The advent of biosimilars: challenges and risks
DOI: https://doi.org/10.4414/smw.2014.13980
Rüdiger
Müller, Christoph
Renner, Cem
Gabay, Giuseppe
Cassata, Andreas
Lohri, Paul
Hasler
Summary
Biosimilars represent a new class of medicinal products that will have significant impact on clinical use. They are identical on an amino acid sequence level to existing reference biopharmaceutical products (originals). However, they may exhibit differences on a protein level. This paper provides a brief overview of biosimilar development and describes the risk and challenges that should be considered during the admission of biosimilars into the clinic.
Key words: biosimilars; monoclonal antibodies; manufacturing process; extrapolation; interchangeability; switching; Swissmedic; approval process
Introduction
Biopharmaceuticals are a cornerstone of therapy for a wide spectrum of disorders, from cancer to autoimmune or autoinflammatory diseases. Currently, the patents for several well-established biopharmaceuticals have expired or are approaching expiration [1]. In the wake of these patent expirations, numerous biosimilar products are or have been under development. Given the increase in potential choices of therapies, the physicians should be aware of the biochemical and clinical parameters that distinguish biosimilars from the original products.
It is important to differentiate between biosimilars and the follow-on compounds of small molecules, called generics. Identical generic copies of the active substance of small molecule drugs can be produced using well-defined chemical synthetic procedures [1]. Unlike generics, however, identical copies of biopharmaceuticals cannot be produced [2]. Not only they can be 100–1000 times larger than a small molecule, biopharmaceuticals also possess highly complex three-dimensional structures and the full spectrum of their functions (as for small molecules) is often not completely understood [3, 4].
Due to their nature and complexity, these therapeutic molecules are products of highly controlled biological processes. Although identical in protein sequence, significant differences between biosimilar and original may arise at various steps in the manufacturing process, all of which could result in heterogeneities in the structure of the final molecule [5, 6].
Complex biopharmaceuticals, such as monoclonal antibodies (mAb) frequently used for cancer treatment and auto-immune/inflammatory diseases, are characterised not only by the amino acid sequence, but also by their three-dimensional structure, the degree and location of their glycosylation sites, their isoform profiles (the proportions of different isoforms of the product), and the degree of protein aggregation [1, 7, 8]. Another important aspect is the pharmaceutical formulation of the final product, which not only affects the three-dimensional structure of the active protein, but also its aggregation status [3, 6]. These properties, in turn, will determine tissue distribution and binding with the target molecules, as well as interaction with other factors, such as cell-surface receptors and nucleic acids. Thus, the structural properties of a biopharmaceutical are key determinants that dictate its pharmacokinetic and pharmacodynamic profile, including biological activity, clinical efficacy and safety in patients [3].
This review summarises the opinion of a Swiss expert panel on the various aspects of biosimilar development, and highlights the key points that should be taken into account when facing the choice between biosimilar and original.
A word on terminology: what is a biosimilar?
The European Union (EU) defines a biosimilar as a medicinal product which is a copy of a biological product (the reference product) that has already received authorisation [9–11]. The term ‘biogenerics’ has previously been used to refer to the follow-on products of biopharmaceuticals that have gone off-patent. This term, however, is inaccurate as it implies that the follow-on product is identical to the original, in accordance with the principle of chemical generics. It should be noted that the term ‘biogenerics’ has widespread use in developing countries. Other terms used to denote biosimilars are ‘follow-on biologicals’, ‘similar biotherapeutic products’, or ‘subsequent-entry biologicals’. These should not be confused with ‘second-generation biologicals’ or ‘biobetters’, which refer to drugs that undergo a full development programme demonstrating clinical advantages over the previous-generation product [12].
The following sections examine the key steps of the biological manufacturing process, and explain how these give rise to structural variability that can ultimately affect clinical efficacy and safety [2, 13, 14].
