Selective manipulation of aging: a novel strategy for the treatment of neurodegenerative disorders
DOI: https://doi.org/10.4414/smw.2014.13917
Ehud
Cohen, Lorna
Moll, Tayir
El-Ami
Summary
Aging is the major risk factor for the development of human neurodegenerative maladies such as Alzheimer’s, Huntington’s and Parkinson’s diseases, and prion disorders, all of which stem from toxic protein aggregation. Although sporadic cases typically onset during the patient’s seventh decade of life or later, mutation-linked, familial disorders manifest during the fifth or sixth decade of life. This common temporal emergence pattern suggests that slowing aging can postpone the onset of these maladies and alleviate their symptoms once emerged. Studies in worms and flies that express disease-linked aggregative proteins revealed that reducing the activity of the insulin / insulin-like growth factor (IGF) signalling (IIS), a prominent aging regulatory pathway, protects these animals from toxic protein aggregation. The therapeutic potential of this approach has been tested and confirmed in mammals as reducing the activity of the IGF1 signalling cascade partially protects Alzheimer’s-model mice from premature death, and behavioural and pathological impairments associated with the disorder. Here we review the recent advances in the field, describe the known mechanistic links between toxic protein aggregation, neurodegenerative disorders and the aging process and delineate recent studies that point at IGF1 signalling inhibitors as promising therapies for the treatment of various late-onset neurodegenerative disorders.
Abbreviations
Aβ amyloid β
AD Alzheimer’s disease
ALS amyotrophic lateral sclerosis
APP amyloid precursor protein
DNA deoxyribonucleic acid
DR dietary restriction
ETC electron transport chain
FOXO forkhead box class O
HD Huntington’s disease
HSF-1- heat shock factor 1
Hsp heat shock protein
IGF1 insulin-like growth factor-1
IGF1r insulin-like growth factor-1 receptor
IIS insulin/IGF signalling
IRS insulin receptor substrate
MJD Machado–Joseph disease
PD Parkinson’s disease
polyQ polyglutamine
PS1 presenilin-1
RNA ribonucleic acid
RNAi RNA interference
SOD1 superoxide dismutase-1
TAR transactivation response
TDP-43 TAR DNA binding protein 43
The maintenance of protein homeostasis is vital for life
Homeostasis at the molecular, cellular and organismal levels is required for viability and functionality of all living organisms. Specialised mechanisms act to maintain the integrity of the proteome by assisting newly synthesised molecules to attain their desired, functional spatial structures and by restoring the correct confirmation of unfolded mature proteins (reviewed in [1]). Polypeptides that fail to fold properly and damaged mature proteins are designated for degradation by the ubiquitin proteasome system [2] or by autophagy [3]. The clearance of these molecules prevents deleterious effects associated with protein misfolding and aggregation (proteotoxicity). These coordinated cellular activities of protein folding, post-translational modification, quality control and degradation maintain functional protein homeostasis that was termed “proteostasis” [4]. Despite the activity of the proteostasis network, subsets of aggregation-prone proteins fail to fold properly, escape degradation and form insoluble aggregates within the cell. Cells tend to gather nondegradable protein aggregates in deposition sites that either serve as dynamic quality control structures or accumulate terminally aggregated proteins [5]. Although cellular deposition sites are often protective [6], it is possible that under certain circumstances they turn to be sources of toxicity (reviewed in [7]). In some cases, protein aggregation leads to the development of maladies that were collectively termed “conformational diseases” [8]. Late-onset neurodegenerative maladies such as Alzheimer’s disease (AD), Huntington’s (HD) disease, amyotrophic lateral sclerosis (ALS) and prion disorders are conformational diseases. Accordingly, the presence of protein aggregate-containing deposition sites is a unifying pathological hallmark of these neurodegenerative disorders [9].
The mechanisms of neurodegenerative disorders
The widely accepted amyloid hypothesis suggests that AD, the most prevalent neurodegenerative disorder, stems from a dual digestion of the amyloid precursor protein (APP) by two proteases, β and γ secretases. This cleavage of APP releases a family of short peptides that are known as “Aβ peptides” including Aβ1-40and Aβ1-42. Owing to their hydrophobic nature, Aβ peptides have the tendency to form aggregates of various sizes [10]. Small Aβ aggregative structures (oligomers) have been shown to be the most toxic species and to correlate best with the development of AD [11, 12]. The disease is characterised by neuronal loss, neuroinflammation, cognitive failure and eventually death [10]. Hitherto, the mechanistic details of how Aβ oligomers lead to the manifestation of AD are poorly understood; however, mutations in presenilin-1, the active component of the γ secretase complex, that increase the production of Aβ have been shown to be associated with the development of familial AD [13]. The discovery that a mutation in APP that reduces Aβ production decreases the risk of developing AD supports the idea that Aβ aggregation is a key aetiological cause of the disorder [14]. However, a careful analysis revealed that not all familial AD-associated mutations in the sequence of presenilin-1 increased Aβ production [15], suggesting that in some cases loss of presenilin-1 function underlies the development of AD [16]. Among other functions, presenilin-1 has key roles in autophagy [17, 18] and in the formation of functional interactions between the endoplasmic reticulum and the mitochondria [19]. Together these studies propose that more than one mechanism is accountable for the development of AD.
