Circadian rhythms – from genes to physiology and disease
DOI: https://doi.org/10.4414/smw.2014.13984
Thomas
Bollinger, Ueli
Schibler
Summary
Most physiological processes in our body oscillate in a daily fashion. These include cerebral activity (sleep-wake cycles), metabolism and energy homeostasis, heart rate, blood pressure, body temperature, renal activity, and hormone as well as cytokine secretion. The daily rhythms in behaviour and physiology are not just acute responses to timing cues provided by the environment, but are driven by an endogenous circadian timing system. A central pacemaker in the suprachiasmatic nucleus (SCN), located in the ventral hypothalamus, coordinates all overt rhythms in our body through neuronal and humoral outputs. The SCN consists of two tiny clusters of ~100,000 neurones in humans, each harbouring a self-sustained, cell-autonomous molecular oscillator. Research conducted during the past years has shown, however, that virtually all of our thirty-five trillion body cells possess their own clocks and that these are indistinguishable from those operative in SCN neurones. Here we give an overview on the molecular and cellular architecture of the mammalian circadian timing system and provide some thoughts on its medical and social impact.
Introduction
Owing to the rotation of the earth around its own axis all living beings are exposed to 24-hour light-dark and temperature cycles. In the course of phylogeny, most organisms from light-sensitive bacteria to mammals have evolved internal timing systems, known as circadian clocks, which allow them to anticipate the recurring daily changes of their environment. The word “circadian” is derived from the Latin words “circa diem”, meaning “about a day”. Thus, when organisms are kept under constant conditions (i.e. constant darkness and constant temperature) they display cycles of behaviour and physiology of approximately, but not exactly 24 hours. For example, in humans and mice the average free-running period length of the circadian clock is about 24.3 hours and 23.7 hours, respectively. These cycles are driven by molecular oscillators in the suprachiasmatic nucleus (SCN) located in the ventral hypothalamus, which harbours the circadian pacemaker [1, 2]. In order for the SCN to stay in synchrony with the outside world, its circadian clocks must be readjusted every day by a few minutes. This is accomplished mainly by the photoperiod. Light signals, perceived by photoreceptors and intrinsically photosensitive ganglion cells in the retina, are transmitted to SCN neurones via the retinohypothalamic tract. The SCN neurones convert the electrical into chemical signals that reset their molecular oscillators by influencing the phase of clock gene expression. How do we know that the SCN is really the conductor of the clock orchestra? The most convincing demonstration comes from stereotaxic lesion and transplantation experiments between wild-type and tau mutant hamsters, which in constant darkness display locomotor activity rhythms with period lengths of 24 hours and 20 hours, respectively [3]. Both wild-type and mutant animals with a precise ablation of their SCN are completely arrhythmic when kept in constant darkness. However, when foetal SCN brain tissue is implanted into the region of the ablated SCN, animals of both genotypes become rhythmic again after a few days. More importantly, lesioned wild-type animals with an SCN implant of the tau mutant hamster show rest-activity cycles with a period length of the latter, and vice versa. As the SCN dictates the length of the daily oscillation, it must be on top of the hierarchy of the circadian system.
It was initially believed that the SCN is the only mammalian tissue capable of circadian rhythm generation. However, Tosini and Menaker showed that cultured retina secreted melatonin, the so called “night hormone” (mostly produced by the pineal gland), in a circadian fashion [4]. Moreover, these melatonin secretion cycles could be synchronised in vitro by light-dark cycles. With the discovery of circadian clock genes in the late nineties it became possible to examine circadian oscillators in any cell type. Balsalobre and coworkers demonstrated that even cultured fibroblasts harbour circadian clocks [5], and two years later Yamazaki and coworkers made a similar finding for various peripheral rat tissues [6]. In the meantime circadian cellular oscillators have been described in virtually all cell types, suggesting that we have nearly as many circadian oscillators as we have cells (i.e. 3–4 x 1013 [7]). The cellular circadian oscillators in all cell types share the same molecular mechanism. As revealed by single cell recordings of circadian gene expression, these clocks work in a self-sustained, cell-autonomous manner [8, 9]. These observations beg the question of how all of these cellular clocks are coordinated in the body. As outlined below, the circadian system has a hierarchic architecture, in which the SCN is believed to synchronise peripheral clocks through a wide variety of mechanisms.
In mammals, including humans, virtually all aspects of physiology are rhythmic [10, 11]. Thus, cerebral activity, the cardiovascular system (heart rate and blood pressure) [12], renal activity (plasma flow, urine production, blood pressure, electrolyte and water homeostasis) [13], the endocrine system (production and secretion of hormones) [14], metabolism [15], the immune system [16], and body temperature [17] display well-coordinated daily fluctuations. Importantly, these rhythms persist under constant conditions under which the organisms do not receive time cues from the environment, and they are therefore most likely driven by the endogenous circadian timing system. In this article we will give an overview on the molecular and cellular makeup of this timekeeping system and outline prominent examples illustrating the social and medical relevance of our internal clocks.