The process of manufacturing biological medicinal products
"Biologics" denotes the diverse class of medicines that are produced and isolated from living systems, such as bacteria, yeast and mammalian cells. The manufacturing of a biologic agent requires multiple complex steps (fig. 1). First, the appropriate genetic sequence has to be selected and cloned into a suitable expression vector. Next, the protein of interest has to be produced by a suitable cell expression system. These basic steps are followed by quality control, scale-up and purification, up to formulation of the end product. Each step in the process has a profound impact on the structure of the biological molecule [5]. Fundamental parameters, such as temperature, pH, agitation and the type of containers used, are a few basic factors that can influence the quality of the final product [6]. Another variable is the type of expression system used for recombinant protein production, which dictates the final protein structure. For example, recombinant human granulocyte colony-stimulating factor (G-CSF) is expressed as a nonglycosylated form in Escherichia coli, whereas the use of the Chinese hamster ovary cell expression system results in a glycosylated protein [15]. Similar heterogeneity in glycosylation patterns has also been seen with interferon-gamma produced using different expression systems [8]. The end product is often a mixture of several different isoforms or structural variants. Unlike small molecule drugs, which exist as homogenous structures (>98%), biologics can exist as a mixture of different isoforms. Their enormous size relative to small molecules and the ensuing complexity in secondary, tertiary and quaternary structure renders it nearly impossible to verify the optimal isoform proportions of the biological product [3]. Nevertheless, it is crucial to understand which variants are impurities and how these can impact on the efficacy, safety and immunogenicity profile of the final drug product [16].
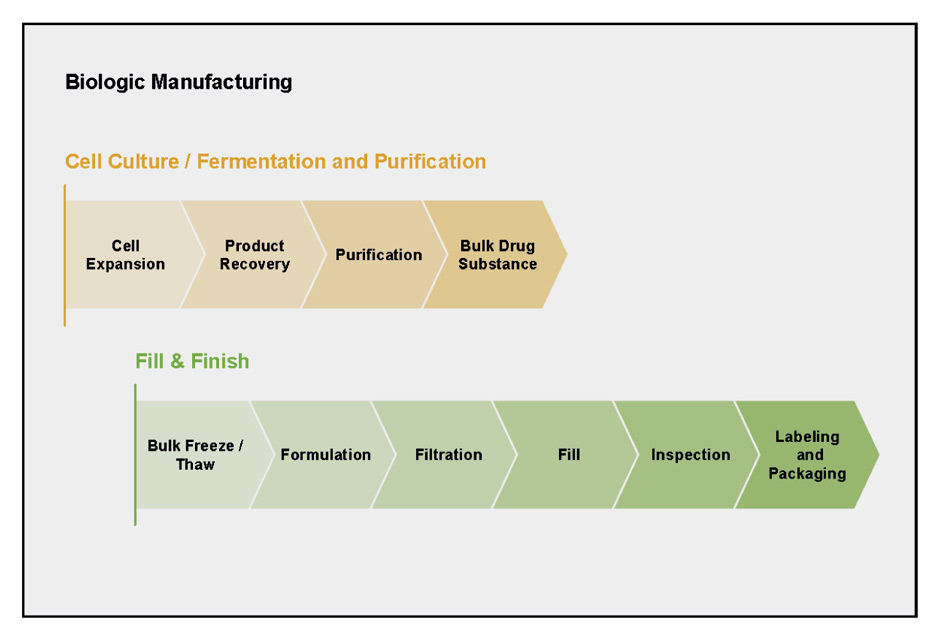
Figure 1
The process of manufacturing a biological medicinal product is highly complex and requires strict control and adherence to quality procedures. Adapted from: Lee JF, Litten JB, Grampp G. Comparability and biosimilarity: considerations for the healthcare provider. Curr Med Res Opin. 2012;28:1053–8. Copyright © 2012, Informa Healthcare. Reproduced with permission of Informa Healthcare.
For biological medicinal products whose clinical properties depend upon their structural properties, and whose structural properties are in turn dictated by the manufacturing process, great care is needed during their manufacture to ensure the quality and integrity of the final product.
Exercises in comparability and changes to the manufacturing process
For various reasons a manufacturer may introduce a change to the manufacturing process of an approved biologic, such as the need to improve product quality and yield, adherence with regulatory commitments, or to improve the efficiency and reliability of the manufacturing process [5]. Routine quality control or analytical measures, however, may not be able to assess the impact of a particular change on the final quality, efficacy and safety of the product. Depending on the extent of the modification, further nonclinical and clinical evaluations may be necessary for a proper evaluation of the product [17, 18].