The aggregation of mutant huntingtin bearing an abnormally long poly-glutamine stretch (polyQ) causes HD, a disease that solely appears as a familial disorder, is characterised by impaired movement and cognition and eventually leads to death [20].
Like HD, Machado–Joseph disease (MJD, also known as spinocerebellar ataxia 3) is caused by a CAG trinucleotide expansion within the sequence of the SCA3 gene that encodes the ataxin-3 protein. This results in aberrantly expanded polyQ stretch within the deubiquitinating enzyme ataxin-3, which in turn acquires the tendency to form toxic aggregates. In addition, it has been suggested that mutated ataxin-3, carrying a polyQ expansion, initiates MJD by directing the E3-ubiquitin ligase parkin to autophagic-mediated degradation [21].
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease that was found to be associated with frontotemporal dementia and characterised by muscle atrophy and spasticity (reviewed in [22]). Mutations in the transactivation response (TAR) deoxyribonucleic acid (DNA) binding protein 43 (TDP-43) [23] and in the Cu/Zn superoxide dismutase 1 (SOD1) [24] were found to destabilise these proteins, enhance their propensity to aggregate and to be accountable for certain familial ALS cases. Many neurodegenerative disorders, including Alzheimer’s diseases and ALS, exhibit surprisingly similar temporal emergence patterns. Although most cases manifest sporadically during the seventh decade of life or later, the rarer, familial, mutation-linked cases appear during the patient’s fifth or sixth decade of life. This common feature of different neurodegenerative maladies defines aging as the major risk factor for the development of these disorders [25] and suggests that the aging process enables the emergence of neurodegeneration in late life stages by actively reducing the activity of proteostasis-maintaining mechanisms. Thus, these mechanisms, which protect the young organism from diseases, fail to prevent their onset late in life. Accordingly, this theme raises the prospect that the alteration of aging could serve as a novel strategy for the treatment of neurodegeneration. The exploration of aging regulating pathways during the last two decades allowed significant progress in this research avenue.
Aging regulating mechanisms
For decades, aging was thought to emanate from a random deterioration in the integrity of cellular maintenance mechanisms (reviewed in [26]). Although random events probably play important roles in the progression of aging, this process is also amenable to metabolic and genetic regulation. The first indication that aging can be manipulated was provided by McCay and colleagues [27], who discovered that rats that consumed reduced amounts of food had an extended lifespan compared with their counterparts that had unlimited access to food. This effect of dietary restriction (DR) was later extended to a variety of organisms ranging from yeast to monkeys (reviewed in [28]). The discovery that DR-associated longevity is dependent upon the activity of transcription factors [29, 30] indicated that genetic pathways are involved in the mediation of this effect.
Reducing the activity of the electron transport chain (ETC) can also alter the aging process as RNA interference (RNAi) mediated knockdown of genes that encode ETC components extend the lifespan of Caenorhabditis elegans (C. elegans) [31, 32]. Interestingly, to achieve this lifespan extension effect the activity of the ETC has to be mitigated during development [31]. This mechanism involves intertissue communication that is promoted by signalling mediating molecules [33].
Perhaps the most prominent and best characterised aging-regulating genetic pathway is the insulin/IGF signalling pathway (IIS). IIS reduction by RNAi or mutations extends the lifespans of flies [34], worms [35] and mice [36]. In the nematode C. elegans,DAF-2, the lone insulin/IGF receptor, initiates a signalling cascade that promotes the phosphorylation of its downstream transcription factors, DAF-16/FOXO [37] and SKN-1/NRF [38]. These phosphorylation events prevent the transcription factors from entering the nucleus and from regulating their target gene networks. Similarly, the IIS negatively regulates the activity of the heat shock factor 1 (HSF-1) by preventing the phosphorylation of the HSF-1 interacting protein DDL-1. Nonphosphorylated DDL-1 binds HSF-1 and retains it in the cytosol [39]. The IIS also governs the cellular localisation of the transcription factor PQM-1, which responds to IIS in opposition to DAF-16 and has a role in the IIS-controlled lifespan mechanism [40]. Thus, IIS reduction by daf-2 knockdown modifies the cellular localisation of a nexus of downstream transcription factors creating youthful, long-lived, stress resistant worms (reviewed in [41]).
The mammalian signalling pathway downstream of IGF1 shares a high rate of similarity with the worm’s IIS. Upon activation, the IGF1 receptor (IGF1r), the closest daf-2 orthologue in mammals [42], undergoes auto-phosphorylation, followed by the recruitment and phosphorylation of the insulin receptor substrates (IRS) 1 and 2 on tyrosine residues. These events lead to the activation of the kinase Akt which phosphorylates its downstream transcription factors, including the family of the forkhead box class O (FOXO). Phosphorylated FOXO molecules are prevented from entering the nucleus and from regulating their target genes [43].
