Figure 1
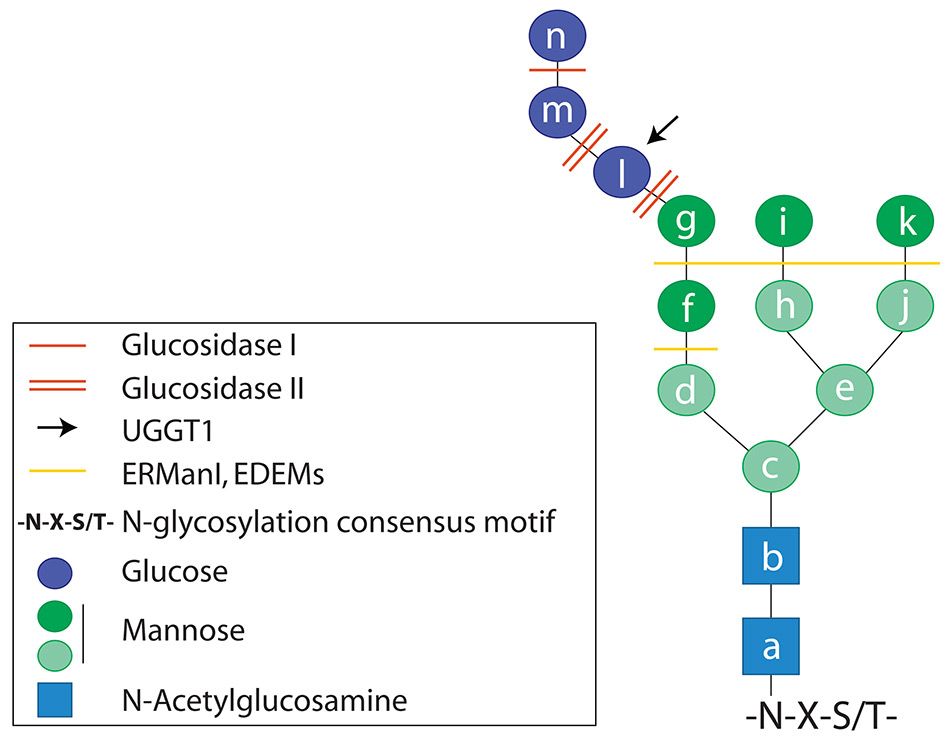
Structure of the N-linked oligosaccharide. The oligosaccharide is covalently bound to the side chain of asparagine residues in asparagine(N)-any aminoacid(X)-serine(S) or threonine(T) consensus motives.
DOI: https://doi.org/10.4414/smw.2014.14001
The majority of the nascent polypeptides emerging in the ER lumen are rapidly modified by covalent attachment of pre-assembled oligosaccharides to asparagine residues within a specific consensus motif (N-glycosylation) [1]. The transfer of the 14–units of oligosaccharide (three glucoses, nine mannoses and two N-acetylglucosamines, fig. 1) from a lipid donor in the ER membrane to nascent proteins is mediated by the oligosaccharyltransferase complex (OST) (fig. 2) [2]. N-glycosylation increases the solubility of unstructured nascent polypeptide chains and supports the recruitment of lectin (i.e. sugar-binding) molecular chaperones and associated folding enzymes that catalyse rate-limiting reactions during the folding process [3–5].
Figure 1
Structure of the N-linked oligosaccharide. The oligosaccharide is covalently bound to the side chain of asparagine residues in asparagine(N)-any aminoacid(X)-serine(S) or threonine(T) consensus motives.
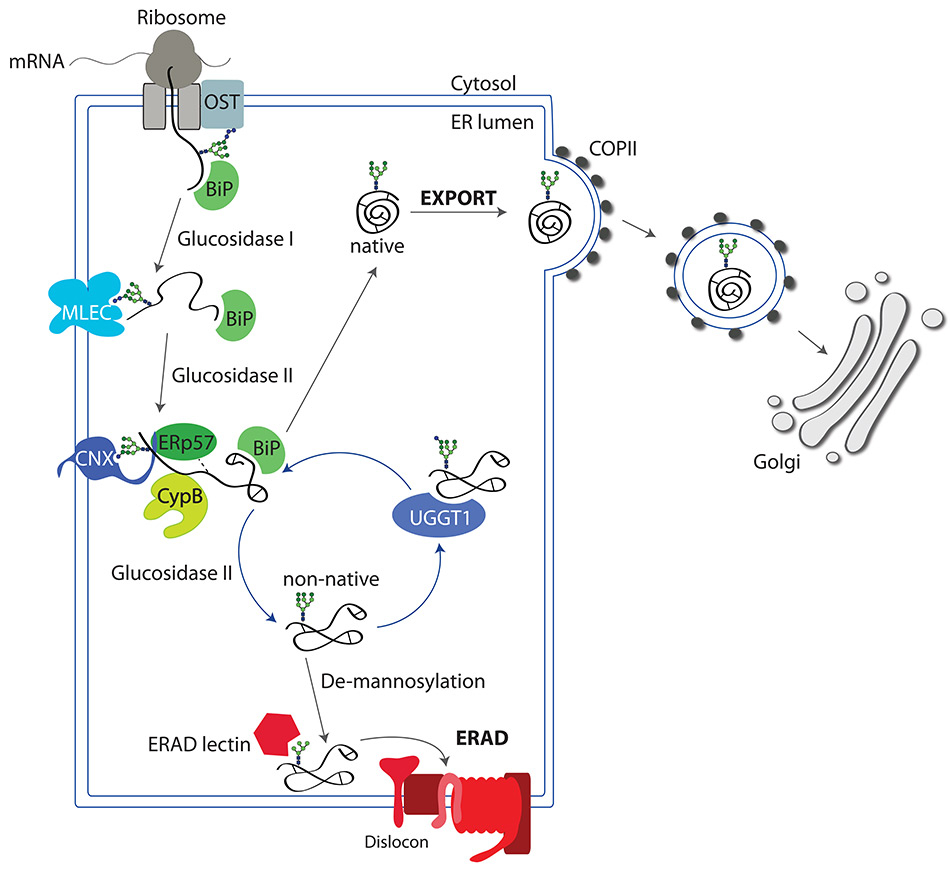
Figure 2
Folding cycle in the ER. The nascent polypeptide arising from the ribosome is translocated into the ER lumen. The polypeptide is co-translationally N-glycosylated by the OST. Hydrophobic patches exposed by the not-yet native polypeptide are shielded by molecular chaperones such as BiP to prevent aggregation. Upon removal of the outermost glucose residue by glucosidase I, the lectin malectin binds to the N-glycan. Removal of the second glucose residue by glucosidase II, allows recruitment of CNX (or CRT) and the associated folding enzymes (the oxidoreductase ERp57 and the peptidyl-prolyl cis/trans isomerase CypB that catalyse the rate-limiting steps of the folding programme leading to the attainment of the correct configuration of disulfide and peptidyl-prolyl bonds, respectively). The polypeptide is eventually released from CNX, the third glucose residue is removed by glucosidase II, and the polypeptide structure is inspected by the folding sensor UGGT1. Native proteins are exported to the Golgi in COPII-coated vesicles. Non-native polypeptides are re-glucosylated by UGGT1 and sent back to the CNX folding platform. Terminally misfolded polypeptides are extensively de-mannosylated and dislocated across the ER membrane to be degraded by cytosolic proteasomes in processes collectively defined as ER-associated degradation (ERAD).