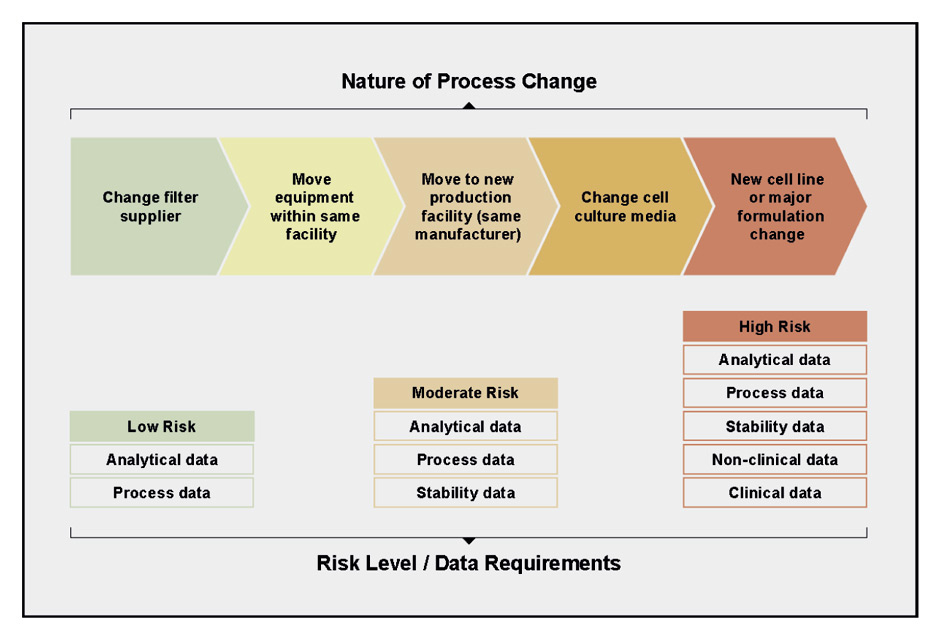
Figure 2
The extent and nature of a change to the manufacturing process determines the amount and type of data needed to evaluate comparability of the final product. Changes to the manufacturing process are governed by the ICH Q5E comparability guidance. Adapted from Lee et al. and sources therein [5, 19, 20]. Copyright © 2012, Informa Healthcare. Reproduced with permission of Informa Healthcare.
In its early days, the production of recombinant biologics was considered so complex that manufacturers avoided making any changes, particularly to critical steps of the production process. Today, however, regulatory agencies have accumulated sufficient experience with analytical characterisation, allowing extrapolation of the structure-function relationship of approved biologicals, to define "comparability exercises" for validating a manufacturing process change [17, 18]. The International Conference on Harmonization comparability guidance ICH Q5E [19, 20] is a result of the cumulative efforts of the US Food and Drug Administration (FDA) [21], European Medicines Agency (EMA) and other regulatory bodies to provide a comparability guideline [5].
For all products, patient safety is the primary consideration, and any evaluations of comparability are performed in consultation with regulatory authorities. A comparability evaluation allows a manufacturer to ensure that there is no negative impact on the product’s quality (fig. 2). This involves a complete risk assessment that includes knowledge of the product, the scope of the process change and its potential impact on the product [5]. If analytical studies are deemed insufficient to assess the impact of the changes, nonclinical and clinical bridging studies may be required to confirm clinical efficacy and safety. Table 1 depicts a few examples of manufacturing changes and highlights the necessity of clinical studies to assess the true impact of seemingly minor alterations to the manufacturing process.
|
Table 1: The consequences of manufacturing process changes for registered drugs [5]. |
|
Product
|
Process change
|
Consequences
|
| Avonex® (interferon beta-1a) |
New cell bank used. |
This change in the manufacturing process was not pursued, due to unexpected clinical outcomes. |
| Raptiva® (efalizumab) |
Manufacturing transferred to a new organization. |
Additional clinical studies needed. |
| Myozyme® (alglucosidase alfa) |
Manufacturing process scale-up within the same organisation. |
Additional clinical studies needed. |
Biosimilars: leaping backwards across the knowledge gap
The bulk of the innovator manufacturer’s product information is proprietary, including the manufacturing process (even after patent loss of the product). The complexity of these processes may be interpreted as a certain advantage for the companies involved. This information is not always available in the public domain; thus, the manufacturer of a biosimilar begins with a knowledge gap. Starting with only the identity of the final product, a biosimilar manufacturer must work backwards to establish a protein expression system, scale-up, purification, formulation and packaging. The manufacturer must demonstrate the biological similarity of the follow-on product against the originator, in terms of analytical properties, and preclinical and clinical aspects.
In the EU, clinical studies to demonstrate biosimilarity are required prior to regulatory approval [5]. This is not surprising, given the knowledge gap that exists between the manufacturing of the originator product and the biosimilar. Despite these requirements, some of the biosimilars approved in the EU have exhibited different physical, chemical or immunogenicity profiles compared to their innovator products [22]. Examples were seen in a biosimilar filgrastim, which showed unexpected differences in exposure, and a biosimilar epoetin, which showed differences in titrated dosing [23]. However, for these two biosimilars, regulatory authorities ruled that these differences would not affect the overall safety or efficacy of the products. Nevertheless, the unexpected clinical findings in molecules that are supposed to be copies of the originators highlight an important point: clinical studies are a critical aspect of biosimilar development and evaluation, and are important for detecting clinical differences between structurally related molecules [22].