The functional roles of the IIS as a lifespan and stress resistance regulator have been shown to be conserved from worms to mammals. First, female mice carrying only one copy of Igf1r are long-lived and exhibit elevated resistance to oxidative stress [36]. Similarly, mice lacking IRS1, and thus exhibiting impaired IGF1 signalling, are also long-lived [44]. Studies that compared components of the IGF1 signalling mechanism of human centenarians and individuals members of cohorts that do not exhibit extended lifespans, revealed association of mutations in either the IGF1r or the FOXO transcription factors with extreme longevity [45–47].
These observations indicate that the aging regulating functions of the IIS are conserved from worms to mice and strongly suggest that IGF1 signalling is also a lifespan determinant in humans.
IIS-regulated target gene networks
The identification of the IIS and its downstream transcription factors as pivotal regulators of aging enabled the characterisation of the gene networks that are involved in setting the pace of aging. Several approaches have been adopted to identify these genes; DNA microarrays have been used to compare the gene expression patterns of worms that carry weak daf-2 alleles or mutated daf-16 with these of wild-type animals [48, 49]. The most prominent group of genes identified in these experiments was the family of heat shock proteins (Hsps) and genes that encode stress response proteins such as catalase. However, the expression levels of many other genes that possess various biological activities have been found to be IIS-regulated. Serial analysis of gene expression (SAGE) [50] and chromatin immunoprecipitation (ChIP) [51] have also been utilised to identify DAF-16 regulated genes. These techniques yielded partially overlapping results, confirming that stress resistance mediating genes are regulated by the IIS. Finally, the differences in soluble protein content among worms with mitigated IIS and wild-type animals has also been analysed by quantitative mass spectrometry [52]. This study revealed that the levels of catalases and Hsps are increased consequently to IIS knockdown, supporting the previous discoveries. Together these findings raised the prospect that DAF-16 and HSF-1 promote longevity via the preservation of proteostasis in late stages of life and suggest that their activation by IIS reduction can protect from proteotoxicity-related diseases.
IIS reduction protects model nematodes from proteotoxicity
To explore the possible links between proteotoxicity and aging, and to test whether IIS reduction ameliorates the toxic phenotypes of neurodegeneration-associated proteotoxicity, a series of transgenic C. elegans strains, each expressing a fluorescently tagged, polyQ stretch of different length, was created [53]. The visualisation of polyQ-YFP fluorescent dots in these worm strains revealed that at least 40 glutamine repeats were needed to promote efficient aggregation in young (day 2 of adulthood) nematodes. Interestingly, 40 glutamine repeats is also the threshold number required to initiate the development of HD in humans [20]. Visualisation of polyQ-YFP deposits over time revealed that the number of polyQ repeats required for efficient aggregation declines with age. Fluorescent foci containing polyQ35-YFP aggregates were first visible at day 4 of adulthood, whereas polyQ29-YFP deposits could not be detected earlier than day 9 of adulthood. These findings directly linked polyQ-YFP aggregation to the animal’s aging process, and corroborated the notion that the alteration of aging could slow protein aggregation and mitigate proteotoxicity. To directly test this theme, IIS activity was reduced by the knockdown of age-1,a gene that encodes a key IIS component. This manipulation reduced the level of polyQ82-YFP aggregation in worm embryos and protected adult nematodes from polyQ aggregation-associated motility impairment [53].
To further explore the molecular mechanisms by which IIS reduction protects from proteotoxicity we employed worms that express human Aβ in their body wall muscles (Aβ worms [54]). Aβ aggregation in the muscles of these animals results in a progressive paralysis within the worm population. Following this phenotype we found that IIS reduction by daf-2 RNAi protects worms from Aβ aggregation in DAF-16 and HSF-1 dependent manners. Surprisingly, these transcription factors were found to mediate opposing activities: HSF-1 promotes disaggregation whereas DAF-16 facilitates protective active aggregation [55]. These transcription factors exhibit dissimilar temporal patterns of activity; whereas HSF-1 was found to execute its counter-proteotoxic activity during development, DAF-16 is foremost important for protection from proteotoxicity during adulthood [56].
Interestingly, DR was also shown to protect Aβ worms from proteotoxicity in an HSF-1 dependent manner [57] and to attenuate Aβ deposition in the brains of AD-model mice [58], indicating that aging manipulation other than IIS reduction also counters proteotoxicity.