Upon addition of the N-glycan, the outermost glucose residue is immediately removed by the glucosidase I. The resulting di-glucosylated N-glycan is a ligand for the ER lectin malectin [6] that preferentially associates with newly synthesised polypeptides that have entered off-pathways of the folding programme and must be selected for rapid destruction (fig. 2) [7].
Subsequent trimming of the second glucose residue by glucosidase II generates a mono-glucosylated N-linked glycan that attracts CNX, a type I integral membrane lectin chaperone, and its luminal paralogue CRT. Binding of CNX and CRT as well as association with other molecular chaperones such as BiP/GRP78 and GRP94 protect nascent polypeptide chains from aggregation in the crowded ER environment and initiate the folding programme (fig. 2) [8, 9].
CNX and CRT form functional complexes with ERp57 and CypB [10]. These are members of the protein disulfide isomerase (PDI) and of the peptidyl-prolyl cis/trans isomerase families that promote two rate-limiting steps during protein folding, namely the formation of disulfide bonds (i.e., of inter- and intra-molecular covalent bonds between the side chains of cysteine residues) [11] and the cis/trans isomerisation of peptidyl-prolyl bonds (fig. 2).
Substrate association with CNX and CRT and the enzymatic action of ERp57 and CypB are concluded by the removal of the third glucose residue from the oligosaccharide displayed on newly synthesised proteins, which is operated by glucosidase II (fig. 2) [12]. After release from the lectin chaperones, the folding protein collapses into a three-dimensional structure, which is carefully inspected by the ER quality control system.
The folding status of newly synthesised glycoproteins released by CNX and CRT is checked by the UDP-glucose:glycoprotein glucosyltransferase 1 (UGGT1) [13]. Native proteins are ignored by the UGGT1 and can be exported from the ER to their final intra- or extracellular destination (fig. 2). Non-native polypeptides that display structural defects such as surface-exposed hydrophobic patches are recognised by the UGGT1 which then adds a terminal glucose residue to their N-glycans thereby re-generating a binding site for CNX/CRT (fig. 2) [8, 13]. Re-association of non-native polypeptides with CNX/CRT exposes them again to the action of folding enzymes that will re-arrange disulfide and peptidyl-prolyl bonds, eventually promoting the attainment of the native structure. Depending on the nature of the polypeptide, only one or several cycles of release/re-binding are necessary to obtain the native 3D structure [14].
In some cases, the newly synthesised proteins are folding-defective because of mutations in the DNA or in the mRNA that cause amino acids substitutions/deletions or premature interruption of the polypeptide chain (fig. 2, fig. 3, step 1). For glycosylated proteins, the signal for destruction is activated following prolonged retention in the ER lumen, which is a symptom of problems in attaining the native structure and consists in the extensive de-mannosylation of the N-glycan by the ER-resident mannosidases ERManI, EDEM1, EDEM2, EDEM3. These remove α1,2–bonded mannose residues (fig. 1, dark green circles) from oligosaccharides displayed on terminally misfolded polypeptides [15]. De-mannosylated N-glycans recruit the ERAD lectins OS-9 and XTP3–B that shuttle the misfolded protein to large supramolecular complexes integrated in the ER membrane, the dislocons (fig. 2, fig. 3, step 2) [16]. Dislocons are built around membrane-embedded E3 ubiquitin ligases and comprise, amongst other components, of PDIs [17, 18] and PPIs [19] that unfold the polypeptide chain to be dislocated across the ER membrane. The ER membrane contains several distinct E3 ligase complexes with peculiar substrate specificity that tag terminally misfolded polypeptides for degradation upon addition of long chains of the small molecule ubiquitin. Extraction from the ER and channelling of misfolded proteins to cytosolic proteasomes is regulated by the ATP-driven chaperone VCP/p97 (fig. 3, step 2) [16, 20, 21].
It has been observed that misfolded proteins may accumulate in a specialised sub compartment of the ER upon proteasomal inhibition, the so-called ER quality control compartment (ERQC) [22]. The ERQC appears to function as a deposit site for the toxic non-native protein species under conditions of proteasome dysfunction. Additionally, misfolded proteins that are retro-translocated from the ER can be deposited in cytosolic aggresomes to prevent interferences with cellular functions [23].
Accumulation of non-native polypeptides in the ER can have detrimental consequences if not resolved. The cell can encounter this situation following increased secretory demands (e.g. insulin production in β cells of the pancreas, antibody production of plasma cells), upon defective function of the chaperone machinery, environmental changes such as redox, ions, nutrient imbalances, pathogen infection, differentiation, aging or, more relevant in this context, the expression of mutated gene products [20].
In healthy cells, ERAD activity is tightly regulated. This prevents the inappropriate destruction of non-native intermediates of protein folding programmes that could occur when un-physiologically high ERAD activity interferes with ongoing folding programmes, or the intracellular accumulation of misfolded polypeptides as a consequence of insufficient ERAD activity. A well-studied example of pathologic consequences of de-regulated ERAD is the inappropriate destruction of the tumour suppressor KAI1 in certain tumour cell types characterised by hyperactivity of the E3 ubiquitin ligase gp78 that results in enhanced metastatic potential [24].
Recent evidence reveals that adaptation of ERAD activity is an early response following fluctuations of ER cargo load, leading to a rise in immature polypeptides in the ER lumen. This early response occurs at the post-translational level, has been named ERAD tuning (fig. 3, step 3) and is thoroughly described elsewhere [21, 25–27].
