are caused by the different cell tropisms of frequent cardiotropic viruses and post-infectious (auto)immunity. Optimal heart failure treatment according to guidelines have to be administered to all patients. The mode of treatment of persisting cardiac infections is virus type specific. Reliable treatment data for acute cardiac infections do not exist. Proposed treatment strategies differ from the management of acute systemic viral infections. Post-infectious autoimmunity is treated by immunosuppression.
Acute parvovirus B19 (erythrovirus genotype 1, B19V) infection is a common acute childhood disease infrequently observed in adults [36]. Erythrovirus infection and replication are primarily restricted to erythroid progenitor cells in the bone marrow, but consistent with the presence of the primary erythrovirus receptor P antigen as well as its co-receptors (Integrins, KU80) on vascular endothelial cells, persistent latent infection is detected in the vascular endothelium (EC) of different organs, including the heart in which the virus has been localised in endothelial cells of venuoles, small arteries or arterioles of children and adults with fulminant myocarditis or sudden onset heart failure [36–38].
In the majority of cases, latent infection is asymptomatic. If the virus becomes reactivated, transcriptional active erythrovirus is often associated with symptomatic endothelial dysfunction [7]. Cardiovascular impairment caused by erythrovirus infected endothelial cells may also explain the earlier graft loss and the premature development of advanced transplant coronary artery disease in paediatric cardiac transplant recipients [15, 16].
Human herpesvirus type 6 is another widespread latent virus infection frequently detected in endomyocarial tissue specimens (fig. 2) [14]. Clinical isolates form two genetically related but biologically distinct groups (HHV-6A and HHV-6B) which, similar to B19V, persists in >70% of the adult population after primary infection in childhood [39]. HHV6 is a lymphotropic virus which also infects various other cell types including cardiomyocytes and the vascular endothelium even though infectious virus cannot be isolated from the peripheral blood and the virus genome remains below the detection limit in these patients [40–42]. Intriguingly, HHV-6 is able to integrate its genomes into telomeres of human chromosomes (ciHHV6), which allows transmission of ciHHV-6 via the germ line in about 0.4–0.8% of the US and European populations [19, 43].
Similar to the other herpes viruses, HHV-6 and ciHHV6 become frequently reactivated with subacute clinical presentations. Recently, HHV-6 has been detected in the myocardium of patients with myocarditis and clinically suspected dilated cardiomyopathy (DCM) by PCR. Short-term follow-ups have revealed an association with the clinical course of the disease [14, 42].
If the antiviral immunity has elaborated fast and efficiently with subsequent rapid resolution of cellular processes, residual damage of the myocardium may be minor and the remaining myocardium can compensate sufficiently for the partial loss of contractile tissue (fig. 1 and 3). Consequently, 60% to 70% of patients recover completely within 2 to 12 months with no or only minor residual clinical signs of heart injury. During or after recovery, follow-up biopsy will be consistent with healed myocarditis. Even complete recovery and inconspicuous histology, however, do not prove optimal long-term outcome, since a group of those patients will develop slowly progressive heart failure or compromising arrhythmias even after years of asymptomatic intervals [33].
Depending on the severity of initial cardiac damage, other patients may retain residual myocardial impairment. Moderate loss of contractile tissue with more pronounced remodelling of the myocardial matrix accounts for the course of those 25% to 30% of patients who only partially recover (fig. 1 and 3). In the longer run many of these patients experience progressive heart failure despite regular heart failure medication. At this time point, idiopathic DCM is diagnosed, histologically.
The resulting clinical presentation is, however, not only influenced by the severity of irreversible matrix alterations and the potential of the myocardium to compensate for these processes. It may also depend on the effects which are exerted on the cardiac tissue by a persisting lytic virus infection, virus-associated low grade inflammatory processes or autoimmune mechanisms. Under these circumstances, biopsy derived findings will be compatible with inflammatory cardiomyopathy or chronic viral heart disease, respectively (fig. 1).
The transition of myocarditis into DCM following direct virus- or immune-mediated myocardial damage is generally accepted and supported by literature [31, 32]. Continuous myocardial damage caused by persisting virus infection and/or ongoing immune processes, however, has not been proven unambiguously in human disease. A great deal of scepticism stems from the inconsistency of currently available data. This inconsistency is mostly derived from insufficiently diagnosed and inconsequently followed cohorts of patients. There are, however, a number of sound clinical reports which demonstrate that persistent viral infections directly contribute to the progression of heart failure and adverse prognosis in human disease [14, 44, 45].
