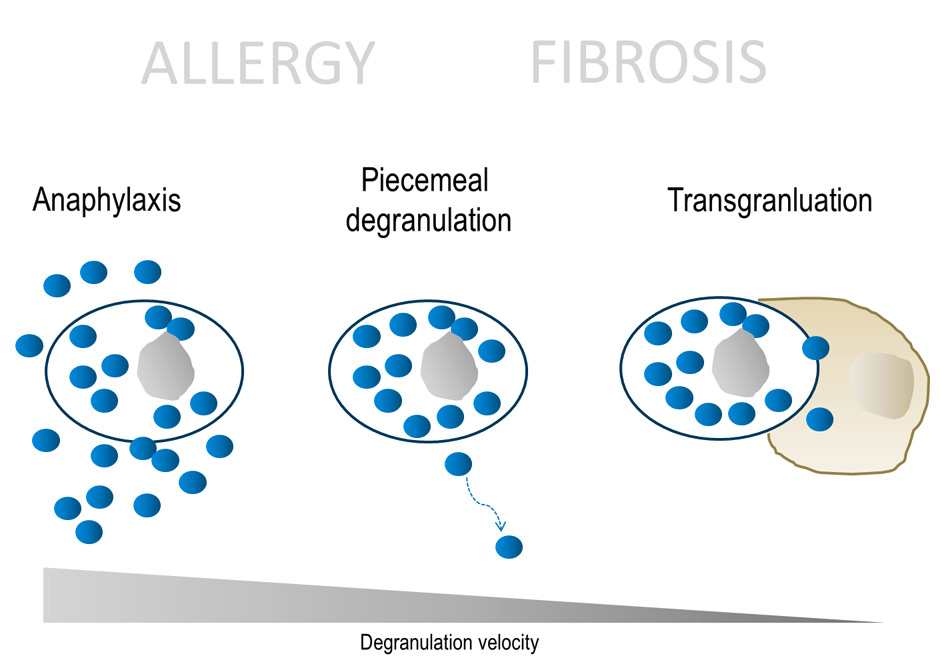
Figure 1
Different forms of degranulation in mast cells.
DOI: https://doi.org/10.4414/smw.2014.13999
By serendipity, the medical student Paul Ehrlich discovered mast cells in 1877 during his dissertation in Freiburg im Breisgau, Germany [1]. He performed histological studies using newly available synthetic stainings based on Anilin, which was produced in large quantities by the chemical industry, e.g., nearby in Basel. Ehrlich described a new cell type in the interstitial tissue, characterised by a high content of cytosolic vesicles. He postulated that those cells are fed by the surrounding fibroblasts. Therefore he named them mast cells from ‚mästen‘, (German: to feed). That the close interaction of mast cells and fibroblasts would be a putative mechanism of fibrosis, only became apparent much later.
Figure 1
Different forms of degranulation in mast cells.
Mast cells are derived from haematopoietic progenitor cells in the bone marrow and mature in the interstitial tissue of most organs [2]. The maturation of mast cells is notably regulated by the soluble stem cell factor (SCF) and its receptor c-kit [3]. Despite originating from different cell lines, mast cells resemble basophils in form and function. Degranulation is regarded as the main function of mast cells with immunoglobuline (Ig)E as the main trigger [4]. Mast cells express large quantities of FcεRI receptor on their surface. As the binding of IgE to FcεRI essentially is irreversible, mast cells are largely covered with IgE. Once an IgE molecule encounters a specific antigen or allergen, IgERI-crosslinking and calcium influx leads to degranulation of the mast cells. As a result, histamine is released and causes the well-known symptoms such as bronchoconstriction or pruritus. The natural role of this very susceptible and efficient mast cell-IgE ‘dual system’ is the immunity against parasites [5]. In the case of pulmonary or intestinal parasites, which of course still occur, mast cell degranulation leads to cough or diarrhoea as an attempt to eliminate the parasites from the body. If a parasite has entered a “dead end” such as in the liver or skin, a fibrotic immune reaction (e.g., liver fibrosis in schistosomiasis) or encapsulation (Echinococcus) occurs in order to prevent further organ damage.