When activation of post-translational programmes is insufficient to relieve the stress situation and the ER load reaches a certain threshold, transcriptional programmes are triggered following signal transmission from the ER to the nucleus (fig. 3, step 4). In mammals, the UPR is regulated by three distinct pathways responding to the activation of three ER-resident transmembrane “stress sensors”: ATF6 (activating transcription factor 6), IRE1 (inositol-requiring enzyme 1) and PERK (protein kinase RNA-activated-like ER kinase) (fig. 3, step 4). Under stress conditions, ATF6 and IRE1 activation results in transcriptional induction of folding, degradation and lipid synthesis factors. PERK activation attenuates cargo protein synthesis and may eventually trigger cell-death pathways [20, 28]. All in all, the activation of the transcriptional UPR serves to re-establish ER proteostasis or, if this fails, to eliminate the suffering cell from the organism. While cells can adapt to mild forms of ER stress [29], severe chronic stress may lead to cell death and thereby to destruction of the affected tissue [20]. Cells in aged individuals show a decreased ability to activate the pro-survival arms of the UPR, which may enhance the risk and worsen the outcome of diseases related to impaired ER proteostasis [30–32]. Interestingly, it has recently been shown in C. elegans that the aging process can be slowed down by ectopic expression of XBP1s in neurons [33]. Crucial for the enhanced lifespan is the secretion of neurotransmitters from XBP1s-expressing neurons to other tissues. Hence, ER stress signalling between tissues appears to be important for the maintenance of ER proteostasis in C. elegans, which might also be the case in higher organisms.
As with every biological process, protein biogenesis is prone to errors. Attempts to determine the average efficiency of protein folding programmes gave contradictory results with values ranging from 70% [34] to substantially more than that [35]. As for individual proteins, one could report the examples of α1–antitrypsin (A1AT), whose folding efficiency approaches the 90% [36], and of the cystic fibrosis trans-membrane regulator (CFTR), whose maturation is much less efficient (20–40 % [37]). These values may dramatically drop upon errors occurring during transcription, translation, post-translational modifications thus determining the onset of so-called conformational diseases (proteopathies, protein misfolding diseases), which result from degradation (loss-of-function) or intra-/extra-cellular accumulation (gain-of-toxic-function) of mutant polypeptides [38, 39]. The cases of hereditary lung emphysema and of cystic fibrosis (CF) are paradigmatic examples of this type of disease. In hereditary lung emphysema, the replacement of a glutamic acid at position 342 with a lysine in the A1AT sequence is sufficient to cause a 90% reduction in circulating A1AT. This results in defective protection of lung tissues from destructive proteases released by neutrophils during inflammations [40]. For CF, a deletion of a single phenylalanine at position 508 virtually abolishes CFTR folding thereby causing its clearance from cells [41] (and see below).
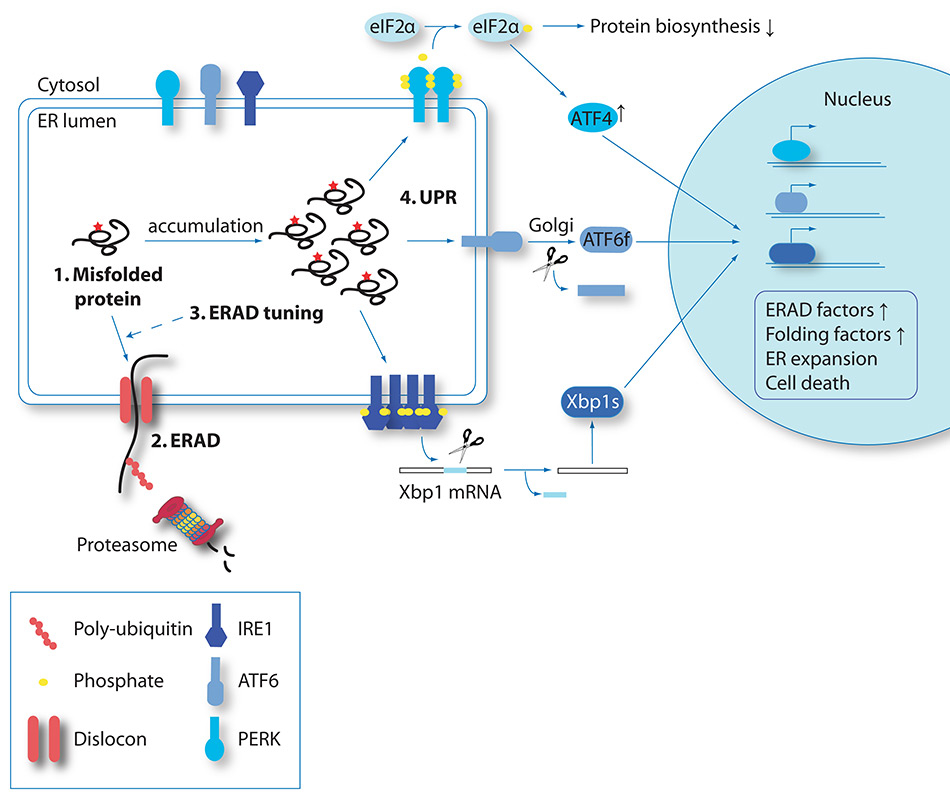
Figure 3
Responses to misfolded proteins in the ER. Misfolded protein species can arise due to the error-proneness of the folding process or due to mutations (red asterisk in step1). They are selected for ERAD (step 2). At steady state, post-translational mechanisms, collectively termed ERAD tuning, are in place to keep ERAD activity under control (step 3). Accumulation of misfolded polypeptides in the ER lumen may trigger the UPR (step 4). IRE1 forms active clusters that remove a short intron from the XBP1 mRNA thus resulting in expression of the active transcription factor XBP1s. ATF6 is transported to the Golgi, where it is cleaved to release the active transcription factor ATF6f. XBP1s and ATF6f are eventually dislocated into the nucleus, where they activate expression of ER stress-induced genes. PERK forms clusters that phosphorylate the elF2α elongation factor thereby attenuating cargo protein synthesis. On the contrary, the translation of the transcription factor ATF4 is specifically enhanced under these conditions and induces the expression of UPR genes.