The clinical importance of persistent enteroviral genomes in the myocardium was investigated by Why and colleagues who demonstrated a higher mortality at 25 months (25% versus 4%) in the 41 patients with persistent enteroviral infection [44].
The data reported by Frustaci et al. from a retrospective analysis of immune suppressively treated patients with inflammatory cardiomyopathy point to a similar direction [5]. In this study patients with persistent viruses did not improve or even deteriorated upon immunosuppression while virus-negative patients improved significantly. Within a short period of 9 months, 7 out of 44 treated virus-positive patients died or were transplanted. In another recent paper, Caforio and co-workers reported on a two year follow-up of patients with active (n = 85) and borderline myocarditis (n = 89) in which virus persistence was an univariate predictor of adverse prognosis, in addition to anti-heart autoantibodies and clinical signs or symptoms of left and right heart failure [30].
These data are in accordance with our own observations. When we initially followed 172 consecutive patients with left ventricular dysfunction and biopsy-proven viral infection by re-analysis of biopsies and haemodynamic measurements after a median period of seven months, viral genomes persisted in 64% of patients with single virus infections [14]. 50% of the enteroviral genomes were cleared spontaneously. Respective data for adenovirus, parvovirus B19 and herpesvirus 6 were 36%, 22% and 44%. These data on spontaneous clearance of the virus infection demonstrate that a single biopsy analysis can never prove virus persistence unambiguously.
Clearance of virus was associated with a significant decrease in left ventricular dimensions and improvement in left ventricular ejection fraction. In contrast, LV function decreased mildly during this short follow-up in patients with persistent viral genomes [46]. About five years later, 41% of the patients with enterovirus persistence had died (10 year mortality rate: 52.5%), whereas 92% of patients who spontaneously had cleared the infection where still alive after 10 years [45].
Respective data on other viruses are not available because most published data do not refer to biopsy-based virus controls and attempts to definitely prove virus persistence for the whole study period by follow-up PCR analysis have predominantly not been carried out in most studies. Since a high percentage of viral infections are cleared at early stages of myocarditis by the antiviral immune response, it is still unknown whether reported adverse prognoses have to be attributed to early and more pronounced tissue damage in initially virus-positive patients (phase 1) or whether it is caused by latent viral infections with smoldering immune processes (phases 2 and 3).
In an attempt to gain more information on this important issue, virus-positive patients with chronic cardiomyopathy (median history: 44 months) were treated with interferon-β in a non-randomised study. Upon treatment enterovirus and adenovirus clearance was successful in all treated patients [47]. Virus clearance was paralleled by a significant decrease of ventricular dimensions and clinical complaints. LV ejection fraction improved significantly in both patients with moderately and severely suppressed ventricular function. Ten year follow-up documented, in contrast to untreated patients, a significantly reduced mortality of treated patients [47].
Erythroviruses including parvovirus B19 and HHV6 are neither cleared by IFN-α nor IFN-β ([48] and unpublished data). Despite this fact, symptomatic B19V-positive patients but not untreated controls benefit from suppression of virus transcriptional activity. We recently have reported that endothelial dysfunction and respective symptoms improve upon antiviral treatment with interferon-beta (IFN-β) although the B19V virus load is barely affected [48]. The underlying mechanisms of how IFN-β delivers such beneficial clinical effects without clearing the virus substantially are unknown but cell culture analyses using infected immortalised human microvascular EC cells (HMEC-1) have shown, that IFN-β inhibits B19V reactivation and improves endothelial cell viability [48]. In B19V infected patients IFN-β reduces endothelial cell apoptosis and improves endothelial dysfunction [48]. In a recent preliminary study, the onset of advanced transplant vasculopathy was delayed in parvovirus positive children who received IVIG in addition to standard immune suppression [16, 49].
Less information is available for HHV6 and ciHHV6. Similar to B19V, HHV6 is not cleared by interferons or ganciclovir. CiHHV6 cannot be cleared due to its chromosomal integration in every cell of the body. Again, symptomatic patients with reactivated HHV6 and ciHHV6 improve symptomatically upon ganciclovir treatment [51].
The above follow-up data and treatment observations indicate that the course of the virus infection predetermines the clinical course of the disease. The data furthermore implicate that ventricular dysfunction in chronic enteroviral heart disease should not be put on a level with irreversibly damaged myocardium even in patients with a chronic history of cardiomyopathy, since haemodynamic improvement occurs in 67% of treated patients with chronic cardiomyopathy [45, 50].