Molecular make-up of mammalian circadian oscillators
During the past years impressive progress has been made in the identification of mammalian clock genes and mechanisms required to keep the clock ticking at the normal pace. The currently held molecular oscillator model is outlined in figure 1 (for review see [1, 18]). It is based on two interconnected feedback loops in clock gene expression. Two rhythmically active transcription factors, CLOCK and BMAL1, lie at the centre of the feedback loops. CLOCK and BMAL1 form heterodimers and activate the genes encoding the Period proteins PER1 and PER2 and the Cryprochrome proteins CRY1 and CRY2. (fig. 1) The PER and CRY proteins form large corepressor complexes with many additional polypeptides, and once these multi-subunit complexes have reached a critical concentration/activity they bind to the CLOCK-BMAL1 heterodimer and thereby attenuate its capacity to stimulate transcription (messenger RNA [mRNA] synthesis). Therefore, CRY and PER mRNAs and proteins are no longer synthesised, and owing to their relatively short half-lives, decrease in cellular concentration until the PER-CRY complexes can no longer interfere with the activity of CLOCK-BMAL1 heterodimers. As a consequence, a new PER/CRY accumulation cycle can ensue. In a second feedback loop CLOCK and BMAL1 regulate their own circadian transcription, by activating the transcription of the genes specifying the two nuclear receptors REV-ERBα and REV-ERBβ. These two repressors oscillate with a large amplitude and periodically occupy RORE DNA elements within the Bmal1 and Clock promoters, on which they compete with RORα, RORβ (which is brain-specific) and RORγ, two nuclear receptors serving as activators of Bmal1 and Clock transcription. REV-ERBα and REV-ERBβ recruit the corepressor NCoR1 and the histone deacetylase HDAC3, which silence the transcription of nearby genes [19]. In addition to these transcriptional mechanisms a plethora of well-coordinated posttranslational modifications on core clock proteins, such as protein phosphorylations and acetylations are required to keep the clocks ticking at a normal speed (for review, see [15, 18, 20]). We wish to emphasise, however, that the complexity of the molecular interactions between core clock components will make it difficult to understand at a satisfactory mechanistic level how this circuitry can generate oscillations with a period length of approximately 24 hours.
Cellular architecture of the circadian timing system
As mentioned above, virtually all body cells have their own circadian oscillator functioning according to the molecular principles described above. Given the dominating role of the SCN in coordinating rhythmic behaviour and physiology, it probably must assume two functions: the coordination of rest-activity cycles and the synchronisation of clocks in peripheral cells. The SCN is composed of two tiny aggregates of neurones located just above the optical chiasma, as implied by the term “suprachiasmatic”. In rats and humans, each of the two nuclei contains about 10,000 and 100,000 neurones, respectively. These neurones are communicating with each other by synaptic and paracrine mechanisms or gap junctions, so as to maintain phase coherence between their clocks. Ca2+ and cyclic adenosine monophosphate (cAMP) signalling [21] as well as signalling by vasointestinal peptide (VIP) through the VPAC2 receptor [22] are well-established paracrine mechanisms through which SCN cells couple their oscillators and keep them in synchrony. At least in mice these coupling mechanisms probably keep phase coherence in the SCN during the entire life time, as in long-term recordings the circadian amplitude of locomotor activity is maintained for many months without any loss of amplitude in animals kept in constant darkness [23].
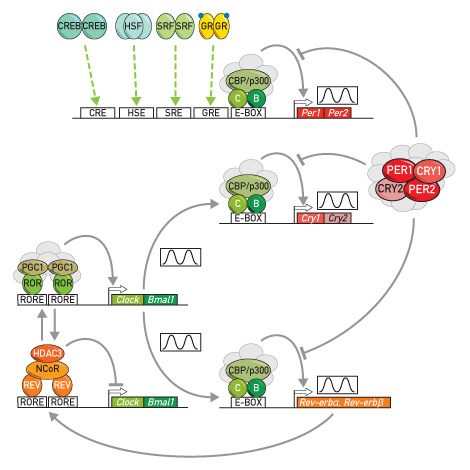
Figure 1
Simplified molecular model of the mammalian circadian oscillator.
The two transcription factors CLOCK (C) (or its closely related paralog NPAS2) and BMAL1 (B) lie at the centre of the two coupled feedback loops in clock gene expression. CLOCK and BMAL1 recruit the coactivator proteins CBP/p300 and a few other polypeptides (shown in grey) and thereby activate the transcription of their target genes in the first feedback loop (Per1, Per2, Cry1, Cry2) and in the second (accessory) feedback loop (Rev-erbα, Rev-erbβ). Together with a large number of additional polypeptides (shown in grey) PER1, PER2, CRY1, and CRY2 assemble into large protein complexes. Once these repressor complexes have reached a threshold concentration and/or activity, they annul the transactivation activity of CLOCK and BMAL1. Under these conditions, the messenger PER and CRY mRNAs and proteins are no longer made, and the repressor complexes diminish until they no longer inhibit the activity of CLOCK and BMAL1. As a consequence, a new 24-hour PER and CRY production cycle can ensue. In a second feedback loop CLOCK and BMAL1 activate and PER/CRY complexes repress the transcription of the genes encoding the two repressors REV-ERBα and REV-ERVβ (REV). The circadian expression of REVs results in the rhythmic repression of the Clock and Bmal1 genes. When REV concentrations are low, ROR transcription factors bind to RORE elements within promoter and enhancer regions of these two genes and activate transcription by recruiting protein complexes containing the coactivator PGC1. When REV concentrations are high, REV competes with RORs for the binding to RORE elements and recruits protein complexes containing the corepressor NCoR and the histone deacetylase HDAC3.
Per1 and Per2 genes not only serve as core clock components, but also as immediate early genes. Their expression can be stimulated by immediate early transcription factors whose activity is controlled by systemic cues, such as hormones, second messengers, temperature, and neurotransmitters. These immediate early transcription factors include cyclic AMP responsive element (CRE) binding protein (CREB), heat shock transcription factor 1 (HSF) binding to heat shock elements (HSE), serum response factor (SRF) binding to serum response elements (SRE), and glucocorticoid receptor (GR) binding to glucocorticoid responsive elements (GRE). The modulation of Per1 and Per2 expression by these signal-sensing transcription factors plays an important role in the synchronisation of circadian clocks.
Arrows and bars at the end of connecting lines indicate activation and repression, respectively, of transcription.
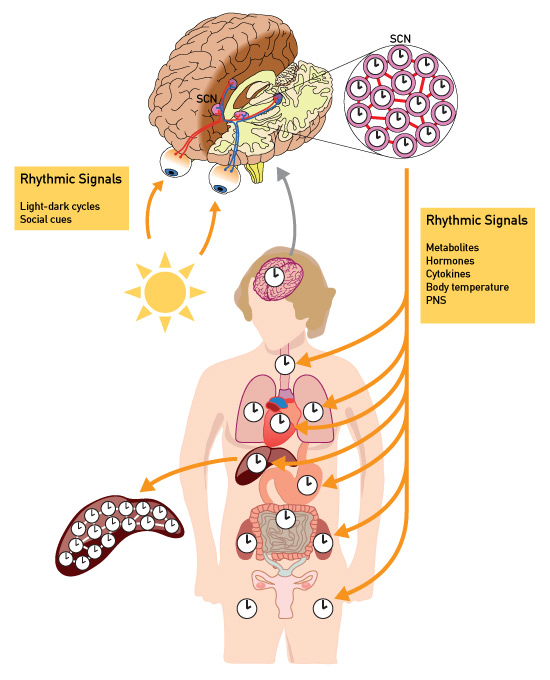
Figure 2
Molecular architecture of the mammalian circadian timing system.