Biosimilars and regulatory requirements
The EMA [14, 24, 25], FDA [26–29] and Swissmedic (Swiss Regulatory Agency) [30, 31] have established guidance on the approval of biosimilars over the past few years. The initial regulatory experience of the EMA involved biologics of relatively small size (insulin, interferon, filgrastim, epoetin and somatropin) [5].
Amongst the biosimilars that underwent EMA review from 2006–2011, six produced unexpected results that were not foreseen during nonclinical testing. Of these biosimilars, four failed to demonstrate comparable efficacy and/or safety against the originator products, and were rejected or withdrawn. For the remaining two manufacturers, one modified the manufacturing process and repeated a clinical trial, and another was denied extension of its label for subcutaneous use, prior to marketing authorisation [5].
For monoclonal antibodies, the EMA "Guideline on similar biological medicinal products containing monoclonal antibodies – non-clinical and clinical issues" came into effect in 2012 [32]. Stepwise nonclinical in vitro and in vivo approaches are recommended to evaluate the similarity of the biosimilar to the reference monoclonal antibody. Subsequently, clinical pharmacokinetic (PK) studies need to be performed in order to demonstrate a similar PK profile, prior to conducting clinical efficacy trials. Due to the nature of monoclonal antibodies, PK is often highly variable even within the same disease, for example in adjuvant versus metastatic breast cancer, where factors such as comorbidities may alter PK. Therefore, PK studies are a necessary component of the clinical programme to establish similar efficacy to the originator antibody.
In addition to PK, pharmacodynamic (PD) studies are also important for assessing comparability. For some drugs, such as filgrastim and epoetin, absolute neutrophil counts and haemoglobin concentration / reticulocyte counts are established and validated markers of drug activity. For other drugs, particularly the antineoplastic antibodies, no validated PD markers exist to indicate antitumour activity [11]. Although the EMA states that the preferred measures of efficacy in oncology indications are progression free, disease free or overall survival, this recommendation is problematic in clinical practice. The EMA acknowledges that parameters such as performance status, tumour burden or previous treatment may be confounding factors that hamper comparisons of efficacy between biosimilar and originator antibodies using these endpoints. It is important to note that the goal of comparability studies is not patient benefit, but merely to show similar safety and efficacy to the originator product. The implicit assumptions are that any difference between the products will be reflected if a sensitive patient population and clinical endpoint is used. The Swiss guidance for approval of biosimilars is largely based on the EMA guidelines and explicitly states that the clinical trial for assessing biosimilarity must be performed using the “most sensitive” and homogenous patient population [11, 30, 32].
Extrapolation across different indications
The acceptance of biosimilars in Europe has been slow, a fact that may most likely reflects uncertainties about the efficacy, safety and interchangeability with their originator products [33]. Compared with biosimilar epoetin, however, the uptake of biosimilar filgrastim is increasing. This may be partly due to the fact that 85% of filgrastim is used for the treatment of chemotherapy-induced neutropenia. The clinical efficacy of this compound can easily be observed in the individual patient. The short treatment period in already immunosuppressed patients minimises the risk of developing antidrug antibodies; furthermore, filgrastim appears to be inherently less immunogenic than epoetin [11]. So, biosimilar filgrastim has received a wide-spread acceptance by clinicians and the controversy about its use remains only in very sensitive areas such as stem cell mobilisation in healthy donors or long-term treatment of patients with chronic neutropenia [34]. Noteworthy, approval for the latter two indications was based on the extrapolation of the results seen in other indications leading to the assumption that a similar mode of action applies to these indications for the biosimilar molecule [11].
At present, biosimilars of monoclonal antibodies such as rituximab and trastuzumab are in development [4, 11]. In the case of rituximab, regulators may allow extrapolation of the biosimilar clinical data across all rituximab indications, including those for which the biosimilar has not been tested. Rituximab is known for its ability to improve survival of patients with aggressive lymphomas [35]. Trastuzumab was one of the first agents able to markedly improve the outcome of Her 2 positive breast cancer patients in the adjuvant setting [36]. In both clinical situations this survival gain can be observed only by studying large cohorts of patients. Clinicians most likely would not feel comfortable to use an agent that has not specifically been tested in those particular situations. They would probably prefer to first use biosimilar drugs in more advanced incurable disease or in elderly patients where cure is not an option. This may cause ethical dilemmas, especially if legislators decide to put financial penalties on the use of an original drug.