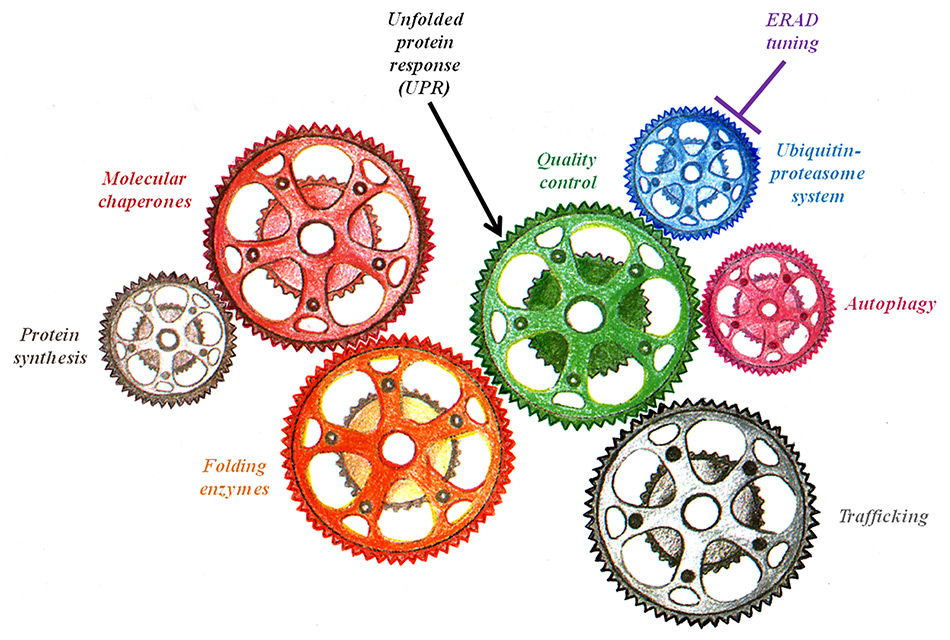
Figure 4
Schematic of the proteostasis network. The main components that regulate and maintain protein homeostasis (proteostasis) are illustrated as gears of a machine.
The terms proteopathies, conformational disorders, protein misfolding diseases refer to a group of disorders in which certain proteins fail to attain their normal conformation, resulting in degradation (loss-of-function) or accumulation (gain-of-toxic-function) of the aberrant products. Here we highlight a few selected examples:
CF is an inherited disorder characterised by the accumulation of mucus at the epithelial surfaces of several organs, such as the lungs, pancreas and gut [42–44]. This hyper-production of mucus predisposes the tissues for inflammation and chronic infection that may lead to respiratory failure, the most common cause of death in CF patients. Although treatments have been improved, the disease may progress to levels of gravity where lung or even multi-organ (liver and pancreas are also affected) transplantation represents the last solution to prolong life expectancy [45]. CF is caused by mutations that affect the folding of the CFTR, a large multi-domain protein that, even in its wild type form, has low folding efficiency [37]. CFTR biogenesis starts in the ER, where the nascent polypeptide associates with CNX. Only about 20–40% of the newly synthesised CFTR properly reaches the cell surface. Most of it is rapidly degraded from the ER. Mutations in the CFTR gene may fully prevent folding or may delay it to such an extent that the protein is selected for disposal before it actually attains the native, transport-competent conformation thus causing this loss-of-function disease [37, 41].
Metabolic storage disorders are caused by mutations that lead to insufficient activity of select enzymes. In particular, metabolic storage disorders include a subgroup of diseases in which the lysosomal activity is impaired, the so-called “lysosomal storage disorders” [46]. The deficiency of a single enzyme may compromise the overall lysosomal degradative capacity thereby resulting in the toxic intracellular accumulation of lysosomal substrates [47]. The most prominent lysosomal storage disorders are Gaucher’s and Fabry’s diseases, which are determined by deficient activity of the housekeeping enzymes beta-glucocerebrosidase (GBA) and alpha-galactosidase A (α-Gal A), respectively [48].
Gaucher’s disease is characterised by glucosylceramide (GC)/glucosylsphingosine (GS) accumulation in visceral organs (type 1) or in the central nervous system (type 2 and 3) [49]. More than 300 mutations have been identified in the GBA gene, leading to premature termination or deletions [50]. Mutant GBA variants are recognised as misfolded proteins inside the ER and thus targeted for ERAD, resulting in glycosphingolipid catabolism interruption.
Fabry’s disease is characterised by progressive accumulation of glycosphingolipids in the lysosomes of vascular endothelial cells. The α-Gal A sequence has high content of Alu sequences, resulting in propensity for gene deletions and duplications [47]. Recently, one novel disease-causing mutation has been described: this mutation is close to the N-glycosylation site, causing premature termination and leading to degradation of the aberrant product [51].
Haemophilia A (HA), also known as “royal disease”, is a genetic disorder characterised by increased bleeding. HA is caused by deficient activity or absence of clotting factor VIII (FVIII) [52]. FVIII is a large glycoprotein that folds in the CNX/CRT cycle [53]. Based on the severity of FVIII deficiency, HA has been divided in three clinical categories: mild (5–40% FVIII activity), moderate (1–5%) and severe deficiency (<1%). Classic HA is determined by different types of mutations in the FVIII gene, such as point mutations, deletions, insertions and intron inversions [52]. Mutations that cause conformational changes in the FVIII protein or lead to truncated versions of FVIII correlate with a severe degree of the pathology.
Alzheimer’s disease (AD) and Parkinson’s disease (PD) represent the most common neurodegenerative disorders characterised by progressive neuronal loss affecting defined areas of the brain [54]. Both pathologies are associated with the deposition and aggregation of misfolded proteins that cause neurodegeneration by a gain-of-toxic function mechanism [55]. Several mechanisms have been proposed to explain the toxicity annexed to the accumulation of protein aggregates, including impairment of proteasomal/lysosomal degradation and induction of ER stress.
In AD, the amyloid precursor protein APP is sequentially cleaved by β- and γ-secretases determining the production of the beta-amyloid peptide (Aβ). APP or secretases mutations may cause an increase in Aβ production leading to its deposition and extracellular amyloid plaques formation [56]. Intracellular neurofibrillary tangles have also been associated to AD. These filamentous aggregates are composed of tau, a protein that is involved both in the assembly and in the stabilisation of microtubules [57]. Tau protein is subjected to several post-translational modifications such as phosphorylation. It has been reported that hyper-phosphorylation of tau causes its instability and drives its sedimentation into the intracellular toxic tangles [54].
The typical hallmark of PD is the presence of the so-called Lewy bodies within the cytoplasm of dopaminergic neurons. The first gene associated to PD is α-synuclein, which encodes for a protein that is widely expressed in the brain and defined as intrinsically disordered [58]. It has been shown that altered expression levels, mutations and hyper-phosphorylation events of α-synuclein correlate with the toxic effects observed in the course of the pathology.
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder affecting motor neurons, which is characterised by gradual paralysis and muscle atrophy. ALS disease is associated with protein aggregates commonly found in spinal motor neurons. These inclusions are ubiquitin-positive and contain several proteins involved in mitochondrial and ER homeostasis [59, 60]. The most common aggregated protein is superoxide dismutase 1 (SOD1), whose mutations found in ALS cause misfolding and instability.