With respect to the clinical management of the patients, data outlined argues for the necessity to identify patients at an early and still reversible stage of virus-associated heart disease (table 2). Depending on the infectious agent, biopsy-guided, tailored antiviral treatment may clear the infection and improve outcome, or be of symptomatic benefit if the virus is not cleared completely. Post-infectious autoimmunity should be treated by immunosuppression (table 2). One has to keep in mind, however, that only patients with still minor or moderate irreversible alterations of the heart tissue will benefit from early and specific treatment and progression of heart failure can only be prevented by in time therapy. Therefore, viral diagnostics and antiviral treatment should be started before irreversible myocardial damage has developed.
Myocarditis is an inflammatory disease of the cardiac muscle tissue caused by myocardial infiltration with immunocompetent cells following any kind of cardiac injury. Infectious aetiologies include a vast number of viruses, bacteria, protozoa or fungi, but most frequently the myocardial inflammatory process is directed against viral pathogens. In the early stage of the disease both the infectious trigger and the resulting immune response may already cause irreversible myocardial injuries that influence acute and long-term outcome. If the infectious agent is rapidly eliminated and the inflammatory process is resolved in a timely manner, the disease will resolve with only minor alterations of the myocardium. At this phase, the true underlying causes of the disease can no longer be identified.
Chronic myocardial injury in viral myocarditis develops if the antiviral immune response fails to eliminate the infectious agent completely or, if the inflammatory process does not resolve properly despite virus clearance. In such conditions, long-term outcome depends on the nature and extent of the virus-affected tissue compartments which varies considerably with the amount and kind of the infectious agent or the number and subtype of the smoldering cellular inflammatory infiltrates. In addition to the initial irreversible tissue alterations, persisting viruses, and post-infectious immune or autoimmune processes may induce persisting or progressive ventricular dysfunction, arrhythmias and symptomatic cardiac complaints.
Viral heart disease often presents as an acute or chronic dilated cardiomyopathy (DCM) but due to its broad spectrum of presentation a solely clinical diagnosis is frequently misleading. Since the pathological conditions in viral myocarditis take place at the cellular level, tissue analysis but not clinical tests are necessary to elucidate the true nature of the underlying acquired disease. If the primary infectious or immune-mediated causes of the disease are carefully defined by clinical and biopsy based tools, specific antiviral treatment options in addition to basic symptomatic therapy are available under certain conditions. This may allow a tailored cause-specific treatment that improves prognosis of patients with acute and chronic disease.
1 Holzmann M, Nicko A, Kühl U, et al. Complication rate in right ventricular endomyocardial biopsy – A retro- and prospective study over a 11 year period analyzing 3048 diagnostic procedures. Circulation. 2008;2008:1722–8.
2 Chimenti C, Frustaci A. Contribution and risks of left ventricular endomyocardial biopsy in patients with cardiomyopathies: retrospective study over a 28–year period. Circulation. 2013;128:1531–41.
3 Caforio AL, Pankuweit S, Arbusti E, et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Europ Heart J. 2013;34:2636–48.
4 Kuehl U, Schultheiss HP. Myocarditis in children. Heart Fail Clin. 2011;6:483–96.
5 Frustaci A, Chimenti C, Calabrese F, et al. Immunosuppressive therapy for active lymphocytic myocarditis: virological and immunologic profile of responders versus nonresponders. Circulation. 2003;107:857–63.
6 Schultheiss HP, Kuehl U, Cooper LT. The Management of Myocarditis. Eur Heart J. 2011;32:2616–65.
7 Kuehl U, Lassner D, Dorner A, et al. A distinct subgroup of cardiomyopathy patients characterized by transcriptionally active cardiotropic erythrovirus and altered cardiac gene expression Basic Research Cardiology. 2013;108:372–82.
8 Blauwet LA, Cooper LT. Myocarditis. Prog Cardiovasc Dis. 2010;52:274–88.
9 Liu PP, Schultheiss HP. Myocarditis. In: Baunwald ed, Heart Disease. 8 ed. Philadelphia: W B Saunders co. 2008:1775–92.
10 Elliott P, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29:270–6.
11 Dec GW, Jr., Palacios IF, Fallon JT, et al. Active myocarditis in the spectrum of acute dilated cardiomyopathies. Clinical features, histologic correlates, and clinical outcome. N Engl J Med. 1985;312:885–90.