Although there is now data on the first biosimilar trastuzumab in metastatic disease [37, 38], the question remains as to whether clinical efficacy results from metastatic breast cancer can be extrapolated to the adjuvant clinical setting as long as no formal trial has been performed. In the malignant lymphomas, such trials most likely will never be performed since many new agents targeting the B cell receptor (ibrutinib, idelalisib, obinotuzumab) are currently entering the market. These are important questions that need to be addressed with the input of professional medical societies [11].
Outside of oncology, rituximab is also used for the treatment of inflammatory disorders such as rheumatoid arthritis, psoriasis, ankylosing spondylitis, Crohn’s disease and antineutrophil cytoplasmic antibody-associated vasculitis. In theory, the results from a rheumatoid arthritis trial with rituximab could be extrapolated to the oncology indications, though the EMA has indicated that this will not be the case [11].
Similarly, CT-P13, the first biosimilar version of infliximab, has now been approved in South Korea and Europe across all six indications of infliximab (psoriasis, Crohn’s disease, ankylosing spondylitis, psoriatic arthritis, rheumatoid arthritis and ulcerative colitis). The biosimilar product underwent clinical testing in rheumatoid arthritis only, and a PK comparison was performed in ankylosing spondylitis. In light of this, Health Canada did not approve extrapolation to all indications for CT-P13 [39], in line with the European Cancer Organisation (ECCO) position statement on biosimilar products for the treatment of inflammatory bowel disease [40].
It’s noteworthy to add that, like the originator Remicade©, Inflectra©, Remsina© the biosimilar is also registered under the same international nonproprietary name (INN): infliximab [41]; this may rise questions on how easy traceability can be assured for pharmacovigilance purposes.
Immunogenicity of biosimilars
In general, immunogenicity remains a relatively difficult phenomenon to predict with antibodies. Experience with several approved originals and biosimilars has underscored concerns over the immunogenicity profiles of these drugs. In the case of the original infliximab in rheumatoid arthritis, a reduction in efficacy was observed, due to the induction of antibodies against the drug [42]. Testing of a biosimilar somatropin during clinical development revealed an increased incidence of antiproduct antibodies. This problem was later resolved through a change in the manufacturing process [43]. A biosimilar epoetin undergoing testing in a postmarketing clinical study was associated with cases of antiproduct neutralising antibodies, when administered to nephrology patients via the subcutaneous route [44].
There is limited information at present on the immunogenicity profiles of biosimilar products. Although opinions on this issue may differ, regulators and biologics manufacturers should bear this in mind when attempting to strike the appropriate balance between preapproval data requirements and postmarketing safety and efficacy monitoring. A key point related to effective monitoring is to ensure clear identification of all drug products, so that the biosimilar can be easily distinguished from the originator. This will be important for attributing any safety incidents to the correct drug product [11].
Interchangeability and substitution
The term "interchangeable" means that the biological medicinal products are fully interchangeable for one another, without loss of efficacy or decrease in safety. The EMA has abstained from providing guidance on interchangeability, leaving this decision to the respective national authorities [11]. To be considered interchangeable, the efficacy and safety risk of a biosimilar should not be greater than that of the originator monoclonal antibody. Swissmedic stated that “The authorisation issued by Swissmedic does not contain any statement regarding whether the biosimilar can be used interchangeably with the reference product. Such a decision must be made exclusively by the prescriber, i.e. the attending physician.”[31] ‘Automatic substitution’ refers to a nationally regulated procedure whereby pharmacists are entitled to substitute the same type of drugs for one another. Whereas in Switzerland for generics, the pharmacists can substitute original preparations on the specialties list by cheaper generics (if not the doctor specifically requested the release of the original preparation and in case of substitution, they have to inform the prescriber over the dispensed preparation), the regulation for substitution for biologics is still unclear [45].
Switching drugs
In this context, the term ‘switching’ means that patients can be treated with the same type of biopharmaceutical produced by different manufacturers during their treatment period. It is important to emphasise that it is not just a question of originator vs biosimilar switches, but also switches between biosimilars. As these biologics and biosimilars are not identical, it is intensely discussed whether these switches between biological products may increase the risk of immunogenic adverse effects or loss of efficacy. This issue is still a matter of debate, with very limited and no hard data to support or refute this stance. So far, no risk of increased side effects has been seen with switching of epoetins [46]; no data are available yet for the monoclonal antibodies. For now, the general consensus among physicians seems to be that a patient should continue to receive successful treatment with the same drug from the same manufacturer, regardless of whether it is a biosimilar or an originator product [11].