Virtually all of our 3.5x1013 cells contain a circadian oscillator with the molecular makeup depicted in figure 1. The suprachiasmatic nucleus (SCN) harbours the central clock, which is responsible for all overt rhythms in behaviour and physiology. It is composed of two tiny agglomerates of neurones, whose oscillators are coupled through synaptic and paracrine signalling mechanisms (represented as red lines connecting the clock neurones). The phase of the SCN is adjusted to geophysical time every day by light signals perceived in the retina. The clocks in peripheral cell types are synchronised by the SCN through a variety of systemic signals, including metabolites (e.g. nicotinamide adenine dinucleotides), hormones (e.g. cortisol), cytokines (e.g. transforming growth factor-β), body temperature, and neuronal signals from the peripheral nervous system. In the liver, the phases of oscillators remain coupled even in the absence of systemic signals from the SCN.
As the SCN can generate cycles of only approximately 24 hours, it must be synchronised daily by the photoperiod in order to stay in resonance with geophysical time. During the day the endogenous clock must be resilient to photic cues, since otherwise its phase would constantly change during the light phase. Indeed, experiments with laboratory rodents show that the SCN clock can only shift its phase during the dark phase. Light signals capable of phase-entrainment are not only perceived by classical rod and cone photoreceptor cells expressing rhodopsin and conopsin, respectively, but also by intrinsically photosensitive ganglion cells in the inner retina layer [24]. These ganglion cells, which express the light receptor melanopsin, constitute about one percent of all retinal ganglion cells. Nevertheless, they are sufficient to synchronise the circadian pacemaker of visually blind mice completely devoid of rods and cones. Melanopsin knockout mice can still adjust their clock to light-dark cycles. However, the genetic ablation of the melanopsin expressing inner ganglion cells completely abolishes the capacity of mice to set their clock. Yet these mice possess a normal visual acuity, which indicates that the classical photoreceptors and their projections into the brain centres processing optical information function normally in these animals. Neuronal tracing studies using the neurotropic pseudorabies virus demonstrated, however, that all rods and cones projecting to the SCN via the retinohypothalamic tract use intrinsically photosensitive inner ganglion cells as relay stations [25]. Hence, in the absence of these cells no photic cues perceived by melanopsin or classical photo pigments can reach the SCN. Photic signals transmitted from the retina to SCN neurones lead to an influx of Ca2+ in postsynaptic neurones, which activates a series of protein kinases and stimulates the activity of cAMP response element binding protein (CREB). CREB is a transcription factor that boosts the transcription of the Per1 and Per2 genes. The sudden increase of PER1 and PER2 attenuates their own expression and that of the other two core clock repressors CRY1 and CRY2. As a consequence the phase of the transcriptional rhythm of period and cryptochrome genes is reset. Thus, Per1 and Per2 are not only integral parts of the clockwork circuitry but also immediate early genes sensing input signals (fig. 1). Immediate early genes are genes which are rapidly transcribed after an adequate stimulus.
The daily phase adaptation capacity of the SCN clock has its limits. In humans kept under a constant routine (i.e. whose clock is free-running), the phase-shifting capacity can be measured by establishing phase response curves after exposure to bright light at different times during the day. Such experiments have shown that the human circadian clock can advance its phase by maximally two hours per day and delay its phase by maximally three hours per day under optimal conditions [26, 27]. Hence, after transatlantic flights encompassing many time zones the human clocks requires several days to adapt its phase to the new time zone. During this adaptation time we suffer from jet lag, since our internal timing system is in conflict with geophysical time. This manifests itself by perturbed sleep-wake cycles, indigestion problems, frequent urination during the night and fatigue.
The implantation of an SCN from wild-type mice into genetically arrhythmic Cry1/Cry2 double knockout mice restores rhythmic locomotor activity. Therefore, peripheral oscillators play only a subordinate role, if any, in establishing rest-activity cycles [28]. Moreover, experiments with encapsulated SCN grafts in hamster suggest that at least some of the output signals involved in this process are likely to be of humoral or paracrine nature [29]. Studies in mice have indicated that SCN derived signalling peptides, such as transforming growth factor alpha (TGFα), prokineticin 2 (PK2) and cardiotrophin-like cytokine (CLC), participate in the temporal regulation of locomotor activity and body temperature rhythms [30].
Many circadian overt output cycles of the circadian timing system involve circadian clocks in peripheral cell types. These peripheral timekeepers are thought to be synchronised by the central pacemaker in the SCN. Experiments with mice and rats revealed that feeding cycles are dominant Zeitgebers (timing cues) for the oscillators operative in most peripheral organs, including liver, pancreas, kidney, heart, lung and skeletal muscles [31, 32]. Thus when feeding cycles are switched from night-time feeding – which is normal in these nocturnal rodents – to daytime feeding, the phase of circadian gene expression in peripheral organs becomes gradually inverted after a week or two. Since feeding time has little impact on the phase of the SCN, peripheral oscillators become uncoupled from the SCN under these conditions. Nonetheless, one would expect that circadian metabolic processes are not severely disturbed in day-fed animals, since the pancreas, liver, intestine and kidney still maintain the same phase, and since this phase is adjusted to that of the feeding rhythm.