It is obvious that the dynamic of mast cell degranulation in fibrogenic reactions differs from anaphylaxis. In IgE-mediated anaphylaxis, literally all vesicles are released from the mast cells within minutes to a few hours (fig. 1). The classical clinical picture is anaphylactic shock with hypotension, bronchoconstriction and exanthema. Non-IgE driven anaphylactoid reactions that cause rapid mast cell degranulation e.g., by staphylococcal toxins or contrast reagents also occur [6]. Probably more frequently, and associated with more subtle symptoms, mast cell vesicles can be released by a slower process called piecemeal degranulation [7] where vesicles can traffic through the interstitial space via lymphatic vessels to distant lymph nodes [8]. Piecemeal degranulation can therefore be regarded as paracrine and even endocrine mechanism of mast cells to initiate immune reactions and possibly also to support fibrosis. A third even less discussed mechanism is the direct release of vesicles to another cell by cell-cell contact, called transgranulation.
It is increasingly recognised that besides IgE, mast cells can also be activated by innate immune receptors such as Fc-receptors, toll-like-receptors (TLR) [9], tumour necrosis factor (TNF) receptors, NOD-like receptors and even physical stimuli [10]. Mast cells contain a variety of cytokines which after activation can be released via degranulation or, similar to other cell types by secretion via the Golgi apparatus. They are a significant reservoir of preformed TNF-alpha in the human body. IL-17, which is also stored in mast cell vesicles, has been shown to be the main source of IL-17 in rheumatoid arthritis [11]. This demonstrates the role of mast cells as central innate immune cells and possible sensor danger signals leading to a subsequent (auto)immune response. Indeed, mast cell numbers often are increased in chronic rheumatologic diseases such as rheumatoid arthritis or systemic sclerosis (SSc) in which chronic inflammation leads to fibrosis [12]. This review aims to describe the pathophysiological role of mast cells in primary organ fibrosis and in the setting of chronic rheumatic diseases.
As described before, mast cells are involved in the parasitic immune response and intrahepatic schistosomiasis is a well-known cause of liver fibrosis [13]. Chronic inflammation and excess of extracellular matrix can be observed in periovular granulomas where an eosinophilic, Th-2–polarised inflammatory reaction leads to IL-4 and IL-10 overexpression. In schistosomiasis infected animals, mast cells are necessary for the development of eosinophils, IL-5 expression and finally liver fibrosis [14].
Mast cell vesicles not only support a profibrotic Th-2–response, they also contain directly profibrotic stimuli such as transforming growth factor (TGF)-beta, platelet derived growth factor (PDGF) or granulocyte macrophage colony-stimulating factor (GM-CSF). Chymase as a main enzymatic component of mast cells can cleave the latent form of TGF-beta from cell membranes [15]. TGF-beta is a main driver for collagen production of fibroblasts and their differentiation into myofibroblasts, respectively. Furthermore, mast cells produce and store negatively charged proteoglycans such as hyaluronic acid or chondroitin sulphate [16]. This release of matrix components in the interstitial space causes extracellular matrix remodelling either directly or by their mitogenic effect on fibroblasts. The negative charge of proteoglycans likely impairs the water content and thus the turgor and elasticity of the tissue. Mast cells also play an active role in wound healing, where they accumulate [17] and secrete histamine, VEGF and lipid mediators which attract different other inflammatory cell types [18, 19]. Typically, mast cells are present in hypertrophic scars, where they are considered as a key stimulus for fibroblasts [20]. It is postulated that in fibrosis, the formation of gap junctions between mast cell and fibroblast leads to reciprocal cell activation [21]. To date, however, it has not been shown that the blockage of gap junctions reduces fibrosis in vivo.