It is noteworthy that, whereas the EMA did not comment nor delegate the question concerning switches, Swissmedic clearly delegated it to the treating physicians [31].
Pricing and cost benefits
The main purpose of producing biosimilars is to reduce the costs of drugs. The global spending on biologicals (including biosimilars) from 2011–2015 is estimated at US $200 billion, of which biosimilars comprise 1% [47]. During this timeframe, however, some blockbusters such as infliximab, adalimumab, rituximab, cetuximab, trastuzumab and bevacizumab will continue to remain at least partly under patent protection. After this time, biosimilar monoclonal antibodies are expected to enter the market. The cost of biosimilars relative to the total costs of biologicals in developed countries such as Japan, Canada and the EU is low (<5%). In emerging nations such as China, Brazil, India, Korea and Mexico, biosimilars comprise 20–60% of the total spending on biologicals. It should be noted that less stringent regulatory criteria govern the approval of biosimilars in the developing world [11].
In Europe, it is expected that biosimilars will offer a 20–30% price reduction against originator drugs. Market competition will also reduce the pricing for originator products. Indeed, a price reduction of 30–40% has already been seen for originator epoetin and filgrastim. The actual drug cost, however, represents only a small proportion of the total costs of, for example, cancer care (10–15%), and this should be kept in mind when making the choice between biosimilar and originator drugs for cancer therapy [11].
Expert opinion: summary of the key points
An expert panel of Swiss physicians (including rheumatologists, oncologists, hematologists and clinical pharmacologists) focused their discussion on the following three major topics.
Drug structure and prediction of efficacy and safety
The panel is aware of the fact that structural differences between the original and the biosimilar could affect the efficacy and safety of the biosimilar. Some members of the panel are surprised by the fact that the expiration of a patent of a certain drug does not necessarily lead to the disclosure of the whole manufacturing process which, in the case of biologicals, may be a central issue regarding the quality of the biosimilar product. Clinical efficacy and patient safety cannot be extrapolated solely from the characterisation of a molecule’s biophysical properties. As a consequence, as long as the structure of the respective compounds are not identical, adequately powered clinical studies with comprehensive efficacy and safety endpoints may be a necessary prerequisite for a biosimilar to pass the regulatory hurdles, especially in treatments with curative intent where the effect of the drug cannot be assessed in an individual patient.
Justification and extent of extrapolations
Generally, the development programmes of biosimilars are not conducted across all the indications for which the original biological was approved. Therefore, the question arises whether extrapolation of the available clinical efficacy and safety data for the biosimilar across all the same indications as the original can be justified.
For monoclonal antibodies, the EMA guidelines state that extrapolation of clinical efficacy and safety data to other indications of the reference monoclonal antibody, even though not specifically studied during the clinical development of the biosimilar monoclonal antibody, is possible based on the overall evidence of comparability provided by the comparability exercise and with adequate justification [32].
The panel recognised the problems associated with the extrapolation of data to clinical settings not specifically investigated. Caution is warranted with respect to assessing the efficacy and safety of a biosimilar to a different degree:
– Some extrapolations may pose less of a dilemma: e.g. epoetins, since for these molecules, the increase in haemoglobin following administration is a PD marker, in accordance with the EMA guideline [32].
– Other situations were judged to be problematic: e.g. the use of G-CSF in bone marrow transplantation, referring to the World Marrow Donor Association position paper which raised a warning flag regarding the extrapolation of efficacy data from the autologous transplant setting to that of allogeneic transplantation [48].
– The following areas may not be acceptable to clinicians:
– Extrapolation across disease entities: e.g. extrapolating data from rheumatoid arthritis to Crohn’s disease (although both are inflammatory diseases).
– Extrapolation across different phases of the same disease (from palliative to curative). Can, i.e. the efficacy of a biosimilar trastuzumab in metastatic disease be extrapolated to its use in early breast cancer?
Pharmacovigilance
Special emphasis should be placed on pharmacovigilance, taking into account the limited number of patients studied during the registration process of biosimilars. Companies introducing biosimilars should therefore set up a risk-management/pharmacovigilance plan. The objective can be achieved through collaboration with patient registries that have been established in several countries. The risk-minimisation activities for the biosimilar should be comparable to those of the reference medicinal product.
Aspects which should be covered in the risk-management/pharmacovigilance plan include:
– Safety assessments, including rare and serious adverse events described and predicted, based on the pharmacology and experience with the original.
– The plan should ensure that any novel safety signals are captured.
– Accurate assessment of immunogenicity data: since immunogenic reactions may arise only in a small number of patients, clinical data monitoring systems must be put in place.