Body temperature rhythms are perhaps unexpected Zeitgebers for peripheral clocks. The rhythmic expression of temperature-dependent genes emerged from a study on the genome-wide analysis of mRNAs whose cyclic expression in the mouse liver is driven by systemic cues rather than local hepatocyte clocks [33]. At times when body temperature is high, the mRNAs encoding several heat shock proteins were strongly up-regulated. In contrast, the cold-induced RNA binding protein CIRP reaches zenith concentrations when body temperature is minimal. Moreover, heat shock transcription factor HSF1, the major regulator of heat shock protein genes, moves from the cytoplasm to the nucleus during the activity phase – when body temperature is elevated – and activates the transcription of its target genes. Importantly, simulated mouse body temperature rhythms oscillating smoothly between 35 °C and 38 °C are sufficient to synchronise circadian oscillators of cultured cells, and HSF1 is required for this process [34–37]. Even more surprisingly, simulated human body temperature cycles fluctuating between 36 °C and 37 °C are able to synchronise the clocks of at least a fraction of cultured cells [37].
Glucocorticoid hormones also participate in the synchronisation of peripheral timekeepers [5, 38, 39]. These hormones (cortisol in humans, corticosterone in rodents) activate the glucocorticoid receptor, which binds to glucocorticoid-responsive elements within promoter and enhancer sequences of the Per1 and Per2 genes. A short treatment of cultured cells with the glucocorticoid receptor agonist dexamethasone transiently synchronises their circadian clocks, and mice with a hepatocyte-specific knockout of the glucocorticoid receptor gene tune their circadian liver clocks more rapidly to inverted feeding cycles than wild-type mice. This suggests that glucocorticoid hormones are used by the SCN as synchronisation signals that antagonise nutrient-dependent signals when the phase of feeding rhythms is in conflict with the phase of the SCN [38]. Another blood-borne signal of proteinaceous nature has recently been shown to govern the diurnal activity of serum response factor (SRF), another immediate early transcription factor activating Per2 gene expression. Immediate early transcription factors are regulatory proteins that are already present in an inactive form before stimulation and that become active immediately after stimulation. The SRF-mediated signalling pathway involves the diurnal regulation of actin dynamics [40]. The genetic disruption of this pathway in the liver results in an accelerated phase adaptation to inverted feeding cycles, similar to what has been observed for glucocorticoid signalling (Pascal Gos, Alan Gerber, Alfred Nordheim, and Ueli Schibler, unpublished results). Hence, signalling routes involving the glucocorticoid receptor and SRF are most likely used by the SCN for the synchronisation of peripheral clocks but not by feeding rhythms. The general architecture of the mammalian circadian timing system is schematically outlined in figure 2.
The so-called “bunker experiments” conducted by Jürgen Aschoff in the 1960s with human subjects who did not get any external timing cues for weeks revealed an unexpected finding. While all individuals displayed body temperature and urine production rhythms of nearly 24 hours even after extended time periods, a sizeable fraction of them moved progressively to extremely long sleep-wake cycles of more than 30 hours after a few days. Put otherwise, these subjects uncoupled their rest-activity cycles from their circadian clock. Aschoff first speculated on the presence of a multioscillatory system in humans (for review, see [41]), but later studies, in which the Swiss scientist Alex Borbély played a pivotal role, suggested another scenario [42]. According to their “sleep homeostasis” model, a sleep homeostat cooperates with the circadian pacemaker to determine the length of rest-activity cycles. The sleep homeostat is an hourglass timer and measures sleep pressure (fig. 3A). The longer we are awake, the more the sleep pressure builds up, until it reaches a threshold value at which we cannot avoid falling asleep. During the sleep phase the pressure diminishes, until we wake up in the morning and are ready to initiate our daily activity. In some human subjects, the sleep homeostat interacts only loosely with the circadian pacemaker and hence can be disconnected from it in the absence of external timing cues.
Circadian rhythms change during the human life span
Human birth rates follow a robust circadian oscillation. Thus, between 2 a.m. and 6 a.m. about 1.8-fold more babies are born than between 2 p.m. and 6 p.m. [43]. However, as all parents can attest, new born babies are nearly arrhythmic or have an ultradian rhythm (i.e. more or less regular episodes of activity lasting a few hours each). The consolidation of circadian sleep-wake cycles and physiological parameters like body temperature and heart rate may take several months to over a year. Surprisingly, circadian sleep-wake cycles and heartbeat rhythms are clearly detectable in human foetuses during the last weeks of pregnancy. The foetal heart rate rhythms require a brain – perhaps the suprachiasmatic nucleus – as demonstrated by the recording of discordant anencephaly twins. In fact, only the intact foetus displayed a clear daily oscillation of heartbeat frequency [44, 45].
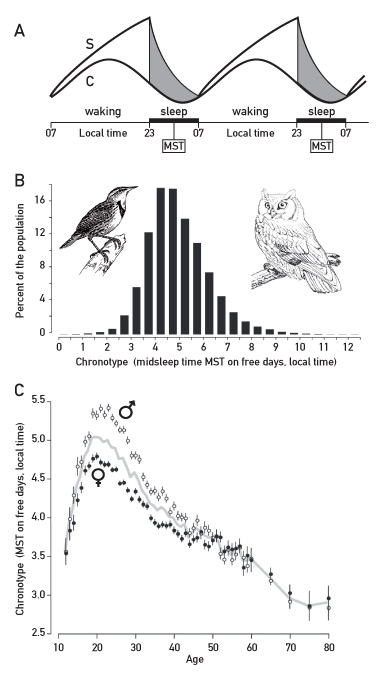
Figure 3
Sleep-wake cycles and human chronotypes.
A. Sleep-wake signals are driven by the circadian clock (C) and a sleep homeostat (S). The sleep homeostat resembles an hourglass timer. Sleep pressure steadily builds up after waking until it is so strong that the individual falls asleep. While sleeping (grey zone / black bar on X-axis), sleep pressure diminishes back to the base level. The sleep homeostat is coupled to the circadian clock under normal conditions, but in some individuals the two time keepers can become disconnected from each other after having been exposed to an extended period without external time cues (see text). MST = midsleep time. This scheme was adapted from: Daan S, Beersma DG, Borbely AA. Timing of human sleep: recovery process gated by a circadian pacemaker. Am J Physiol. 1984;246(2 Pt 2):R161–83 [42] and Borbely AA, Achermann P. Sleep homeostasis and models of sleep regulation. J Biol Rhythms. 1999;14(6):557–68 [72], with permission from the Americal Physiological Society.