Test tube and cell culture experiments contribute to a thorough understanding of processes regulating ER proteostasis (fig. 4), which has helped to develop drugs for the treatment of proteopathies. Although some of these drugs have made it to clinical trials and beyond, more in vitro and in vivo studies are necessary to elucidate their precise mechanism of action and to find novel druggable targets. There are numerous ways to approach conformational diseases including (1) enzyme replacement therapy (ERT) for loss-of-function diseases, (2) enhancement of mutant protein folding capacity with chemical or pharmacological chaperones and (3) activation of the UPR by small molecules [48, 61].
An obvious way to counteract loss-of-function diseases is the replacement of the mutated protein by a functional, recombinant one. A drawback of this therapeutic approach is the dependence of the patients on repeated doses of the recombinant enzymes. Costs are another critical issue, especially for patients with rare diseases because therapy costs inversely correlates with the number of patients. The difficulty of this approach lies in the protocol of administration of the replacing protein. In the case of soluble proteins such as insulin (ERT for diabetes) or β-glucosidase (ERT for Gaucher’s disease), subcutaneous or intravenous administration works [62, 63]. Exogenous insulin will be transported via the bloodstream to its target tissues, exogenous β-glucosidase (or other lysosomal enzymes) will be taken up by cells and delivered via the endocytic pathway to the lysosomes where they operate. Thus, ERT treatments are successfully used for diabetes, Gaucher’s and Fabry’s disease, mucopolysaccaharidose I, II and VI or HA [63–66]. However, ERT can generally not be used to cure or prevent neuropathic diseases as the blood brain barrier does not allow efficient transport of the replacing enzyme [67], or to compensate the defective production of membrane-inserted enzymes such as the CFTR because correct insertion in the membranes, especially for polytopic membrane proteins, can only occur during the synthesis of the polypeptide.
An alternative, more broadly applicable approach to treat loss-of-function disease is to deliver the missing protein in patients by gene therapy. To this end, modified viral vectors containing the gene-of-interest are injected into patients. The use of viral vectors is problematic as it bears the risk of triggering potentially lethal immune reactions (adenovirus-based vectors) or cancer (retrovirus-based vectors) as illustrated by tragic examples in clinical trials [68]. However, improvement of the safety of viral vectors has raised new hope for the use of gene therapy. A successful example is the expression of clotting factor IX in haemophilia B patients with the help of liver-targeted adeno-associated virus (AAV) vectors in the absence of side effects [69, 70]. The first gene therapy protocol that was approved for clinical use in Europe in 2012 is a treatment for familial lipoprotein lipase deficiency based on AAV vector-mediated delivery of lipoprotein lipase [71]. An alternative to the use of viral vectors is gene delivery by liposomes. This approach is currently tested in clinical trial studies for CF [72].
Certain chemical compounds that non-covalently interact with mutant proteins are able to stabilise folding intermediates and have the potential to enhance folding capacity. These are defined as chemical chaperones. Chemical chaperones such as 4–phenylbutyric acid sodium salt (PBA) and tauroursodeoxycholic acid (TUDCA) reduce ER stress in mouse models of type 2 diabetes [73] and, as it is the case for PBA, are approved by the US Food and Drug Administration for clinical use [74, 75]. Another chemical chaperone, betaine, prevents aggregation and improves ER-to-Golgi transport of a trafficking-defective mutant of coagulation factor FVIII, thereby restoring its function in a mouse model of HA [66]. Despite these successful examples of chemical chaperone therapy, the mechanisms of action are largely unknown and unspecific. Furthermore, high concentrations have to be used that may have toxic side effects [76]. Thus, the development of more specifically acting drugs is of great clinical interest.
Pharmacological chaperones are molecules that are designed to enhance folding or to stabilise specific proteins-of-interests. There are various examples of pharmacological chaperones that are performing well in ameliorating conformational diseases. An example is the galactose derivative 1–deoxygalactonojirimycin (DGJ), which is currently in phase III clinical trials for treatment of Fabry’s disease [48]. DGJ stabilises mutant α-Gal A by binding to the active site, thereby facilitating its folding in the ER, preventing its selection for degradation and resulting in a substantial increase of active α-Gal A that eventually traffics to the lysosome. As the interaction is pH-dependent, DGJ dissociates from the α-Gal A active site in the acidic environment of the lysosome leaving a fully active enzyme. Similar compounds that stabilise the disease-causing mutated proteins in Gaucher’s disease and GM1–gangliosidosis (i.e. GBA or β-galactosidase respectively) are in phase I clinical trials [48].
Similar trials have been performed in the context of CF treatments. The pharmacological chaperone Lumacaftor (VX-809) promotes folding and cell surface transport of CFTR ΔF508 in vitro. However, it does not prevent the manifestation of CF in vivo, probably due to the compromised cell surface stability of mutant CFTR [77, 78]. A promising strategy to solve this issue seems to combine Lumacaftor with other stabilising drugs such as Ivacaftor (VX-770), which successfully treats patients with the rare CFTR Gly551Asp mutation by enhancing the opening time of CFTR channels [46, 77, 79].
Another pharmacological chaperone that has entered phase II and III clinical trials is Tafamidis (Vyndaqel®, Pfizer). Tafamidis stabilises mutant transthyretin (Val30Met) tetramers thereby preventing formation of toxic fibrils in patients with transthyretin familial amyloid polyneuropathy [46, 80].
As UPR signalling help cells to re-establish proteostasis by enhancing folding and ERAD capacities, UPR modulation might be instrumental to treat certain loss-of-function conformational diseases such as lysosomal storage disease and CF, and gain-of-toxic function diseases such as AD, PD and prion disease. As an example, small molecule UPR activators or inhibitors are under scrutiny in an attempt to improve ER proteostasis by either global UPR modulation or by manipulation of specific arms of the UPR signalling pathway. Possible risks of pharmacologic UPR induction are the induction of cell death [20] or cancer [81].
The ATF6 and IRE1 arms of the UPR are druggable targets in conformational diseases. Even though only few small molecule activators of ATF6 or IRE1 have been reported so far and their potential use in vivo has yet to be established [61, 82, 83], the aggregation of rhodopsin mutants associated with autosomal dominant retinitis pigmentosa was reduced following over-expression of either ATF6 or IRE1 in cultured primary cells, resulting in improved survival of photoreceptor cells [76, 84, 85]. Furthermore, aggregation of the disease causing PIZ variant of A1AT was reduced following over-expression of the cytosolic domain of ATF6, which is an active inducer of chaperone transcription, due to enhanced ERAD activity [86].