The protective effects of IIS reduction were recently extended to several additional neurodegeneration-linked aggregative proteins in worm models. Nematodes that express an ALS-linked, mutated TDP-43 [59], were protected from motility impairment when their IIS was reduced by the knockdown of daf-2.This protection was dependent on DAF-16 and HSF-1 [60]. The aggregation of ALS-linked mutated SOD-1 and the resulting locomotion impairments were also mitigated by the knockdown of daf-2 [61]. Similarly, the toxicity associated with the aggregation of ataxin-3, an MJD-linked polyQ-containing protein, was lessened by reducing IIS activity [62].
Collectively, these studies clearly indicate that, in the nematode, the aging process and proteotoxicity are directly linked and that IIS reduction can counter proteotoxicity in these animals. However, to assess the therapeutic potential of this approach it was necessary to test whether this link is conserved from worms to the mammalian brain.
IGF1 signalling and proteotoxicity: from the nematode to the mammalian brain
The creation of AD-model mice with altered aging programmes enabled the investigation of whether the alteration of IGF1 signalling protects from neurodegeneration. Killick and colleagues [63] crossed transgenic mice that harbour the familial AD-linked, mutated human APP gene carrying the Swedish mutation (K670N, M671L) (Tg2576 mice) [64] with animals lacking IRS2 to achieve offspring that produce Aβ and have an altered aging programme (strain Tg2576/Irs2-/-). Comparison of 12-month-old Tg2576/Irs2-/-and their age-matched Tg2576counterparts revealed that the deletion ofIrs2 resulted in a significant reduction of the Aβ plaque burden in the animals’ brains.
Adopting a similar approach, the Schubert group crossed Tg2576 mice with animals that lack the IGF1 receptor exclusively in neurones (Tg2576/nIgf1R-/-) and found that both males and females were protected from the premature death typical of Tg2576mice [65]. Aged Tg2576/nIgf1R-/-animals had lower amounts of both peptides, Aβ1-40and Aβ1-42,compared with their age matched Tg2576 siblings. These results revealed that reduction of IGF1 signalling in the mouse brain protect from proteotoxicity.
In order to explore the underlying mechanism by which IGF1 signalling reduction protects mice from AD-like disease we crossed mice that express two mutated AD-linked genes, humanised APPswe and a hyperactive human PS1 (APPswe/PS1ΔE9 mice) [66], with a long-lived mouse strain that harbours only one copy of Igf1r [36]. APPswe/PS1ΔE9 mice develop relatively slow age-dependent neurodegenerative symptoms which resemble those of human AD patients, including behavioural impairments [67], neuroinflammation and Aβ plaque formation [66]. APPswe/PS1ΔE9 mice which carried only one Igf1r copy (APPswe/PS1ΔE9/Igf1R+/-mice) were protected from AD-associated memory and orientation impairments and exhibited reduced rates of neuroinflammation and neuronal loss [68]. Aβ plaques of APPswe/PS1ΔE9/Igf1R+/-mice were smaller in size and of higher density compared with their littermates that hada natural level of IGF1 signalling. Our results proposed that IGF1 signalling reduction confers protection by sequestering toxic Aβ oligomers from the brain [11, 69], packing them in large fibrils of lower toxicity [68].
These mouse-based studies suggest that the manipulation of aging by IGF1 signalling reduction protects the mammalian brain from toxic Aβ aggregation and supported the idea that IIS inhibitors have the promise to serve as counter-neurodegeneration drugs. However, it is important to note that several studies have proposed that circulating IGF1 protects AD-model rodents from proteotoxicity and from Alzheimer’s-like disease.
To test the possible mutual relation of circulating IGF1 on AD-linked pathologies, IGF1was infused to old rats whose brains contain higher levels of Aβ than those of their young counterparts, and the levels of Aβ in the brain were measured. The results showed that elevated levels of IGF1in old rats enhanced the clearance of Aβ to achieve levels similar to those observed in brains of young animals [70]. Recently, it was shown that reduced IGF1 levels in the serum of AD-model mice accelerates the accumulation of Aβ plaques and elevates the load of astrocytes and microglia in the brain [71]. These findings suggested that an increased amount of circulating IGF-1 protects from Aβ toxicity and that hyperactivation of the IIS pathway is neuroprotective (reviewed in [72]).
Although the question of whether decreased or increased IGF1 signalling counters proteotoxicity is under debate, the idea that an increased activity of this pathway protects the mammalian brain is challenged by a few findings. Firstly, reducing IGF1 signalling alters the aging process [36, 44] and the major risk factor for the development of neurodegeneration is aging [25]. Thus, slowing aging by IGF1 signalling reduction rather than increase should ameliorate aging-associated diseases. In addition, increased Aβ load and plaque formation were considered in many studies as a marker of toxicity [71, 73] whereas hyperaggregation may be protective [68]. Lastly, it is possible that increased IGF1 levels in the serum lead to the recycling of the IGF1r and to long-term reduction in the activity of this pathway. Further experimental work is required to resolve this dispute.