B. Humans can be classified into chronotypes. Those active early during the day and late during the night are known as larks and owls, respectively, in chronobiology parlance. The chronotypes are measured by midsleep time (MST, see panel A) on free days. The values on the X-axis have been corrected for gender and age (see panel C). This scheme has been adapted from [49].
C. Age- and sex-dependent changes in chronotypes. Midsleep time (MST) on free days (chronotype) increases and then decreases with age. In teens and young adults, the late chronotype can engender a social jetlag. In females MST is reached some years earlier as compared to males. This scheme has been adapted from: Roenneberg T, Kuehnle T, Juda M, Kantermann T, Allebrandt K, Gordijn M, et al. Epidemiology of the human circadian clock. Sleep Med Rev. 2007;11(6):429–38 [49], with permission from Elsevier.
During the human lifetime two parameters of overt circadian rhythmicity change: the amplitude and the phase. The amplitudes of sleep-wake cycles, hormone secretion, body temperature, and other physiological parameters decrease with progressing age, and in extreme cases, old human subjects can become arrhythmic. For example, in Alzheimer patients the volume and cell number of the SCN can decrease dramatically, until its output is no longer sufficient to drive daily oscillations in behaviour and physiology ([46] and references therein). As suggested by measurements of melatonin rhythms in the saliva, the degeneration of circadian rhythmicity already starts to decrease gradually after puberty [47, 48]. Also, age-related phase changes of our circadian rhythms can have severe social consequences. A common way of assessing the phase of human subjects is to determine mid-sleep time (i.e. the half-way point between sleep-onset and sleep-end) on free days, that is, when there is no pressure to get out of bed early in the morning. Based on this parameter, human subjects can be divided into different chronotypes ranging from very early active “larks” to very late active individuals “owls” (fig. 3B). These studies have revealed two unexpected, if not shocking aspects of circadian rhythmicity [49]. First, extreme larks and owls hardly overlap with regard to their sleep-wake cycles. Second, the chronotype changes significantly during the lifetime of an individual. On average mid-sleep time on free days is about 3:30 a.m. local time for ten year olds, and reaches about 5 a.m. during early adulthood. It then gradually diminishes to earlier than 3 a.m. in 75 to 80 year olds (fig. 3C). With an average sleep duration of 8 hours, this means that the normal wake-up time is roughly 7:30 a.m. for elementary school children and 9 a.m. for apprentices and college students. As adolescents and young adults usually must get up much earlier than at 9 a.m. to fulfil their professional obligations, they chronically suffer from a “social jetlag” in that their endogenous phase is not in synchrony with the socially imposed phase. In addition to adversely affecting the quality of life and professional performance, social jetlag may also contribute to obesity [50].
At least in part, genetic variability may account for the dramatic differences in human chronotypes. The genetics of such complex behaviours is likely to be complex, in that the interaction of many different genes influences the time of sleep-wake cycles. However, in some extreme cases genes – known as advanced and delayed sleep phase syndromes – genes with a major impact on the onset of sleep could be identified. The human clock gene PER2 is the best characterised gene responsible for such a sleep disorder. Studies on a large family in Utah with many individuals affected by a familial advanced sleep phase syndrome (FASPS) culminated in the identification of a point mutation in PER2, which changes the serine at position 662 into a glycine (S662G) [51]. Phosphorylation at S662 by a hitherto unknown protein kinase triggers the phosphorylation of two adjacent serines by casein kinases 1ε and 1δ. These phosphorylations render the PER2 protein more stable [52] and reduce the period length of the circadian clock. It has long been known that changes in the period length frequently have dramatic effects on the phase. Indeed, people carrying one S662G PER2 mutant allele and one wild-type allele fall asleep and wake up about four hours earlier than subjects with two wild-type alleles, demonstrating the dominant) character of this FASPS mutation.
Disorders and disease related to disruption of the circadian timing system
Seasonal affective disorder (SAD) is a mood disorder frequently affecting people living in countries with low light exposures during winter time [53, 54]. In contrast to social jetlag and advanced or delayed sleep phase syndromes, which impair the wellbeing and professional performance of the concerned, SAD causes profound depression with severe comorbidities. It is believed that SAD is caused by the inability to synchronise the clock at the low light intensity of morning light in the winter season. As the average period length of the human circadian clock is somewhat longer than 24 hours, this leads to progressive phase delays and an associated social discomfort. In many cases SAD can be effectively treated with light therapy.
Shift work during extended time periods (decades) has been reported to provoke serious morbidities, including cardiovascular diseases, metabolic syndrome and malignancies such as breast and colon cancer (for review, see [18, 20]). Such claims are particularly conspicuous in grant applications for projects on circadian rhythm research. When reading epidemiological studies concerned with the health consequences of shift work one must be aware of the difficulties in discriminating between direct effects of circadian disruption and effects caused by life style changes associated with the abnormal working schedule (i.e. confounding effects). This can be exemplified by a recent study on lung cancer [55]. The lung cancer incidence (1,455 cases in total) of 78,612 nonsmoking and smoking woman (nurses) performing rotating night-shift work or not was recorded for 20 years (1988–2008). No significant relative risk (RR) of lung cancer incidence was observed among nonsmoking control subjects and rotation-shift workers, even for those having done night-shift work for more than 15 years. In contrast a statistically significant increase in relative risk (RR = 1.61, 95% confidence interval [CI] 1.21‒2.13; p <0.001) was observed among smokers (values corrected for cigarette consumption and age) who performed shift work for at least 15 years. Obviously, cigarette smoking is a major risk factor for lung cancer, so the absolute incidence was higher among smokers. If we assume that the elevated relative risk in smoking night shift nurses is not confounded by residual differences in smoking habits (i.e. inhaling deeper, smoking different brands of cigarettes, etc), this would indicate that shift work constitutes a “second hit” in oncogenesis, perhaps caused by disruption of circadian timing. For example, the arylhydrocarbon receptor, a transcription factor induced by polychlorinated biphenyls, benzopyrene and dioxin-like compounds in cigarette smoke has been associated with carcinogenicity humans and mice [56]. At least in the lungs of mice, arylhydrocarbon receptor accumulation oscillates in a circadian manner [57]. If also true in humans, smoking may be more dangerous at certain day times than at others, and this additional risk may be exacerbated in night-shift workers.