Mast cells have been implicated in the pathogenesis of fibrosis in various organs: mast cells drive cardiac fibrosis after ischaemic cardiopathy or pressure overloaded heart model [22]. Conversely, mast cell deficient mice are protected from cardiac fibrosis during heart failure [23]. In a rat model of hypertensive cardiomyopathy, mast cell stabilisation with neocromil led to normalisation of IL-6 and IL-10 levels [24]. In the kidney, mast cells are not present under normal circumstances. However in renal fibrosis, e.g., due to IgA nephropathy, the number of mast cells is increased and correlates with fibrosis [25]. Similarly, renal fibrosis in kidney rejection after transplant is associated with mast cell infiltration [26]. In patients with idiopathic pulmonary fibrosis (IPF), mast cells are increased in the lung tissue [27] and mast cell products in the bronchoalveolar lavage are higher than in healthy individuals [28]. There are several factors which are released by mast cells and considered to play a pivotal role in lung fibrosis. Expression of mast cell chymase in human IPF is increased [29] and the secretion of histamine or the activation of the renin-angiotensin system stimulates the production of collagen by fibroblasts [30]. Mast cells are often encountered in proximity to IL-4 positive cells, indicating that mast cell infiltration is associated with a Th-2 weighted inflammation. SCF as a survival factor for mast cells is increased in fibrotic lung tissue and mainly produced by fibroblasts [31]. This reciprocal stimulation of mast cells and fibroblasts seems to require direct cell-cell contact. For this interaction, activation of the PAR-2/PKC-α/Raf-1/p44/42 signalling pathway and the presence of SCF are believed to play an important role [32]. Interestingly, mast cells are increased both in non-specific interstitial pneumonia (NSIP), which is considered as a more inflammatory subtype of IPF, and in usual interstitial pneumonia (UIP), the classical form of lung fibrosis with honeycombing and fibroblast foci [33]. This could indicate that mast cell proliferation in fibrosis might occur primarily as a disorder of the innate immune system, without activation of professional immune cells such as lymphocytes. In fact, previous studies have shown that T-regulatory lymphocytes can stabilise mast cells in chronic inflammation, which could explain the more destructive course of UIP compared to NSIP [34]. Whether mast cells present (auto)antigens to lymphocytes in lung fibrosis, or if there is an impaired cross talk between those cells, has not yet been clarified. In autoimmune disorders such as rheumatoid arthritis, it is postulated that activating IgG-receptors on mast cells are engaged by IgG autoantibodies [35] which might explain mast cell activation in secondary lung fibrosis. In any case, besides histamine, tryptase or chymase, mast cells are a large reservoir of preformed TNF-alpha and IL-17, both of which can be associated with fibrosis and definitely play a crucial role in rheumatoid arthritis [11].
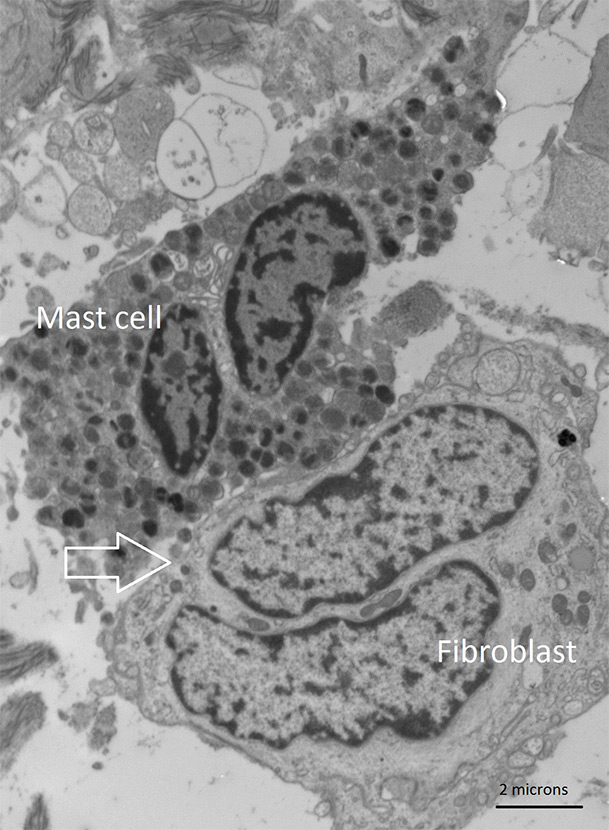
Figure 2
Transmission electron microscopy of the dermis in a patient with systemic sclerosis. The image shows cell-to-cell contact between a mast cell and a fibroblast with transgranulation (arrow).