PERK induction attenuates cargo protein load by phosphorylation of eIF2α thereby reducing the ratio of folding intermediates to chaperones operating in the ER. This might improve the chances of mutated proteins to be properly folded [61]. Thus, prolongation of eIF2α phosphorylation with small molecules seems to be a promising strategy to ameliorate conformational diseases. As prolonged eIF2α phosphorylation might induce cell death, drugs are favoured that specifically act under conditions of ER stress, but not constitutively. One such small molecule is Guanabenz, already approved as a drug for the treatment of hypertension, which specifically inhibits eIF2α de-phosphorylation under conditions of UPR induction. The beneficial effect of Guanabenz on the survival of pancreatic β cell lines expressing mutant insulin, suggests its potential use for treatment of diabetes or other diseases caused by inefficient folding of mutant proteins [87, 88].
In contrast, translational inhibition mediated by PERK can worsen neurological pathologies. Patients with AD or prion disease show hyper-phosphorylation of eIF2α, which has been associated with decreased memory and synaptic plasticity. A recent report by Ma and colleagues suggests that deletion of PERK rescues this phenotype in AD mouse models and that targeting PERK might improve the symptoms of AD patients [88]. On the same lines, Moreno et al. demonstrate the beneficial effects of the specific PERK inhibitor GSK2606414 on prion-infected mice [89].
Production and maintenance of a functional proteome is crucial for cells, tissues and organisms viability. Highly efficient folding, quality control and transport machineries located in specific intracellular compartments such as the ER convert the genetic information stored into the cell nuclei into functional proteins and protein complexes that fulfil the wide array of functions required for life. Paradoxically, mutations that do not affect the function of a given polypeptide may result in debilitating and life threatening diseases if they introduce small structural defects. In fact, the quality control devices that prevent exit of aberrant polypeptides from the biosynthetic compartment and insure their clearance from cells are alerted by non native features such as exposure at the polypeptide surface of hydrophobic patches, unpaired cysteine residues or otherwise unstructured determinants, independent of the capacity of the mutant polypeptide to fulfil its biological activity. Cystic fibrosis, where active CFTR molecules are degraded from the ER because point mutations slightly alter the native 3D structure, is a paradigmatic example thereof. This “quality control paradox” highlights the importance of basic research in cell biology aiming at understanding the molecular basis of retention- and degradation-based mechanisms operating in our cells. Characterisation of these processes at the molecular level is required to develop therapeutic interventions that promote selective export of functional mutant proteins inappropriately segregated for architectural biases or to sustain “unfolded protein responses” that must intervene when misfolded polypeptides start to accumulate in or outside cells. This becomes even more important for aging-related diseases such as many neurodegenerative disorders, which result from gradual impairment of the proteostasis network (fig. 4), as the increased life expectancy is a fact in our society, and the number of patients will ineluctably raise.
1 Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem. 2004;73:1019–49.
2 Kelleher DJ, Gilmore R. An evolving view of the eukaryotic oligosaccharyltransferase. Glycobiology. 2006;16(4):47R-62R.
3 Caramelo JJ, Parodi AJ. How sugars convey information on protein conformation in the endoplasmic reticulum. Sem Cell Dev Biol. 2007;18(6):732–42.
4 Hebert DN, Molinari M. Flagging and docking: dual roles for N-glycans in protein quality control and cellular proteostasis. Trends Biochem Sci. 2012;37(10):404–10.
5 Molinari M. N-glycan structure dictates extension of protein folding or onset of disposal. Nat Chem Biol. 2007;3(6):313–20.
6 Schallus T, Jaeckh C, Feher K, Palma AS, Liu Y, Simpson JC, et al. Malectin: a novel carbohydrate-binding protein of the endoplasmic reticulum and a candidate player in the early steps of protein N-glycosylation. Mol Biol Cell. 2008;19(8):3404–14.
7 Galli C, Bernasconi R, Solda T, Calanca V, Molinari M. Malectin participates in a backup glycoprotein quality control pathway in the mammalian ER. PLoS One. 2011;6(1):e16304.
8 Aebi M, Bernasconi R, Clerc S, Molinari M. N-glycan structures: recognition and processing in the ER. Trends Biochem Sci. 2010;35(2):74–82.
9 Hendershot LM. The ER function BiP is a master regulator of ER function. Mt Sinai J Med. New York. 2004;71(5):289–97.
10 Jansen G, Maattanen P, Denisov AY, Scarffe L, Schade B, Balghi H, et al. An interaction map of endoplasmic reticulum chaperones and foldases. Molecular & cellular proteomics: MCP. 2012;11(9):710–23.
11 Hatahet F, Ruddock LW. Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxidants & redox signaling. 2009;11(11):2807–50.
12 Hammond C, Braakman I, Helenius A. Role of N-linked oligosaccharide recognition, glucose trimming, and calnexin in glycoprotein folding and quality control. Proceed Nat Acad Sci U S A. 1994;91(3):913–7.
13 Sousa MC, Ferrero-Garcia MA, Parodi AJ. Recognition of the oligosaccharide and protein moieties of glycoproteins by the UDP-Glc:glycoprotein glucosyltransferase. Biochemistry. 1992;31(1):97–105.
14 Solda T, Galli C, Kaufman RJ, Molinari M. Substrate-specific requirements for UGT1–dependent release from calnexin. Mol Cell. 2007;27(2):238–49.
15 Olivari S, Molinari M. Glycoprotein folding and the role of EDEM1, EDEM2 and EDEM3 in degradation of folding-defective glycoproteins. FEBS letters. 2007;581(19):3658–64.
16 Olzmann JA, Kopito RR, Christianson JC. The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harb Perspect Biol. 2013;5(9).
17 Molinari M, Galli C, Piccaluga V, Pieren M, Paganetti P. Sequential assistance of molecular chaperones and transient formation of covalent complexes during protein degradation from the ER. J Cell Biol. 2002;158(2):247–57.
18 Tsai B, Rodighiero C, Lencer WI, Rapoport TA. Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell. 2001;104(6):937–48.
19 Bernasconi R, Solda T, Galli C, Pertel T, Luban J, Molinari M. Cyclosporine A-sensitive, cyclophilin B-dependent endoplasmic reticulum-associated degradation. PloS one. 2010;5(9).
20 Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol. 2012;197(7):857–67.