Breast cancer incidence has been claimed to be significantly elevated in woman who performed rotating shift work for decades (e.g. [58, 59]). From a meta-analysis of ten studies the authors calculated that the risk increase is 3% for every five year of rotating shift work and or 13% for every 500 night shifts [59]. Nonetheless, the relationship between shift work and breast cancer is still somewhat controversial, as other meta-analysis studies question the robustness of the evidence [60, 61]. However, assessing relatively small risks is difficult and it thus appears fair to conclude that the final word is not yet out. Even if there were a significant relationship between shift work and cancer incidence, it would still be unclear whether the effect is caused by the internal disruption of circadian timing or with life style changes and stress factors associated with shift work. This uncertainty even applies to animal studies conclusively showing that exposure to chronic jetlag increases tumour growth [62, 63] and reduces longevity [64]. It also has to be stated that it is not clear to what extend the circadian synchrony of the peripheral metabolic organs (excluding synchrony with the SCN) is disrupted. So far, chronic jetlag experiments did not demonstrate a clear loss of phase coherence between different peripheral organs, although the amplitude of circadian liver gene expression is dampened in chronically jetlagged mice [65]. Nevertheless, it would be premature to ascertain that shift work really reflects a model for circadian disruption of peripheral organs.
Conclusions and perspectives
The first mammalian core clock genes have been identified only in the late 1990s. Since then, impressive progress has been made in understanding how the endogenous clockwork circuitry operates and how it drives overt cycles in behaviour and physiology in laboratory rodents and humans. Nevertheless, we are still in the opening game, when expressing the state of circadian research in chess parlance. Thus, the inventory of genes participating in circadian time keeping is still incomplete and the mechanisms by which they establish interconnected feedback loops are superficially understood at best. Even less is known about the medical consequences of circadian disruption on health and longevity. Rotating night-shift work disrupts the normal course of a healthy life not only by perturbing sleep-wake cycles, but also by affecting eating and smoking habits [66, 67]. These lifestyle changes may increase the incidence of metabolic syndrome, cancer and cardiovascular disease. To what extent circadian disruption – that is, the discordance between circadian gene expression/physiology and human activities ‒ contributes to the insults caused by shift work remains to be determined. In fact, cyanobacteria and plants are still the only organisms for which a lack of resonance between environmental and circadian cycles has been unequivocally demonstrated to reduce fitness dramatically [68, 69].
We wish to conclude this article on an optimistic note with regard to medical applications. The knowledge on circadian gene expression and physiology holds promises for improving regimens of medical treatments both with regard to drug efficacy and tolerability. Recently, low-dose glucocorticoid chronotherapy has been shown to yield considerably better results in the treatment of rheumatoid arthritis than conventional glucocorticoid administration [70]. An oral prednisone tablet has been designed to release the drug at 2 a.m., the time of best efficacy. This formulation can be ingested before going to sleep, so that the patient’s sleep does not have to be interrupted. Cancer chronotherapy, based on the circadian activity of the xenobiotic detoxification system and on daytime-dependent drug tolerability, has also delivered its first promising results (for review, see [71]). In a comparison of overall survival of male patients with colorectal cancer there was a clear advantage of applying a chronomodulated regimen of a combination of 5-fluorouracil, leucovorin and oxaliplatin over a conventional infusion of these drugs. Surprisingly, however, no statistically significant overall survival difference between these treatments was observed for the time-adapted treatment of female patients. Clearly more clinical studies are required to assess the virtues of chronotherapeutic regimens in fighting cancer and other diseases. A major problem is that only a few physicians are currently involved in such endeavours. We hope that this article will help to motivate clinicians to take circadian physiology into account when designing new therapeutic regimens.
References
1 Dibner C, Schibler U, Albrecht U. The mammalian circadian timing system: organization and coordination of central and peripheral clocks. Annu Rev Physiol. 2010;72:517–49.
2 Partch CL, Green CB, Takahashi JS. Molecular architecture of the mammalian circadian clock. Trends Cell Biol. 2014;24(2):90–9.
3 Ralph MR, Foster RG, Davis FC, Menaker M. Transplanted suprachiasmatic nucleus determines circadian period. Science. 1990;247(4945):975–8.
4 Tosini G, Menaker M. Circadian rhythms in cultured mammalian retina. Science. 1996;272(5260):419–21.
5 Balsalobre A, Damiola F, Schibler U. A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell. 1998;93(6):929–37.
6 Yamazaki S, Numano R, Abe M, Hida A, Takahashi R, Ueda M, et al. Resetting central and peripheral circadian oscillators in transgenic rats. Science. 2000;288(5466):682–5.
7 Bianconi E, Piovesan A, Facchin F, Beraudi A, Casadei R, Frabetti F, et al. An estimation of the number of cells in the human body. Ann Hum Biol. 2013;40(6):463–71.
8 Nagoshi E, Saini C, Bauer C, Laroche T, Naef F, Schibler U. Circadian gene expression in individual fibroblasts: cell-autonomous and self-sustained oscillators pass time to daughter cells. Cell. 2004;119(5):693–705.
9 Welsh DK, Yoo SH, Liu AC, Takahashi JS, Kay SA. Bioluminescence imaging of individual fibroblasts reveals persistent, independently phased circadian rhythms of clock gene expression. Curr Biol. 2004;14(24):2289–95.
10 Dijk DJ, Czeisler CA. Contribution of the circadian pacemaker and the sleep homeostat to sleep propensity, sleep structure, electroencephalographic slow waves, and sleep spindle activity in humans. J Neurosci. 1995;15(5 Pt 1):3526–38.