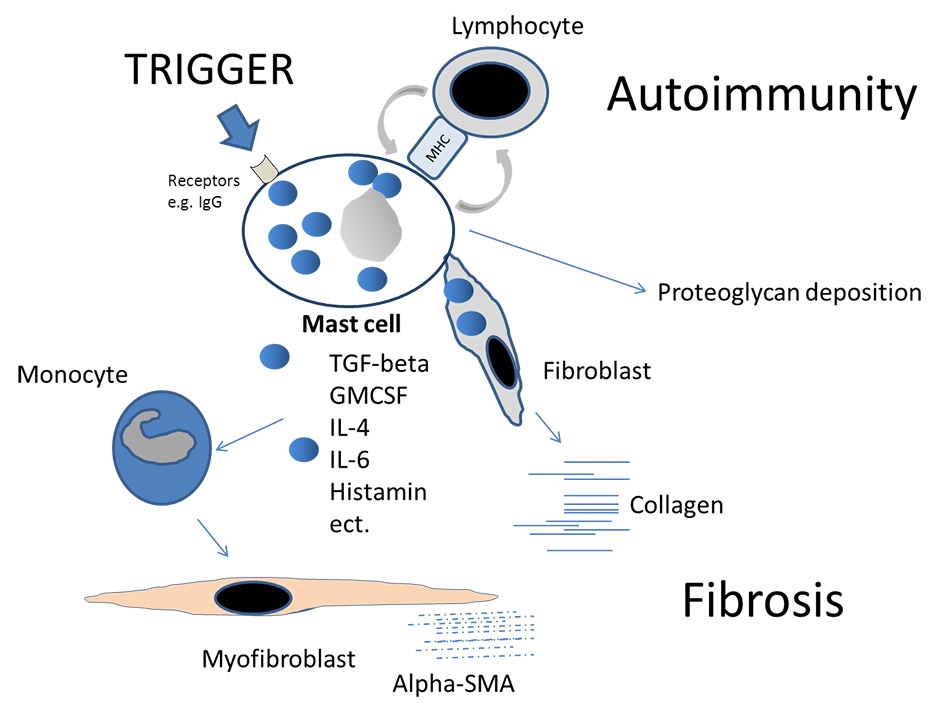
Figure 3
Schematic illustration of mast cells as key players in fibrosis. After stimulation by a trigger, e.g., IgG molecules, mast cells induce profibrotic cytokines or growth factors. Fibroblasts are stimulated and monocytes differentiate into myofibroblasts. Proteoglycans can be directly released by the mast cell in the surrounding matrix. Antigen-presentation to lymphocytes might induce autoimmunity. The suppression of mast cell degranulation by T-regulatory cells under physiological conditions can be impaired in chronic inflammation.
Due to its systemic nature, SSc can be considered as the prototype of all fibrotic diseases. It has been shown that the number of mast cells in the skin of SSc patients increased [36] and that mast cells in SSc are activated, as seen by their heterochrome vesicle colour. The fact that SSc patients often suffer from pruritus, notably in the early phase of the disease, suggests mast cell activation and the release of histamine, at least in an early stage. However, biomarkers for mast cell degranulation such as serum tryptase usually are not elevated in patients with SSc or other forms of fibrosis. Possible reasons for this can be the slow dynamic of mast cell degranulation or different expression patterns compared to allergic reaction. In the skin (and likewise in other tissues) of SSc patients, mast cells are the main source of TGF-beta. In immuno-gold labelled electron microscopy experiments, we have shown that mast cell vesicles contain a substantial part of all TGF-beta molecules [37] and that TGF-beta in the vesicles is being transferred from the mast cell to the fibroblasts (fig. 2). The likely underestimated phenomenon of transgranulation via cell-cell contacts as mechanism of fibrosis has previously been demonstrated in vitro,by the transfer of dye from mast cells to fibroblasts [38] and by our group in the dermis of SSc patients [39] (fig. 3). In SSc, IL-4 and GM-CSF, both which are stored in mast cells, participate in the differentiation of peripheral blood-derived monocytes into functionally active and contractile myofibroblasts [40].