Reducing IGF1 signalling as a novel strategy to combat neurodegeneration
To assess the therapeutic potential of IGF1 signalling inhibitors it was first necessary to test whether this manipulation provides protection from proteotoxicity when applied late in life, the stage in which neurodegenerative disorders emerge and are diagnosed. Employing the Aβ worm model and conditional daf-2 knockdown, we found that late-life IIS reduction was sufficient to protect the worms from Aβ toxicity when it can no longer extend lifespan [56]. Interestingly, this temporal study showed that in the worm the counter-proteotoxic effect of IIS reduction is separable from its longevity effects, proposing that protection from toxic protein aggregation can be achieved without lifespan extension.
Next, it was necessary to develop compounds that stabilise proteostasis and assess whether they can mitigate proteotoxicity. To identify such molecules, Calamini and colleagues screened a set of small molecules using model worms and cultured cells. They identified compounds that modulate proteostasis, which act through the activation of HSF-1, FOXO, Nrf-2 [74] and the induction of Hsp encoding genes.
Several additional compounds were shown to alleviate proteotoxicity in worm models. Trehalose, which activates DAF-16 and HSF-1, was shown to increase lifespan and protect C. elegans from polyQ-associated proteotoxicity [75]. Similarly, the amyloid binding dye, Thioflavin T, was also shown to slow aging, increase lifespan and protect nematodes from Aβ-mediated toxicity [76]. The counter-proteotoxic compound psammaplysene A, which has been tested in cell culture and in a fly model of proteotoxicity, drives FOXO3a into the neuronal nuclei and protects motor neurones and fly eyes from proteotoxicity and death [77]. These studies highlighted the pivotal roles of IIS-regulated transcription factors for the maintenance of proteostasis and strengthened the hypothesis that the inhibition of IIS by small molecules has a counter-neurodegeneration potential.
Potent IIS inhibitors offer the advantage of concurrently enhancing the activity of several protective transcription factors and preserving proteostasis. Recently, we found that NT219, a newly developed, highly efficient IIS inhibitor reduces the activity of the mammalian IGF1 signalling cascade by a dual step mechanism. It inhibits the auto-phosphorylation of the IGF1 receptor, confers the phosphorylation of IRS1 and IRS2 and promotes their degradation. NT219 elevates the expression of IIS target genes and protects model worms from the toxic effects of Aβ and polyQ aggregation [78] (fig. 1). These observations confirm that IIS inhibitors can serve as a panacea for the treatment of distinct proteotoxicity worm models that express neurodegeneration-linked, aggregative proteins.
Future prospects
The development of counter-proteotoxic compounds and their evaluation as counter-neurodegeneration therapies has become an important research avenue. However, key issues should be addressed before this research avenue yields true therapy. Although it was shown that IGF1 signalling reduction by genetic manipulations is capable of protecting mice from AD-associated phenotypes it will be crucial to test whether IGF1 signalling inhibitors that alleviate proteotoxicity in worms can protect the mouse brain from neurodegeneration. To address this question it will be necessary to ensure proper penetration of the drug into the brain, define the optimal administration regimen and characterise possible side effects. It is plausible that the highest therapeutic competence will be achieved by a combination of proteostasis enhancing compounds which will generate a synergistic protective effect that slows the progression of neurodegenerative disorders. Accordingly, we foresee an effort to test the efficiency of proteostasis-modifying compounds as counter-neurodegeneration drugs in model mice, prior to clinical trials. It is plausible that this new strategy of selective alteration of aging will eventually provide care givers with new tools for the treatment of hitherto incurable neurodegenerative disorders and new hope for patients and their families.
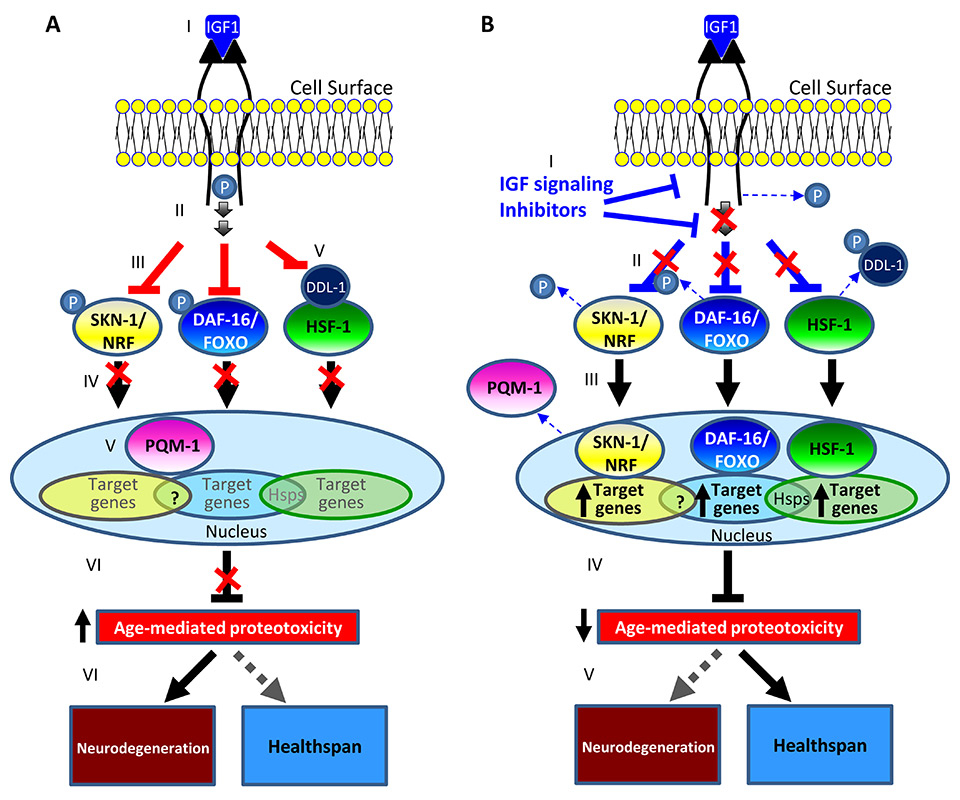
Figure 1
A model for the prevention of neurodegeneration by IIS reduction in worms and mammals.