11 Richards J, Gumz ML. Mechanism of the circadian clock in physiology. Am J Physiol Regul Integr Comp Physiol. 2013;304(12):R1053–64.
12 Morris CJ, Yang JN, Scheer FA. The impact of the circadian timing system on cardiovascular and metabolic function. Prog Brain Res. 2012;199:337–58.
13 Firsov D, Tokonami N, Bonny O. Role of the renal circadian timing system in maintaining water and electrolytes homeostasis. Mol Cell Endocrinol. 2012;349(1):51–5.
14 Tsang AH, Barclay JL, Oster H. Interactions between endocrine and circadian systems. J Mol Endocrinol. 2014;52(1):R1–16.
15 Asher G, Schibler U. Crosstalk between components of circadian and metabolic cycles in mammals. Cell Metab. 2011;13(2):125–37.
16 Scheiermann C, Kunisaki Y, Frenette PS. Circadian control of the immune system. Nat Rev Immunol. 2013;13(3):190–8.
17 Refinetti R. The circadian rhythm of body temperature. Front Biosci (Landmark Ed). 2010;15:564–94.
18 Buhr ED, Takahashi JS. Molecular components of the Mammalian circadian clock. Handb Exp Pharmacol. 2013(217):3–27.
19 Feng D, Liu T, Sun Z, Bugge A, Mullican SE, Alenghat T, et al. A circadian rhythm orchestrated by histone deacetylase 3 controls hepatic lipid metabolism. Science. 2011;331(6022):1315–9.
20 Reischl S, Kramer A. Kinases and phosphatases in the mammalian circadian clock. FEBS Lett. 2011;585(10):1393–9.
21 O’Neill JS, Reddy AB. The essential role of cAMP/Ca2+ signalling in mammalian circadian timekeeping. Biochem Soc Trans. 2012;40(1):44–50.
22 Harmar AJ. An essential role for peptidergic signalling in the control of circadian rhythms in the suprachiasmatic nuclei. J Neuroendocrinol. 2003;15(4):335–8.
23 Richardson GS, Moore-Ede MC, Czeisler CA, Dement WC. Circadian rhythms of sleep and wakefulness in mice: analysis using long-term automated recording of sleep. Am J Physiol. 1985;248(3 Pt 2):R320–30.
24 Schmidt TM, Chen SK, Hattar S. Intrinsically photosensitive retinal ganglion cells: many subtypes, diverse functions. Trends Neurosci. 2011;34(11):572–80.
25 Chen SK, Badea TC, Hattar S. Photoentrainment and pupillary light reflex are mediated by distinct populations of ipRGCs. Nature. 2011;476(7358):92–5.
26 Khalsa SB, Jewett ME, Cajochen C, Czeisler CA. A phase response curve to single bright light pulses in human subjects. J Physiol. 2003;549(Pt 3):945–52.
27 Ruger M, et al. Human phase response curve to a single 6.5 h pulse of short-wavelength light. J Physiol. 2013;591(Pt 1):353–63.
28 Sujino M, St Hilaire MA, Brainard GC, Khalsa SB, Kronauer RE, Czeisler CA, et al. Suprachiasmatic nucleus grafts restore circadian behavioral rhythms of genetically arrhythmic mice. Curr Biol. 2003;13(8):664–8.
29 Silver R, LeSauter J, Tresco PA, Lehman MN. A diffusible coupling signal from the transplanted suprachiasmatic nucleus controlling circadian locomotor rhythms. Nature. 1996;382(6594):810–3.
30 Li JD, Hu WP, Zhou QY. The circadian output signals from the suprachiasmatic nuclei. Prog Brain Res. 2012;199:119–27.
31 Damiola F, Le Minh N, Preitner N, Kornmann B, Fleury-Olela F, Schibler U. Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes Dev. 2000;14(23):2950–2961.
32 Stokkan KA, Yamazaki S, Tei H, Sakaki Y, Menaker M. Entrainment of the circadian clock in the liver by feeding. Science. 2001;291(5503):490–3.
33 Kornmann B, Schaad O, Bujard H, Takahashi JS, Schibler U. System-driven and oscillator-dependent circadian transcription in mice with a conditionally active liver clock. PLoS Biol. 2007;5(2): p. e34.
34 Brown SA, Zumbrunn G, Fleury-Olela F, Preitner N, Schibler U. Rhythms of mammalian body temperature can sustain peripheral circadian clocks. Curr Biol. 2002;12(18):1574–83.
35 Buhr ED, Yoo SH, Takahashi JS. Temperature as a universal resetting cue for mammalian circadian oscillators. Science. 2010;330(6002):379–85.
36 Morf J, Rey G, Schneider K, Stratmann M, Fujita J, Naef F, et al. Cold-inducible RNA-binding protein modulates circadian gene expression posttranscriptionally. Science. 2012;338(6105):379–83.
37 Saini C, Morf J, Stratmann M, Gos P, Schibler U. Simulated body temperature rhythms reveal the phase-shifting behavior and plasticity of mammalian circadian oscillators. Genes Dev. 2012;26(6):567–80.
38 Le Minh N, Damiola F, Tronche F, Schütz G, Schibler U. Glucocorticoid hormones inhibit food-induced phase-shifting of peripheral circadian oscillators. Embo J. 2001;20(24):7128–7136.
39 Reddy AB, Maywood ES, Karp NA, King VM, Inoue Y, et al. Glucocorticoid signaling synchronizes the liver circadian transcriptome. Hepatology. 2007;45(6):1478–88.
40 Gerber A, Esnault C, Aubert G, Treisman R, Pralong F, Schibler U. Blood-Borne Circadian Signal Stimulates Daily Oscillations in Actin Dynamics and SRF Activity. Cell. 2013;152(3):492–503.
41 Aschoff J, Wever R. Human circadian rhythms: a multioscillatory system. Fed Proc. 1976;35(12):236–32.
42 Daan S, Beersma DG, Borbely AA. Timing of human sleep: recovery process gated by a circadian pacemaker. Am J Physiol. 1984;246(2 Pt 2):R161–83.