12 Pauschinger M, Bowles NE, Fuentes-Garcia FJ, et al. Detection of adenoviral genome in the myocardium of adult patients with idiopathic left ventricular dysfunction. Circulation. 1999;99:1348–54.
13 Bowles NE, Ni J, Kearney DL, et al. Detection of viruses in myocardial tissues by polymerase chain reaction. evidence of adenovirus as a common cause of myocarditis in children and adults. J Am Coll Cardiol. 2003;42:466–72.
14 Kühl U, Pauschinger M, Noutsias M, et al. High prevalence of viral genomes and multiple viral infections in the myocardium of adults with “idiopathic” left ventricular dysfunction. Circulation. 2005;111:887–93.
15 Breinholt JP, Moulik M, Dreyer WJ, et al. Viral epidemiologic shift in inflammatory heart disease: the increasing involvement of parvovirus B19 in the myocardium of pediatric cardiac transplant patients. J Heart Lung Transplant. 2010;29:739–46.
16 Towbin JA, Ware SM, Jefferies JL. Heart transplants in pediatric patients: viral infection as a loss predictor. Future Cardiology. 2010;6:735–41.
17 Kandolf R. Virus etiology of inflammatory cardiomyopathy. Dtsch Med Wochenschr 2004;129:2187–92.
18 Norja P, Hokynar K, Aaltonen LM, et al. Bioportfolio: Lifelong persistence of variant and prototypic erythrovirus DNA genomes in human tissue. Proc Natl Acad Sci U S A. 2006;103:7450–53.
19 Pellett PE, Ablashi DV, Ambros PF, et al. Chromosomally integrated human herpesvirus 6: questions and answers. Rev Med Virol. 2012;22:144–55.
20 Kereiakes DJ, Parmley WW. Myocarditis and cardiomyopathy. Am Heart J. 1984;108:1318–26.
21 Mason JW. Myocarditis and dilated cardiomyopathy: an inflammatory link. Cardiovasc Res. 2003;60:5–10.
22 Liu PP, Opavsky MA. Viral myocarditis: receptors that bridge the cardiovascular with the immune system? Circ Res. 2000;86:253–4.
23 Bergelson JM, Cunningham JA, Droguett G, et al. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science. 1997;275:1320–3.
24 Kandolf R, Ameis D, Kirschner P, Canu A, Hofschneider PH. In situ detection of enteroviral genomes in myocardial cells by nucleic acid hybridization: an approach to the diagnosis of viral heart disease. Proc Natl Acad Sci U S A. 1987;84:6272–6.
25 Fairweather D, Frisancho-Kiss S, Rose NR. Viruses as adjuvants for autoimmunity: evidence from Coxsackievirus-induced myocarditis. Rev Med Virol. 2005;15:17–27.
26 Streitz M, Noutsias M, Volkmer R, et al. NS1 specific CD8+ T-cells with effector function and TRBV11 dominance in a patient with parvovirus B19 associated inflammatory cardiomyopathy. PLoS ONE. 2008;3:e2361.
27 Mason JW. Immunopathogenesis and treatment of myocarditis: the United States Myocarditis Treatment Trial. J Card Fail. 1996;2:S173–77.
28 Pauschinger M, Chandrasekharan K, Schultheiss HP. Myocardial remodeling in viral heart disease: possible interactions between inflammatory mediators and MMP-TIMP system. Heart Fail Rev. 2004;9:21–31.
29 Heymans S, Pauschinger M, De Palma A, et al. Inhibition of urokinase-type plasminogen activator or matrix metalloproteinases prevents cardiac injury and dysfunction during viral myocarditis. Circulation. 2006;114:565–73.
30 Caforio AL, Calabrese F, Angelini A, et al. A prospective study of biopsy-proven myocarditis: prognostic relevance of clinical and aetiopathogenetic features at diagnosis. Eur Heart J. 2007;28:1326–33.
31 Baboonian C, Treasure T. Meta-analysis of the association of enteroviruses with human heart disease. Heart. 1997;78:539–43.
32 D’Ambrosio A, Patti G, Manzoli A, et al. The fate of acute myocarditis between spontaneous improvement and evolution to dilated cardiomyopathy: a review. Heart. 2001;85:499–504.
33 Towbin JA, Lowe AM, Colan SD, et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA. 2006;296:1867–76.
34 Noutsias M, Fechner H, de Jonge H, et al. Human coxsackie-adenovirus receptor is colocalized with integrins alpha(v)beta(3) and alpha(v)beta(5) on the cardiomyocyte sarcolemma and upregulated in dilated cardiomyopathy: implications for cardiotropic viral infections. Circulation. 2001;104:275–80.