A. The binding of IGF1 to its receptor leads to the dimerisation and activation of the IGF1 signalling cascade (I). Upon activation the IGF1 receptor undergoes auto-phosphorylation (II) followed by the recruitment of downstream components and the phosphorylation of its downstream transcription factors DAF-16/FOXO and SKN-1/NRF (III). Phosphorylated DAF-16/FOXO and SKN-1/NRF are prevented from entering the nucleus and from regulating their target genes (IV). Similarly, IIS activity prevents the phosphorylation of DDL-1 which retains the heat shock factor 1 (HSF-1) in the cytosol, preventing it from controlling its target gene network (V). IIS activity enables PQM-1 to stay in the nucleus and repress gene activity. The modulated activity levels of IIS-regulated transcription factors promote proteostasis collapse (VI) allowing the development of neurodegenerative disorders late in life.
B. Reducing IIS activity by pharmacological agents (I) reduces the rate of DAF-16/FOXO and of SKN-1/NRF phosphorylation and promotes the phosphorylation of DDL-1 (II). The migration of DAF-16/FOXO, SKN-1/NRF and HSF-1 into the nucleus and the exit of PQM-1 to the cytosol, activate the expression of their target gene networks (III) which reduce the rate of aging-mediated proteostasis collapse (IV). The maintenance of proteostasis prevents the accumulation of toxic protein aggregates (V) and preserves healthspan, delaying the manifestation of late-life neurodegenerative maladies.
Acknowledgment:We thank Dr. Michal Bejerano-Sagie and Ms. Filipa Carvalhal Marques for critical reading of this manuscript.
References
1 Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475:324–32.
2 Ciechanover A. The ubiquitin proteolytic system: from a vague idea, through basic mechanisms, and onto human diseases and drug targeting. Neurology. 2006;66:S7–19.
3 Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci. 2010;13:805–11.
4 Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–9.
5 Kaganovich D, Kopito R, Frydman J. Misfolded proteins partition between two distinct quality control compartments. Nature. 2008;454:1088–95.
6 Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;31:805-–10.
7 Ben-Gedalya T, Cohen E. Quality control compartments coming of age. Traffic. 2012;13:635–42.
8 Kopito RR, Ron D. Conformational disease. Nat Cell Biol. 2000;2:E207–209.
9 Soto C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci. 2003;4:49–60.
10 Selkoe DJ. Alzheimer’s disease. Cold Spring Harb Perspect Biol. 2011;3(7).
11 Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, et al. (2008) Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14(8):837‒42.
12 O’Nuallain B, Freir DB, Nicoll AJ, Risse E, Ferguson N, et al. Amyloid beta-protein dimers rapidly form stable synaptotoxic protofibrils. J Neurosci. 2010;30:14411–9.
13 Campion D, Flaman JM, Brice A, Hannequin D, Dubois B, Martin C, et al. Mutations of the presenilin/gene in families with early-onset Alzheimer’s disease. Hum Mol Genet. 1995;4:2373–7.
14 Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 2012;488(7409):96‒9.
15 Chavez-Gutierrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, et al. The mechanism of gamma-Secretase dysfunction in familial Alzheimer disease. Embo J. 2012;31:2261–74.
16 Shen J, Kelleher RJ, 3rd. The presenilin hypothesis of Alzheimer’s disease: evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci U S A 2007;104:403–9.
17 Coen K, Flannagan RS, Baron S, Carraro-Lacroix LR, Wang D, et al. Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. J Cell Biol. 2012;198:23–35.
18 Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–58.
19 Area-Gomez E, Del Carmen Lara Castillo M, Tambini MD, Guardia-Laguarta C, de Groof AJ, et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. Embo J. 2012;31:4106–23.