Unlike allergic disorders, mast cell activation in fibrosis does not seem to be IgE-driven and histamine only plays a minor role. To this end, antihistamines or IgE-targeting monoclonal antibodies are unlikely to succeed as a treatment for fibrosis. Mast cell stabilisers such as ketotifen appear more attractive. Ketotifen has reduced capsule fibrosis in post-traumatic joint contraction in animal models [41] and in the tight skin mouse model, both ketotifen and cromoglycate prevented skin fibrosis [42, 43]. However, unlike positive results previously obtained in case series, ketotifen was not superior to a placebo in a double-blind controlled trial in patients with early SSc [44]. Possibly, the number of 24 patients in this study was too low and 6–months follow-up too short for this highly heterogeneic disease. In early SSc or other fibrotic disorders with concomitant autoimmune inflammation, a combination between a mast cell stabiliser and a compound acting on (autoimmune) lymphoproliferation such as methotrexate or steroids might be more promising. Another promising approach might be tyrosine blockage by masitinib which specifically inhibits the receptor tyrosine kinase c-kit and is used for mast cell tumours. Masitinib is currently tested in multiple sclerosis (ClinicalTrials.gov Identifier: NCT01450488) but not yet in SSc. Other tyrosine kinase inhibitors such as imatinib have recently shown positive effects in animal models and case series, but failed in controlled trials [45]. The probable reason for this discrepancy might be the low expression of tyrosine kinase inhibitor targets (PDGF or C-Abl) in skin tissue from SSc patients compared with certain animal models [46].
Mast cells are natural biosensors of the interstitial tissue orchestrating vasotonus and extracellular matrix turn-over. Slow release of mast cell vesicles with profibrotic cytokines and growth factors induce collagen expression in fibroblasts. Transgranulation via cell-cell contact seems to underline this phenomenon. Mast cells also interact with other immune cells such as monocytes or lymphocytes and monocyte differentiation into myofibroblasts has been described in SSc. Activated mast cells also release proteoglycans as direct matrix components. Therefore, fibrosis could be regarded as unspecific result of chronic mast cell activation rather than being specific for one disease. Further studies are necessary to understand this vesicle transport and, more importantly, to identify triggers for mast cell degranulation to develop new efficient treatment options.
Acknowledgement: I thank Dr. Jeroen Geurts and Ben Pippenger for critically reviewing this manuscript.
1 Ehrlich P. Beiträge zur Kenntnis der Anilinfärbungen und ihrer Verwendung in der mikroskopischen Technik. Archiv fuer mikroskopische Anatomie. 1877;13:263–78.
2 Kitamura Y, Tamai M, Miyano Y, Shimada M. Development of hematopoietic spleen colonies in nonirradiated genetically normal mice. Blood. 1977;50(6):1121–7.
3 Kirshenbaum AS, Goff JP, Semere T, Foster B, Scott LM, Metcalfe DD. Demonstration that human mast cells arise from a progenitor cell population that is CD34(+), c-kit(+), and expresses aminopeptidase N (CD13). Blood. 1999;94(7):2333–42.
4 Gould HJ, Sutton BJ. IgE in allergy and asthma today. Nat Rev Immunol. 2008;8(3):205–17.
5 Prussin C, Metcalfe DD. 4. IgE, mast cells, basophils, and eosinophils. J Allergy Clin Immunol. 2003;111(2 Suppl):S486–S494.
6 Nakamura Y, Oscherwitz J, Cease KB, Chan SM, Munoz-Planillo R, Hasegawa M, et al. Staphylococcus delta-toxin induces allergic skin disease by activating mast cells. Nature. 2013;503(7476):397–401.
7 Dvorak AM, Kissell S. Granule changes of human skin mast cells characteristic of piecemeal degranulation and associated with recovery during wound healing in situ. J Leukoc Biol. 1991;49(2):197–210.
8 Wang HW, Tedla N, Lloyd AR, Wakefield D, McNeil PH. Mast cell activation and migration to lymph nodes during induction of an immune response in mice. J Clin Invest. 1998;102(8):1617–26.