35 Fujioka S, Kitaura Y, Ukimura A, et al. Evaluation of viral infection in the myocardium of patients with idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 2000;36:1920–6.
36 Bultmann BD, Klingel K, Sotlar K, et al. Fatal parvovirus B19–associated myocarditis clinically mimicking ischemic heart disease: an endothelial cell-mediated disease. Hum Pathol. 2003;34:92–5.
37 Bock CT, Klingel K, Aberle S, et al. Human Parvovirus B19: A new emerging pathogen of inflammatory cardiomyopathy. J Vet Med B. 2005;52:340–3.
38 Klingel K, Sauter M, Bock CT, et al. Molecular pathology of inflammatory cardiomyopathy. Med Microbiol Immunol (Berl). 2004;193:101–7.
39 Di Luca D, Mirandola P, Ravaioli T, Bigoni B, Cassai E. Distribution of HHV-6 variants in human tissues. Infect Agents Dis. 1996;5:203–14.
40 Caruso A, Rotola A, Comar M, et al. HHV-6 infects human aortic and heart microvascular endothelial cells, increasing their ability to secrete proinflammatory chemokines. J Med Virol. 2002;67:528–33.
41 Fotheringham J, Akhyani N, Vortmeyer A, et al. Detection of active human herpesvirus-6 infection in the brain: correlation with polymerase chain reaction detection in cerebrospinal fluid. J Infect Dis. 2007;195:450–4.
42 Krueger GRF, Rojo J-, Buja LM, Lassner D, Kuehl U. Human herpesvirus-6 (HHV-6) is a possible cardiac pathogen: An immunihistological and ultrastructural study. Hospital General. 2008;71:187–91.
43 Arbuckle JH, Medveczky MM, Luka J, et al. The latent human herpesvirus-6A genome specifically integrates in telomeres of human chromosomes in vivo and in vitro. Proc Natl Acad Sci U S A. 2010;107:5563–8.
44 Why HJ, Meany BT, Richardson PJ, et al. Clinical and prognostic significance of detection of enteroviral RNA in the myocardium of patients with myocarditis or dilated cardiomyopathy. Circulation. 1994;89:2582–9.
45 Kuehl U, Lassner D, von Schlippenbach J, Poller W, Schultheiss H-P. Interferon-beta- Improves Survival in Enterovirus-Associated Cardiomyopathy. J Am Coll Cardiol. 2012;60:1295–6.
46 Kühl U, Pauschinger M, Seeberg B, et al. Viral persistence in the myocardium is associated with progressive cardiac dysfunction. Circulation. 2005;112:1965–70.
47 Kühl U, Pauschinger M, Schwimmbeck PL, et al. Interferon-beta treatment eliminates cardiotropic viruses and improves left ventricular function in patients with myocardial persistence of viral genomes and left ventricular dysfunction. Circulation. 2003;107:2793–8.
48 Schmidt-Lucke C, Spillmann F, Bock T, et al. Interferon Beta Modulates Endothelial Damage in Patients with Cardiac Persistence of Human Parvovirus B19 Infection. J Infect Dis. 2010;201:6.
49 Moulik M, Breinholt JP, Dreyer WJ, et al. Viral endomyocardial infection is an independent predictor and potentially treatable risk factor for graft loss and coronary vasculopathy in pediatric cardiac transplant recipients. J Am Coll Cardiol. 2010;56:582–92.
50 Chen J, Kuhlencordt P, Urano F, et al. Effects of chronic treatment with L-arginine on atherosclerosis in apoE knockout and apoE/inducible NO synthase double-knockout mice. Arterioscler Thromb Vasc Biol. 2003;23:97–103.
51 Kuehl U, Lassner D, Wallaschek N, Gross U, Krueger GRF, et al. Chromosomally integrated human herpesvirus 6 in heart failure – prevalence and treatment. Eur J Heart Failure. 2014; in press.
Funding / potential competing interests: Part of this work was supported by grants of the German Research Foundation (DFG), Transregional Collaborative Research Centre “Inflammatory Cardiomyopathy – Molecular Pathogenesis and Therapy” (SFB TR 19 04, Project Z1) and the Federal Ministry of Education and Research (BMBF, Germany) for KMU innovative program (No. 616 0315296). For their excellent technical assistance, we thank Mrs. K. Winter, S. Ochmann, C. Seifert, M. Willner and E. Hertel, Berlin, Germany.