21 Bernasconi R, Molinari M. ERAD and ERAD tuning: disposal of cargo and of ERAD regulators from the mammalian ER. Curr Opin Cell Biol. 2011;23(2):176–83.
22 Kamhi-Nesher S, Shenkman M, Tolchinsky S, Fromm SV, Ehrlich R, Lederkremer GZ. A novel quality control compartment derived from the endoplasmic reticulum. Mol Biol Cell. 2001;12(6):1711–23.
23 Ben-Gedalya T, Lyakhovetsky R, Yedidia Y, Bejerano-Sagie M, Kogan NM, Karpuj MV, et al. Cyclosporin-A-induced prion protein aggresomes are dynamic quality-control cellular compartments. J Cell Sci. 2011;124(Pt 11):1891–902.
24 Tsai YC, Mendoza A, Mariano JM, Zhou M, Kostova Z, Chen B, et al. The ubiquitin ligase gp78 promotes sarcoma metastasis by targeting KAI1 for degradation. Nat Med. 2007;13(12):1504–9.
25 Merulla J, Fasana E, Solda T, Molinari M. Specificity and regulation of the endoplasmic reticulum-associated degradation machinery. Traffic. 2013;14(7):767–77.
26 Leitman J, Ron E, Ogen-Shtern N, Lederkremer GZ. Compartmentalization of ER quality control and ERAD factors. DNA Cell biol. 2013;32(1):2–7.
27 Nakatsukasa K, Brodsky JL, Kamura T. Astalled retrotranslocation complex reveals physical linkage between substrate recognition and proteosomal degradation during ERAD. Mol Biol Cell. 2013;24811):1765–1775.
28 Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334(6059):1081–6.
29 Rutkowski DT, Kaufman RJ. That which does not kill me makes me stronger: adapting to chronic ER stress. Trends Biochem Sci. 2007;32(10):469–76.
30 Hussain SG, Ramaiah KV. Reduced eIF2alpha phosphorylation and increased proapoptotic proteins in aging. Biochem Biophys Res Commun. 2007;355(2):365–70.
31 Paz Gavilan M, Vela J, Castano A, Ramos B, del Rio JC, Vitorica J, et al. Cellular environment facilitates protein accumulation in aged rat hippocampus. Neurobiol Aging. 2006;27(7):973–82.
32 Naidoo N, Ferber M, Master M, Zhu Y, Pack AI. Aging impairs the unfolded protein response to sleep deprivation and leads to proapoptotic signaling. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2008;28(26):6539–48.
33 Taylor RC, Dillin A. XBP-1 is a cell-nonautonomous regulator of stress resistance and longevity. Cell. 2013;153(7):1435–47.
34 Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature. 2000;404(6779):770–4.
35 Vabulas RM, Hartl FU. Protein synthesis upon acute nutrient restriction relies on proteasome function. Science. 2005;310(5756):1960–3.
36 Bernasconi R, Pertel T, Luban J, Molinari M. A dual task for the Xbp1–responsive OS-9 variants in the mammalian endoplasmic reticulum: inhibiting secretion of misfolded protein conformers and enhancing their disposal. J Biol Chem. 2008;283(24):16446–54.
37 Kopito RR. Biosynthesis and degradation of CFTR. Physiological reviews. 1999;79(1 Suppl):S167–73.
38 Hebert DN, Molinari M. In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiological reviews. 2007;87(4):1377–408.
39 Wilke CO, Drummond DA. Signatures of protein biophysics in coding sequence evolution. Curr Opin Struct Biol. 2010;20(3):385–9.
40 Silverman GA, Pak SC, Perlmutter DH. Disorders of protein misfolding: alpha-1–antitrypsin deficiency as prototype. J Pediatr. 2013;163(2):320–6.
41 Ledford H. Drug bests cystic-fibrosis mutation. Nature. 2012;482(7384):145.
42 Cant N, Pollock N, Ford RC. CFTR structure and cystic fibrosis. Int J Biochem Cell Biol. 2014 Feb 15.
43 Balch WE, Roth DM, Hutt DM. Emergent properties of proteostasis in managing cystic fibrosis. Cold Spring Harb Perspect Biol. 2011;3(2).
44 Oberdorf J, Pitonzo D, Skach WR. An energy-dependent maturation step is required for release of the cystic fibrosis transmembrane conductance regulator from early endoplasmic reticulum biosynthetic machinery. J Biol Chem. 2005;280(46):38193–202.
45 Corris PA. Lung transplantation for cystic fibrosis and bronchiectasis. Sem Respir Crit Care Med. 2013;34(3):297–304.
46 Lindquist SL, Kelly JW. Chemical and biological approaches for adapting proteostasis to ameliorate protein misfolding and aggregation diseases: progress and prognosis. Cold Spring Harb Perspect Biol. 2011;3(12).
47 Gieselmann V. Lysosomal storage diseases. Biochim Biophys Acta. 1995;1270(2–3):103–36.
48 Suzuki Y. Chaperone therapy update: Fabry disease, GM1–gangliosidosis and Gaucher disease. Brain Dev. 2013;35(6):515–23.
49 Xu YH, Xu K, Sun Y, Liou B, Quinn B, Li RH, et al. Multiple pathogenic proteins implicated in neuronopathic Gaucher disease mice. Human Mol Genet. 2014 Mar 18.
50 Hruska KS, LaMarca ME, Scott CR, Sidransky E. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Human mutation. 2008;29(5):567–83.
51 Celtikci B, Topcu M, Ozkara HA. Two novel alpha-galactosidase A mutations causing Fabry disease: A missense mutation M11V in a heterozygote woman and a nonsense mutation R190X in a hemizygote man. Clin Biochem. 2011;44(10–11):809–12.
52 Oldenburg J, El-Maarri O. New insight into the molecular basis of hemophilia A. Int J Hematol. 2006;83(2):96–102.
53 Kaufman RJ, Pipe SW, Tagliavacca L, Swaroop M, Moussalli M. Biosynthesis, assembly and secretion of coagulation factor VIII. Blood coagulation & fibrinolysis: an international journal in haemostasis and thrombosis. 1997;8(Suppl 2):S3–14.
54 Winklhofer KF, Tatzelt J, Haass C. The two faces of protein misfolding: gain- and loss-of-function in neurodegenerative diseases. EMBO J. 2008;27(2):336–49.
55 Tenreiro S, Eckermann K, Outeiro TF. Protein phosphorylation in neurodegeneration: friend or foe? Front Mol Neurosci. 2014;7:42.
56 Drouet B, Pincon-Raymond M, Chambaz J, Pillot T. Molecular basis of Alzheimer’s disease. Cellular and molecular life sciences: CMLS. 2000;57(5):705–15.