9 Sandig H, Bulfone-Paus S. TLR signaling in mast cells: common and unique features. Front Immunol. 2012;3:185.
10 Zhang D, Spielmann A, Wang L, Ding G, Huang F, Gu Q, et al. Mast-cell degranulation induced by physical stimuli involves the activation of transient-receptor-potential channel TRPV2. Physiol Res. 2012;61(1):113–24.
11 Hueber AJ, Asquith DL, Miller AM, Reilly J, Kerr S, Leipe J, et al. Mast cells express IL-17A in rheumatoid arthritis synovium. J Immunol. 2010;184(7):3336–40.
12 Claman HN. On scleroderma. Mast cells, endothelial cells, and fibroblasts. JAMA. 1989;262(9):1206–9.
13 Andrade ZA. Schistosomiasis and liver fibrosis. Parasite Immunol. 2009;31(11):656–63.
14 Sabin EA, Kopf MA, Pearce EJ. Schistosoma mansoni egg-induced early IL-4 production is dependent upon IL-5 and eosinophils. J Exp Med. 1996;184(5):1871–8.
15 Taipale J, Lohi J, Saarinen J, Kovanen PT, Keski-Oja J. Human mast cell chymase and leukocyte elastase release latent transforming growth factor-beta 1 from the extracellular matrix of cultured human epithelial and endothelial cells. J Biol Chem. 1995;270(9):4689–96.
16 Eggli PS, Graber W. Cytochemical localization of hyaluronan in rat and human skin mast cell granules. J Invest Dermatol. 1993;100(2):121–5.
17 Trautmann A, Toksoy A, Engelhardt E, Brocker EB, Gillitzer R. Mast cell involvement in normal human skin wound healing: expression of monocyte chemoattractant protein-1 is correlated with recruitment of mast cells which synthesize interleukin-4 in vivo. J Pathol. 2000;190(1):100–6.
18 Takato H, Yasui M, Ichikawa Y, Waseda Y, Inuzuka K, Nishizawa Y, et al. The specific chymase inhibitor TY-51469 suppresses the accumulation of neutrophils in the lung and reduces silica-induced pulmonary fibrosis in mice. Exp Lung Res. 2011;37(2):101–8.
19 Weller CL, Collington SJ, Brown JK, Miller HR, Al-Kashi A, Clark P, et al. Leukotriene B4, an activation product of mast cells, is a chemoattractant for their progenitors. J Exp Med. 2005;201(12):1961–71.
20 Gailit J, Marchese MJ, Kew RR, Gruber BL. The differentiation and function of myofibroblasts is regulated by mast cell mediators. J Invest Dermatol. 2001;117(5):1113–9.
21 Ehrlich HP. A Snapshot of Direct Cell-Cell Communications in Wound Healing and Scarring. Adv Wound Care. (New Rochelle) 2013;2(4):113–21.
22 Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci. 2013;71:549–74.
23 Hara M, Ono K, Hwang MW, Iwasaki A, Okada M, Nakatani K, et al. Evidence for a role of mast cells in the evolution to congestive heart failure. J Exp Med. 2002;195(3):375–81.
24 Levick SP, McLarty JL, Murray DB, Freeman RM, Carver WE, Brower GL. Cardiac mast cells mediate left ventricular fibrosis in the hypertensive rat heart. Hypertension. 2009;53(6):1041–7.
25 Roberts IS, Brenchley PE. Mast cells: the forgotten cells of renal fibrosis. J Clin Pathol. 2000;53(11):858–62.
26 Ishida T, Hyodo Y, Ishimura T, Takeda M, Hara I, Fujisawa M. Mast cell numbers and protease expression patterns in biopsy specimens following renal transplantation from living-related donors predict long-term graft function. Clin Transplant. 2005;19(6):817–24.
27 Tuder RM. A pathologist’s approach to interstitial lung disease. Curr Opin Pulm Med. 1996;2(5):357–63.
28 Pesci A, Bertorelli G, Gabrielli M, Olivieri D. Mast cells in fibrotic lung disorders. Chest. 1993;103(4):989–96.
29 Hirata K, Sugama Y, Ikura Y, Ohsawa M, Inoue Y, Yamamoto S, et al. Enhanced mast cell chymase expression in human idiopathic interstitial pneumonia. Int J Mol Med. 2007;19(4):565–70.