– Clear traceability and identification of the drug (original or biosimilar) associated with an adverse event.
Conclusions
The development and introduction of biosimilars creates several opportunities and challenges. Physicians, pharmaceutical companies, regulatory agencies and health authorities should collaborate closely to ensure equal efficacy and safety of both the original biologicals and biosimilars that will allow the accessibility of these powerful agents to a broader number of patients in less privileged areas of our planet.
References
1 Crommelin D, Bermejo T, Bissig M, et al. Pharmaceutical evaluation of biosimilars: important differencies from generic low-molecular-weight pharmaceuticals. The European Journal of Hospital Pharmacy Science. 2005;11(1):11–7.
2 Weise M, Bielsky MC, De Smet K, et al. Biosimilars: what clinicians should know. Blood. 2012;120:5111–7.
3 Kleinberg M, Mosdell KW. Current and future considerations for the new classes of biologicals. American journal of health-system pharmacy: Am J Health Syst Pharm. 2004;61:695–708; quiz 9–10.
4 Vital EM, Kay J, Emery P. Rituximab biosimilars. Expert Opin Biol Ther. 2013;13:1049–62.
5 Lee JF, Litten JB, Grampp G. Comparability and biosimilarity: considerations for the healthcare provider. Curr Med Res Opin. 2012;28:1053–8.
6 Rathore N, Rajan RS. Current perspectives on stability of protein drug products during formulation, fill and finish operations. Biotechnol Prog. 2008;24:504–14.
7 Hoglund M. Glycosylated and non-glycosylated recombinant human granulocyte colony-stimulating factor (rhG-CSF) – what is the difference? Med Oncol. 1998;15:229–33.
8 Hooker A, James D. The glycosylation heterogeneity of recombinant human IFN-gamma. J Interferon Cytokine Res. 1998;18:287–95.
9 EMA. Guideline on Similar Biological Medicinal Products. http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000408.jsp 2013.
10 EMA. DIRECTIVE 2001/83/EC OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL. http://eceuropa.eu/health/files/eudralex/vol-1/dir_2001_83_cons2009/2001_83_cons2009_en.pdf 2009.
11 Mellstedt H. Anti-neoplastic biosimilars--the same rules as for cytotoxic generics cannot be applied. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO 2013;24(Suppl 5):v23–8.
12 Weise M, Bielsky MC, De Smet K, et al. Biosimilars-why terminology matters. Nat Biotechnol. 2011;29:690-3.
13 FDA. U.S. Food and Drug Administration. Draft guidance on biosimilar product development. http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/default.htm 2012.
14 EMA. Guideline On Similar Biological Medicinal Products Containing Biotechnology-Derived Proteins As Active Substance: Quality Issues. http://www.ema.europa.eu/ema/pages/includes/document/open_documentjsp?webContentId=WC500127960 2006.
15 Hoglund M. Glycosylated and non-glycosylated recombinant human granulocyte colony-stimulating factor (rhG-CSF) – what is the difference? Med Oncol. 1998;15:229–33.
16 Sharma B. Immunogenicity of therapeutic proteins. Part 3: impact of manufacturing changes. Biotechnol Adv. 2007;25:325–31.
17 Lewis RM, Cosenza ME. Summary of DIA Workshop: Comparability Challenges: Regulatory and Scientific Issues in the Assessment of Biopharmaceuticals. Drug Inf J. 2010;44:485–504.
18 Putnam WS, Prabhu S, Zheng Y, Subramanyam M, Wang YM. Pharmacokinetic, pharmacodynamic and immunogenicity comparability assessment strategies for monoclonal antibodies. Trends Biotechnol. 2010;28:509–16.
19 FDA. Q5E Comparability of biotechnological/biological products subject to changes in their manufacturing process. http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm122879.htm 2005.
20 ICH. Comparability of biotechnological/biological products subject to changes in their manufacturing process, Q5E. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q5E/Step4/Q5E_Guideline.pdf 2004.
21 FDA. Demonstration of comparability of human biological products, including therapeutic biotechnology-derived products. http://www.fda.gov/drugs/guidancecomplianceregulatoryinformation/guidances/ucm122879.htm 2005.
22 Miletich J, Eich G, Grampp G, Mounho B. Biosimilars 2.0: guiding principles for a global “patients first” standard. mAbs. 2011;3:318–25.
23 Schellekens H, Moors E. Clinical comparability and European biosimilar regulations. Nat Biotech. 2010;28:28–31.
24 EMA. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. http://www.ema.europa.eu/ema/pages/includes/document/open_documentjsp?webContentId=WC500144124 2013.