57 Lee G, Leugers CJ. Tau and tauopathies. Prog Mol Biol Transl Sci. 2012;107:263–93.
58 Goldberg MS, Lansbury PT, Jr. Is there a cause-and-effect relationship between alpha-synuclein fibrillization and Parkinson’s disease? Nat Cell Biol. 2000;2(7):E115–9.
59 Al-Chalabi A, Jones A, Troakes C, King A, Al-Sarraj S, van den Berg LH. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol. 2012;124(3):339–52.
60 Schwenk BM, Edbauer D. The ER under rapid fire. EMBO J. 2014;33(11):1195–7.
61 Ryno LM, Wiseman RL, Kelly JW. Targeting unfolded protein response signaling pathways to ameliorate protein misfolding diseases. Curr Opin Chem Biol. 2013;17(3):346–52.
62 Leahy JL, Cefalu WT. Insulin physiology and therapy. Preface. Endocrinol Metab Clin North Am. 2012;41(1):xiii–xiv.
63 Brady RO. Emerging strategies for the treatment of hereditary metabolic storage disorders. Rejuvenation Res. 2006;9(2):237–44.
64 Giugliani R. Mucopolysacccharidoses: From understanding to treatment, a century of discoveries. Genet Mol Biol. 2012;35(4 (suppl)):924–31.
65 Noh H, Lee JI. Current and potential therapeutic strategies for mucopolysaccharidoses. J Clin Pharm Ther. 2014 Feb 25.
66 Roth SD, Schuttrumpf J, Milanov P, Abriss D, Ungerer C, Quade-Lyssy P, et al. Chemical chaperones improve protein secretion and rescue mutant factor VIII in mice with hemophilia A. PloS one. 2012;7(9):e44505.
67 Valayannopoulos V. Enzyme replacement therapy and substrate reduction therapy in lysosomal storage disorders with neurological expression. Handb Clin Neurol. 2013;113:1851–7.
68 Mullard A. Gene therapies advance towards finish line. Nat Rev Drug Discov. 2011;10(10):719–20.
69 Nathwani AC, Tuddenham EG, Rangarajan S, Rosales C, McIntosh J, Linch DC, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365(25):2357–65.
70 Cancio MI, Reiss UM, Nathwani AC, Davidoff AM, Gray JT. Developments in the treatment of hemophilia B: focus on emerging gene therapy. Appl Clin Genet. 2013;6:91–101.
71 Ferreira V, Petry H, Salmon F. Immune Responses to AAV-Vectors, the Glybera Example from Bench to Bedside. Front Immunol. 2014;5:82.
72 Alton EW, Boyd AC, Cheng SH, Cunningham S, Davies JC, Gill DR, et al. A randomised, double-blind, placebo-controlled phase IIB clinical trial of repeated application of gene therapy in patients with cystic fibrosis. Thorax. 2013;68(11):1075–7.
73 Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313(5790):1137–40.
74 de Almeida SF, Picarote G, Fleming JV, Carmo-Fonseca M, Azevedo JE, de Sousa M. Chemical chaperones reduce endoplasmic reticulum stress and prevent mutant HFE aggregate formation. J Biol Chem. 2007;282(38):27905–12.
75 Yam GH, Gaplovska-Kysela K, Zuber C, Roth J. Sodium 4–phenylbutyrate acts as a chemical chaperone on misfolded myocilin to rescue cells from endoplasmic reticulum stress and apoptosis. Investigative ophthalmology & visual science. 2007;48(4):1683–90.
76 Sauer T, Patel M, Chan CC, Tuo J. Unfolding the Therapeutic Potential of Chemical Chaperones for Age-related Macular Degeneration. Expert Rev Ophthalmol. 2008;3(1):29–42.
77 Boyle MP, De Boeck K. A new era in the treatment of cystic fibrosis: correction of the underlying CFTR defect. Lancet Respir Med. 2013;1(2):158–63.
78 Okiyoneda T, Barriere H, Bagdany M, Rabeh WM, Du K, Hohfeld J, et al. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science. 2010;329(5993):805–10.
79 Okiyoneda T, Veit G, Dekkers JF, Bagdany M, Soya N, Xu H, et al. Mechanism-based corrector combination restores DeltaF508–CFTR folding and function. Nat Chem Biol. 2013;9(7):444–54.
80 Said G, Grippon S, Kirkpatrick P. Tafamidis. Nat Rev Drug Discov. 2012;11(3):185–6.
81 Dejeans N, Manie S, Hetz C, Bard F, Hupp T, Agostinis P, et al. Addicted to secrete – novel concepts and targets in cancer therapy. Trends Mol Med. 2014 Jan 20.
82 Wang L, Perera BG, Hari SB, Bhhatarai B, Backes BJ, Seeliger MA, et al. Divergent allosteric control of the IRE1alpha endoribonuclease using kinase inhibitors. Nat Chem Biol. 2012;8(12):982–9.
83 Kudo T, Kanemoto S, Hara H, Morimoto N, Morihara T, Kimura R, et al. A molecular chaperone inducer protects neurons from ER stress. Cell Death Differ. 2008;15(2):364–75.
84 Chiang WC, Hiramatsu N, Messah C, Kroeger H, Lin JH. Selective activation of ATF6 and PERK endoplasmic reticulum stress signaling pathways prevent mutant rhodopsin accumulation. Investigative ophthalmology & visual science. 2012;53(11):7159–66.
85 Chiang WC, Messah C, Lin JH. IRE1 directs proteasomal and lysosomal degradation of misfolded rhodopsin. Mol Biol Cell. 2012;23(5):758–70.
86 Smith SE, Granell S, Salcedo-Sicilia L, Baldini G, Egea G, Teckman JH, et al. Activating transcription factor 6 limits intracellular accumulation of mutant alpha(1)-antitrypsin Z and mitochondrial damage in hepatoma cells. J Biol Chem. 2011;286(48):41563–77.
87 Tsaytler P, Harding HP, Ron D, Bertolotti A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science. 2011;332(6025):91–4.
88 Ma T, Trinh MA, Wexler AJ, Bourbon C, Gatti E, Pierre P, et al. Suppression of eIF2alpha kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat Neurosci. 2013;16(9):1299–305.
89 Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Science translational medicine. 2013;5(206):206ra138.
Funding / potential competing interests: MM is supported by Signora Alessandra, by the Foundation for Research on Neurodegenerative Diseases, the Swiss National Science Foundation and the Comel, Gabriele and Gelu Foundations.