43 Mirmiran M, Lunshof S. Perinatal development of human circadian rhythms. Prog Brain Res. 1996;111:217–26.
44 Mirmiran M, Ariagno RL. Influence of light in the NICU on the development of circadian rhythms in preterm infants. Semin Perinatol. 2000;24(4):247–57.
45 Mirmiran M, Maas YG, Ariagno RL. Development of fetal and neonatal sleep and circadian rhythms. Sleep Med Rev. 2003;7(4):321–34.
46 Hofman MA, Swaab DF. Living by the clock: the circadian pacemaker in older people. Ageing Res Rev. 2006;5(1):33–51.
47 Zhou JN, Liu RY, van Heerikhuize J, Hofman MA, Swaab DF. Alterations in the circadian rhythm of salivary melatonin begin during middle-age. J Pineal Res. 2003;34(1):11–6.
48 Karasek M. Melatonin, human aging, and age-related diseases. Exp Gerontol. 2004;39(11–12):1723–9.
49 Roenneberg T, Kuehnle T, Juda M, Kantermann T, Allebrandt K, Gordijn M, et al. Epidemiology of the human circadian clock. Sleep Med Rev. 2007;11(6):429–38.
50 Roenneberg T, Allebrandt KV, Merrow M, Vetter C. Social jetlag and obesity. Curr Biol. 2012;22(10):939–43.
51 Toh KL, Jones CR, He Y, Eide EJ, Hinz WA, Virshup DM, et al. An hPer2 phosphorylation site mutation in familial advanced sleep phase syndrome. Science. 2001;291(5506):1040–3.
52 Shanware NP, Hutchinson JA, Kim SH, Zhan L, Bowler MJ, Tibbetts RS. Casein kinase 1–dependent phosphorylation of familial advanced sleep phase syndrome-associated residues controls PERIOD 2 stability. J Biol Chem. 2011;286(14):12766–74.
53 Kurlansik SL, Ibay AD. Seasonal affective disorder. Am Fam Physician. 2012;86(11):1037–41.
54 Levitan RD. The chronobiology and neurobiology of winter seasonal affective disorder. Dialogues Clin Neurosci. 2007;9(3):315–24.
55 Schernhammer ES, Feskanich D, Liang G, Han J. Rotating night-shift work and lung cancer risk among female nurses in the United States. Am J Epidemiol. 2013;178(9):1434–41.
56 Portal-Nunez S, Shankavaram UT, Rao M, Datrice N, Atay S, Aparicio M, et al. Aryl hydrocarbon receptor-induced adrenomedullin mediates cigarette smoke carcinogenicity in humans and mice. Cancer Res. 2012;72(22) 5790–800.
57 Tanimura N, Kusunose N, Matsunaga N, Koyanagi S, Ohdo S. Aryl hydrocarbon receptor-mediated Cyp1a1 expression is modulated in a CLOCK-dependent circadian manner. Toxicology. 2011;290(2–3):203–7.
58 Grundy A, Richardson H, Burstyn I, Lohrisch C, SenGupta SK, Lai AS, et al. Increased risk of breast cancer associated with long-term shift work in Canada. Occup Environ Med. 2013;70(12):831–8.
59 Wang F, Yeung KL, Chan WC, Kwok CC, Leung SL, et al. A meta-analysis on dose-response relationship between night shift work and the risk of breast cancer. Ann Oncol. 2013;24(11):2724–32.
60 Ijaz S, Verbeek J, Seidler A, Lindbohm ML, Ojajärvi A, Orsini N, et al. Night-shift work and breast cancer – a systematic review and meta-analysis. Scand J Work Environ Health. 2013;39(5):431–47.
61 Kamdar BB, Tergas AI, Mateen FJ, Bhayani NH, Oh J. Night-shift work and risk of breast cancer: a systematic review and meta-analysis. Breast Cancer Res Treat. 2013;138(1):291–301.
62 Filipski E, Li XM, Levi F. Disruption of circadian coordination and malignant growth. Cancer Causes Control. 2006;17(4):509–14.
63 Li XM, Delaunay F, Dulong S, Claustrat B, Zampera S, Fujii Y, et al. Cancer inhibition through circadian reprogramming of tumor transcriptome with meal timing. Cancer Res. 2010;70(8):3351–60.
64 Davidson AJ, Sellix MT, Daniel J, Yamazaki S, Menaker M, Block GD. Chronic jet-lag increases mortality in aged mice. Curr Biol. 2006;16(21):R914–6.
65 Filipski E, Innominato PF, Wu M, Li XM, Iacobelli S, Xian LJ, et al. Effects of light and food schedules on liver and tumor molecular clocks in mice. J Natl Cancer Inst. 2005;97(7):507–17.
66 Amani R, Gill T. Shiftworking, nutrition and obesity: implications for workforce health- a systematic review. Asia Pac J Clin Nutr. 2013;22(4):505–15.
67 Knutsson A, Boggild H. Shiftwork and cardiovascular disease: review of disease mechanisms. Rev Environ Health. 2000;15(4):359–72.
68 Dodd AN, Salathia N, Hall A, Kévei E, Tóth R, Nagy F, et al. Plant circadian clocks increase photosynthesis, growth, survival, and competitive advantage. Science. 2005;309(5734):630–3.
69 Johnson CH, Mori T, Xu Y. A cyanobacterial circadian clockwork. Curr Biol. 2008;18(17):R816–R825.
70 Buttgereit F, Mehta D, Kirwan J, Szechinski J, Boers M, Alten RE, et al. Low-dose prednisone chronotherapy for rheumatoid arthritis: a randomised clinical trial (CAPRA-2). Ann Rheum Dis. 2013;72(2):204–10.
71 Ortiz-Tudela E, Mteyrek A, Ballesta A, Innominato PF, Lévi F. Cancer chronotherapeutics: experimental, theoretical, and clinical aspects. Handb Exp Pharmacol. 2013;(217):261–88.
72 Borbely AA, Achermann P. Sleep homeostasis and models of sleep regulation. J Biol Rhythms. 1999;14(6):557–68.