20 Bates G. Huntingtin aggregation and toxicity in Huntington’s disease. Lancet. 2003;361:1642–4.
21 Durcan TM, Fon EA. Ataxin-3 and its e3 partners: implications for machado-joseph disease. Front Neurol. 2013;4:46.
22 Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci. 2013;14:248–64.
23 Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–3.
24 Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62.
25 Amaducci L, Tesco G. Aging as a major risk for degenerative diseases of the central nervous system. Curr Opin Neurol. 1994;7:283–6.
26 Gems D, Partridge L. Genetics of longevity in model organisms: debates and paradigm shifts. Annu Rev Physiol. 2013;75:621‒44.
27 McCay CM, Crowell MF, Maynard LA. The effect of retarded growth upon the length of life span and upon the ultimate body size. 1935. Nutrition 1989;5:155–71; discussion 172.
28 Mair W, Dillin A. Aging and Survival: The Genetics of Life Span Extension by Dietary Restriction. Annu Rev Biochem. 2008;77:727‒54.
29 Bishop NA, Guarente L. Two neurons mediate diet-restriction-induced longevity in C. elegans. Nature. 2007;447:545–9.
30 Panowski SH, Wolff S, Aguilaniu H, Durieux J, Dillin A. PHA-4/Foxa mediates diet-restriction-induced longevity of C. elegans. Nature. 2007;447:550–5.
31 Dillin A, Hsu AL, Arantes-Oliveira N, Lehrer-Graiwer J, Hsin H, et al. Rates of behavior and aging specified by mitochondrial function during development. Science. 2002;298:2398–401.
32 Feng J, Bussiere F, Hekimi S. Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev Cell. 2001;1:633–44.
33 Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144:79–91.
34 Giannakou ME, Goss M, Jacobson J, Vinti G, Leevers SJ, et al. Dynamics of the action of dFOXO on adult mortality in Drosophila. Aging Cell. 2007;6(4):429‒38.
35 Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461–4.
36 Holzenberger M, Dupont J, Ducos B, Leneuve P, Geloen A, et al. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421:182–7.
37 Lee RY, Hench J, Ruvkun G. Regulation of C. elegans DAF-16 and its human ortholog FKHRL1 by the daf-2 insulin-like signaling pathway. Curr Biol. 2001;11:1950–7.
38 Tullet JM, Hertweck M, An JH, Baker J, Hwang JY, et al. Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell. 2008;132:1025–38.
39 Chiang WC, Ching TT, Lee HC, Mousigian C, Hsu AL. HSF-1 Regulators DDL-1/2 Link Insulin-like Signaling to Heat-Shock Responses and Modulation of Longevity. Cell. 2012;148:322–34.
40 Tepper RG, Ashraf J, Kaletsky R, Kleemann G, Murphy CT, et al. PQM-1 Complements DAF-16 as a Key Transcriptional Regulator of DAF-2-Mediated Development and Longevity. Cell. 2013;154:676–90.
41 Kenyon C. The plasticity of aging: insights from long-lived mutants. Cell. 2005;120:449–60.
42 Kimura KD, Tissenbaum HA, Liu Y, Ruvkun G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science. 1997;277:942–6.
43 Partridge L, Bruning JC. Forkhead transcription factors and ageing. Oncogene. 2008;27:2351–63.
44 Selman C, Lingard S, Choudhury AI, Batterham RL, Claret M, et al. Evidence for lifespan extension and delayed age-related biomarkers in insulin receptor substrate 1 null mice. Faseb J. 2008;22:807–18.
45 Flachsbart F, Caliebe A, Kleindorp R, Blanche H, von Eller-Eberstein H, et al. Association of FOXO3A variation with human longevity confirmed in German centenarians. Proc Natl Acad Sci U S A 2009;106:2700–5.
46 Suh Y, Atzmon G, Cho MO, Hwang D, Liu B, et al. Functionally significant insulin-like growth factor I receptor mutations in centenarians. Proc Natl Acad Sci U S A. 2008;105:3438–42.
47 Willcox BJ, Donlon TA, He Q, Chen R, Grove JS, et al. FOXO3A genotype is strongly associated with human longevity. Proc Natl Acad Sci U S A. 2008;105:13987–92.
48 McElwee J, Bubb K, Thomas JH. Transcriptional outputs of the Caenorhabditis elegans forkhead protein DAF-16. Aging Cell. 2003;2:111–21.
49 Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, et al. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature. 2003;424:277–83.
50 Halaschek-Wiener J, Khattra JS, McKay S, Pouzyrev A, Stott JM, et al. Analysis of long-lived C. elegans daf-2 mutants using serial analysis of gene expression. Genome Res. 2005;15:603–15.
51 Oh SW, Mukhopadhyay A, Dixit BL, Raha T, Green MR, et al. Identification of direct DAF-16 targets controlling longevity, metabolism and diapause by chromatin immunoprecipitation. Nat Genet. 2006;38:251–7.