25 EMA. Guideline On Similar Biological Medicinal Products. http://www.ema.europa.eu/ema/pages/includes/document/open_documentjsp?webContentId=WC500003517 2005.
26 FDA. Guidance for Industry: Quality Considerations in Demonstrating Biosimilarity to a Reference Protein Product. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291134.pdf 2012.
27 FDA. Guidance for Industry: Scientific Considerations in Demonstrating Biosimilarity to a Reference Product. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf 2012.
28 FDA. Guidance for Industry: Biosimilars: Questions and Answers Regarding Implementation of the Biologics Price Competition and Innovation Act of 2009 http://www.fda.gov/downloads/Drugs/Guidances/UCM273001.pdf 2012.
29 FDA. Biologics Price Competition and Innovation Act, 2009. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/ucm216146.pdf 2009.
30 Swissmedic. Verwaltungsverordnung / Anleitung / Zulassung ähnlicher biologischer Arzneimittel (Biosimilars). German. https://www.swissmedic.ch/ueber/00134/00519/ 2014.
31 Swissmedic. Verwaltungsverordnung Anleitung Zulassung ähnlicher biologischer Arzneimittel (Biosimilars) ab 1. Februar 2014. German. https://www.swissmedic.ch/zulassungen/00153/00189/00197/01810/index.html?lang=de 2014.
32 EMA. Guideline on similar biological medicinal products containing monoclonal antibodies – non-clinical and clinical issues. http://www.ema.europa.eu/ema/pages/includes/document/open_documentjsp?webContentId=WC500128686 2012.
33 Minghetti P, Rocco P, Cilurzo F, Vecchio LD, Locatelli F. The regulatory framework of biosimilars in the European Union. Drug Discov Today. 2012;17:63–70.
34 Niederwieser D, Schmitz S. Biosimilar agents in oncology/haematology: from approval to practice. Eur J Haematol. 2011;86:277–88.
35 Sehn LH. A decade of R-CHOP. Blood. 2010;116:2000–1.
36 Joensuu H, Kellokumpu-Lehtinen PL, Bono P, et al. Adjuvant docetaxel or vinorelbine with or without trastuzumab for breast cancer. N Engl J Med. 2006;354:809–20.
37 Celltrion. Herzuma. http://www.biosimilarnews.com/celltrions-herzuma-trastuzumab-now-approved-in-s-korea 2014.
38 Celltrion. Demonstrate Efficacy and Safety of Metastatic Breast Cancer (Compare). clinicaltrialsgov/ 2014;NCT01084876.
39 Werble C. Canadian Biosimilar Approvals for Remicade: Time to Restart the Bus? http://www.elsevierbi.com/publications/rpm-report/first-take/2014/1/canadian-biosimilar-approvals-for-remicade 2014.
40 Danese S, Gomollon F. ECCO position statement: The use of biosimilar medicines in the treatment of inflammatory bowel disease (IBD). J Crohns Colitis. 2013;7:586–9.
41 EMA. INFLECTRA SMPC. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002778/WC500151489.pdf 2013.
42 van den Bemt BJ, den Broeder AA, Wolbink GJ, et al. Anti-infliximab antibodies are already detectable in most patients with rheumatoid arthritis halfway through an infusion cycle: an open-label pharmacokinetic cohort study. BMC Musculoskelet Disord. 2011;12:12.
43 FDA. Omnitrope Summary Basis for Approval Medical Review. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2006/021426s000TOCcfm 2006.
44 Schellekens H. Biosimilars: the long and winding road to clinical equivalence. Hosp Pharm Europe. 2009;47:40–2.
45 KVG. Schweizer Bundesgesetz über die Krankenversicherung: Art.52a Substitutionsrecht. http://www.admin.ch/opc/de/classified-compilation/19940073/index.html 2000.
46 Ebbers HC, Muenzberg M, Schellekens H. The safety of switching between therapeutic proteins. Expert Opin Biol Ther. 2012;12:1473–85.
47 Blackstone EA, Fuhr JP, Jr. Innovation and Competition: Will Biosimilars Succeed?: The creation of an FDA approval pathway for biosimilars is complex and fraught with hazard. Yes, innovation and market competition are at stake. But so are efficacy and patient safety. Biotechnol Healthc. 2012;9:24–7.
48 Shaw BE, Confer DL, Hwang WY, Pamphilon DH, Pulsipher MA. Concerns about the use of biosimilar granulocyte colony-stimulating factors for the mobilization of stem cells in normal donors: position of the World Marrow Donor Association. Haematologica. 2011;96:942–7.