30 Veerappan A, O'Connor NJ, Brazin J, Reid AC, Jung A, McGee D, et al. Mast cells: a pivotal role in pulmonary fibrosis. DNA Cell Biol. 2013;32(4):206–18.
31 Fireman E, Kivity S, Shahar I, Reshef T, Mekori YA. Secretion of stem cell factor by alveolar fibroblasts in interstitial lung diseases. Immunol Lett. 1999;67(3):229–36.
32 Wygrecka M, Dahal BK, Kosanovic D, Petersen F, Taborski B, von GS, et al. Mast cells and fibroblasts work in concert to aggravate pulmonary fibrosis: role of transmembrane SCF and the PAR-2/PKC-alpha/Raf-1/p44/42 signaling pathway. Am J Pathol. 2013;182(6):2094–108.
33 Hirata K, Sugama Y, Ikura Y, Ohsawa M, Inoue Y, Yamamoto S, et al. Enhanced mast cell chymase expression in human idiopathic interstitial pneumonia. Int J Mol Med. 2007;19(4):565–70.
34 Piconese S, Gri G, Tripodo C, Musio S, Gorzanelli A, Frossi B, et al. Mast cells counteract regulatory T-cell suppression through interleukin-6 and OX40/OX40L axis toward Th17–cell differentiation. Blood. 2009;114(13):2639–48.
35 Merluzzi S, Betto E, Ceccaroni AA, Magris R, Giunta M, Mion F. Mast cells, basophils and B cell connection network. Mol Immunol. 2014;S0161–5890(14)00050–9:Epub ahead of print.
36 Hawkins RA, Claman HN, Clark RA, Steigerwald JC. Increased dermal mast cell populations in progressive systemic sclerosis: a link in chronic fibrosis? Ann Intern Med. 1985;102(2):182–6.
37 Hugle T, Hogan V, White KE, van Laar JM. Mast cells are a source of transforming growth factor beta in systemic sclerosis. Arthritis Rheum. 2011;63(3):795–9.
38 Trautmann A, Krohne G, Brocker EB, Klein CE. Human mast cells augment fibroblast proliferation by heterotypic cell-cell adhesion and action of IL-4. J Immunol. 1998;160(10):5053–7.
39 Hugle T, White K, van Laar JM. Cell-to-cell contact of activated mast cells with fibroblasts and lymphocytes in systemic sclerosis. Ann Rheum Dis. 2012;71:1582.
40 Binai N, O’Reilly S, Griffiths B, van Laar JM, Hugle T. Differentiation potential of CD14+ monocytes into myofibroblasts in patients with systemic sclerosis. PLoS One 2012;7(3):e33508.
41 Monument MJ, Hart DA, Befus AD, Salo PT, Zhang M, Hildebrand KA. The mast cell stabilizer ketotifen reduces joint capsule fibrosis in a rabbit model of post-traumatic joint contractures. Inflamm Res. 2012;61(4):285–92.
42 Walker M, Harley R, LeRoy EC. Ketotifen prevents skin fibrosis in the tight skin mouse. J Rheumatol. 1990;17(1):57–9.
43 Walker MA, Harley RA, LeRoy EC. Inhibition of fibrosis in TSK mice by blocking mast cell degranulation. J Rheumatol. 1987;14(2):299–301.
44 Gruber BL, Kaufman LD. A double-blind randomized controlled trial of ketotifen versus placebo in early diffuse scleroderma. Arthritis Rheum. 1991;34(3):362–6.
45 Beyer C, Distler JH, Distler O. Are tyrosine kinase inhibitors promising for the treatment of systemic sclerosis and other fibrotic diseases? Swiss Med Wkly. 2010;140:w13050.
46 Maurer B, Distler A, Dees C, Khan K, Denton CP, Abraham D, et al. Levels of target activation predict antifibrotic responses to tyrosine kinase inhibitors. Ann Rheum Dis. 2013;72(12):2039–46.
Funding / potential competing interests: No financial support and no other potential conflict of interest relevant to this article were reported.