52 Dong MQ, Venable JD, Au N, Xu T, Park SK, et al. Quantitative mass spectrometry identifies insulin signaling targets in C. elegans. Science. 2007;317:660–3.
53 Morley JF, Brignull HR, Weyers JJ, Morimoto RI. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2002;99:10417–22.
54 Link C. Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc Natl Acad Sci U S A. 1995;92:9368–72.
55 Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–10.
56 Cohen E, Du D, Joyce D, Kapernick EA, Volovik Y, et al. Temporal requirements of insulin/IGF-1 signaling for proteotoxicity protection. Aging Cell. 2010;9:126–34.
57 Steinkraus KA, Smith ED, Davis C, Carr D, Pendergrass WR, et al. Dietary restriction suppresses proteotoxicity and enhances longevity by an hsf-1-dependent mechanism in Caenorhabditis elegans. Aging Cell. 2008;7:394–404.
58 Mouton PR, Chachich ME, Quigley C, Spangler E, Ingram DK. Caloric restriction attenuates amyloid deposition in middle-aged dtg APP/PS1 mice. Neurosci Lett. 2009;464:184–7.
59 Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–11.
60 Zhang T, Mullane PC, Periz G, Wang J. (2011) TDP-43 neurotoxicity and protein aggregation modulated by heat shock factor and insulin/IGF-1 signaling. Hum Mol Genet.
61 Boccitto M, Lamitina T, Kalb RG. Daf-2 signaling modifies mutant SOD1 toxicity in C. elegans. PLoS One. 2012;7:e33494.
62 Teixeira-Castro A, Ailion M, Jalles A, Brignull HR, Vilaca JL, et al. (2011) Neuron-specific proteotoxicity of mutant ataxin-3 in C. elegans: rescue by the DAF-16 and HSF-1 pathways. Hum Mol Genet. 2011;20(15):2996-3009.
63 Killick R, Scales G, Leroy K, Causevic M, Hooper C, et al. Deletion of Irs2 reduces amyloid deposition and rescues behavioural deficits in APP transgenic mice. Biochem Biophys Res Commun. 2009;386:257–62.
64 Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102.
65 Freude S, Hettich MM, Schumann C, Stohr O, Koch L, et al. Neuronal IGF-1 resistance reduces Abeta accumulation and protects against premature death in a model of Alzheimer’s disease. Faseb J. 2009;23:3315–24.
66 Jankowsky JL, Slunt HH, Ratovitski T, Jenkins NA, Copeland NG, et al. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng. 2001;17:157–65.
67 Reiserer RS, Harrison FE, Syverud DC, McDonald MP. Impaired spatial learning in the APPSwe + PSEN1DeltaE9 bigenic mouse model of Alzheimer’s disease. Genes Brain Behav. 2007;6:54–65.
68 Cohen E, Paulsson JF, Blinder P, Burstyn-Cohen T, Du D, et al. Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell. 2009;139:1157–69.
69 Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–12.
70 Carro E, Trejo JL, Gomez-Isla T, LeRoith D, Torres-Aleman I. Serum insulin-like growth factor I regulates brain amyloid-beta levels. Nat Med. 2002;8:1390–7.
71 Poirier R, Fernandez AM, Torres-Aleman I, Metzger F. Early brain amyloidosis in APP/PS1 mice with serum insulin-like growth factor-I deficiency. Neurosci Lett. 2012;509:101–4.
72 Fernandez AM, Torres-Aleman I. The many faces of insulin-like peptide signalling in the brain. Nat Rev Neurosci. 2012;13:225–39.
73 Carro E, Trejo JL, Gerber A, Loetscher H, Torrado J, et al. Therapeutic actions of insulin-like growth factor I on APP/PS2 mice with severe brain amyloidosis. Neurobiol Aging. 2006;27:1250–7.
74 Calamini B, Silva MC, Madoux F, Hutt DM, Khanna S, et al. Small-molecule proteostasis regulators for protein conformational diseases. Nat Chem Biol. 2012;8:185–96.
75 Honda Y, Tanaka M, Honda S. Trehalose extends longevity in the nematode Caenorhabditis elegans. Aging Cell. 2010;9:558–69.
76 Alavez S, Vantipalli MC, Zucker DJ, Klang IM, Lithgow GJ. Amyloid-binding compounds maintain protein homeostasis during ageing and extend lifespan. Nature. 2011;472:226–9.
77 Mojsilovic-Petrovic J, Nedelsky N, Boccitto M, Mano I, Georgiades SN, et al. FOXO3a is broadly neuroprotective in vitro and in vivo against insults implicated in motor neuron diseases. J Neurosci. 2009;29:8236–47.
78 El-Ami T, Moll L, Marques CF, Volovik Y, Reuveni H, et al. (2013) A Novel Inhibitor of the Insulin/IGF Signaling Pathway Protects from Age-Onset, Neurodegeneration-linked Proteotoxicity. Aging Cell. 2014;13(1):165–74.