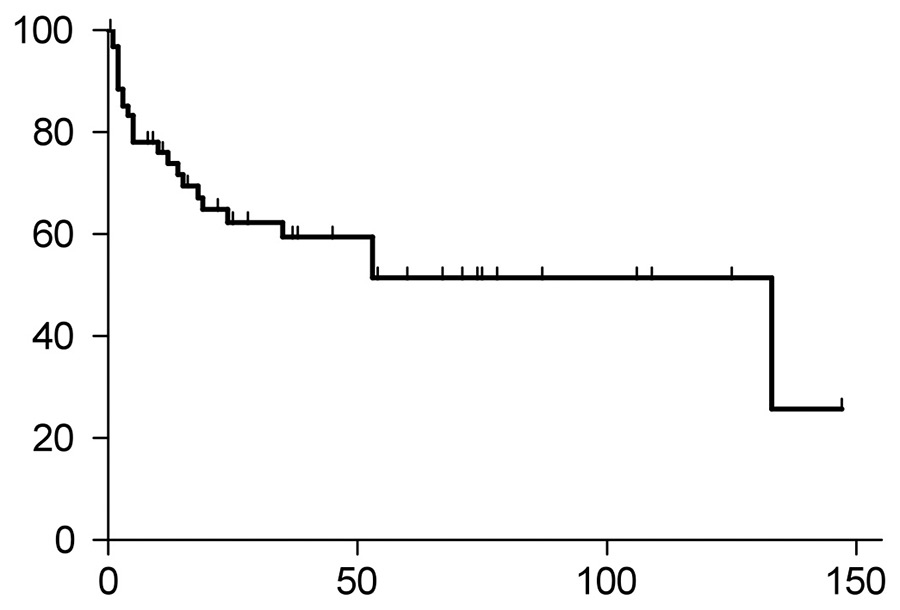
Figure 1A
Overall survival of all AL-amyloidosis patients. The median overall survival of the entire study population was 133 months. Twenty-four patients died. X-axis indicates months, and Y-axis percent survival.
DOI: https://doi.org/10.4414/smw.2014.13922
Systemic AL-amyloidosis (AL) is a rare clonal plasma cell disorder, which is characterised by the deposition of aberrant amyloid protein derived from monoclonal immunoglobulin light chains in a broad variety of soft tissues [1–3]. Although the burden of plasma cells is generally low, the accumulation of the pathogenic amyloid deposits in the heart, kidneys, liver, gastrointestinal tract or nerves may cause substantial morbidity and may lead to rapidly progressive organ failure and ultimately death [4–5].
The prognosis of patients with AL-amyloidosis largely depends on the type and severity of organ involvement, in particular on cardiac involvement [6–7]. Consequently, the current revised prognostic staging system for patients with light chain AL-amyloidosis is based on two cardiac biomarkers – troponin-T and N-terminal-pro-B-type natriuretic peptide (NT-ProBNP) – and on the level of the amyloidogenic light chain synthesis, which largely determine prognosis, but also affect therapeutic options for amyloidosis patients [8].
The introduction of high-dose chemotherapy (HDCT) with autologous stem cell transplantation (ASCT) has improved the outcome of some AL-amyloidosis patients when compared with conventional chemotherapy regimens [9–15]. HDCT with ASCT was reported to confer improved overall survival, particularly in amyloidosis patients achieving a complete remission [16–19], and it has also been associated with improved quality of life [20]. However, a majority of patients with systemic light chain AL-amyloidosis are not considered fit enough to undergo ASCT because of limiting organ involvement [21–24]. Therefore, careful patient selection is crucial to identify amyloidosis patients most likely to benefit from ASCT and to minimise the historically excessive transplant-related mortality of AL-amyloidosis patients [25–26].
Only few data on treatment and outcome of AL-amyloidosis patients in Switzerland are available so far [27]. Therefore, the aim of this study was to examine a single-centre cohort of patients with light chain amyloidosis and to compare the outcome of patients treated with conventional chemotherapy with those treated with HDCT and ASCT. Such a cohort may hopefully be extended at a subsequent stage to initiate a Swiss cohort of AL-amyloidosis patients.
Patients with systemic light chain AL-amyloidosis diagnosed between January 1995 and December 2012 at the Department of Clinical Oncology at the University Hospital in Bern, Switzerland, were included in this retrospective study. Consecutive patients were identified by keyword searching in all databases of this single centre. Patients were required to have a representative tissue biopsy with Congo-red and immunohistochemical staining confirming AL-amyloid disease. The diagnosis of light chain amyloidosis required a documented plasma cell dyscrasia, which was diagnosed by the presence of a clonal plasma cell population in the bone marrow and/or monoclonal gammopathy detected by immunofixation electrophoresis of serum or urine proteins [3, 28–30]. Patients with familial, senile, localised or other forms of amyloidosis were not included in this study [3]. Diagnosis of organ involvement, response to treatment and disease progression were defined according to the international consensus guidelines [31]. For conditioning before ASCT, patients received a total dose of 200 mg/m2 melphalan.
Figure 1A
Overall survival of all AL-amyloidosis patients. The median overall survival of the entire study population was 133 months. Twenty-four patients died. X-axis indicates months, and Y-axis percent survival.
This was an observational study. The study population was divided into two groups, comprising patients treated with conventional chemotherapy or with HDCT and ASCT, representing the two basic therapeutic options for amyloidosis patients. The two groups were compared for differences in clinical characteristics at diagnosis and in outcome. Overall survival was defined as the time from the date of diagnosis until death or last follow-up, whichever occurred first. The time until first progression was the time from diagnosis until first progression or death, whichever occurred earlier, or until last follow-up if patients remained in remission. Curves depicting overall survival and time to progression were plotted using the Kaplan-Meier method. The survival estimates in the two groups were compared using the log-rank test, and differences in the mean values of continuous variables were tested using the t-test. All statistical analyses and graphs were performed using graph pad prism program 5.04 (1992–2010 GraphPad Software, Inc., NC, USA).
We identified 63 patients with systemic light chain amyloidosis, 44 patients having primary AL-amyloidosis and 19 patients having secondary AL-amyloidosis due to multiple myeloma or Waldenström’s macroglobulinaemia. Patients were assigned to either one group including patients treated with conventional chemotherapy regimens (n = 50), or a second group including patients who underwent HDCT with ASCT (n = 13). The reasons not to proceed to HDCT with ASCT were age (n = 12; 24%), relevant organ dysfunction (n = 14; 28%) and multiorgan failure (n = 19; 38%). Five patients (10%) declined HDCT with ASCT.
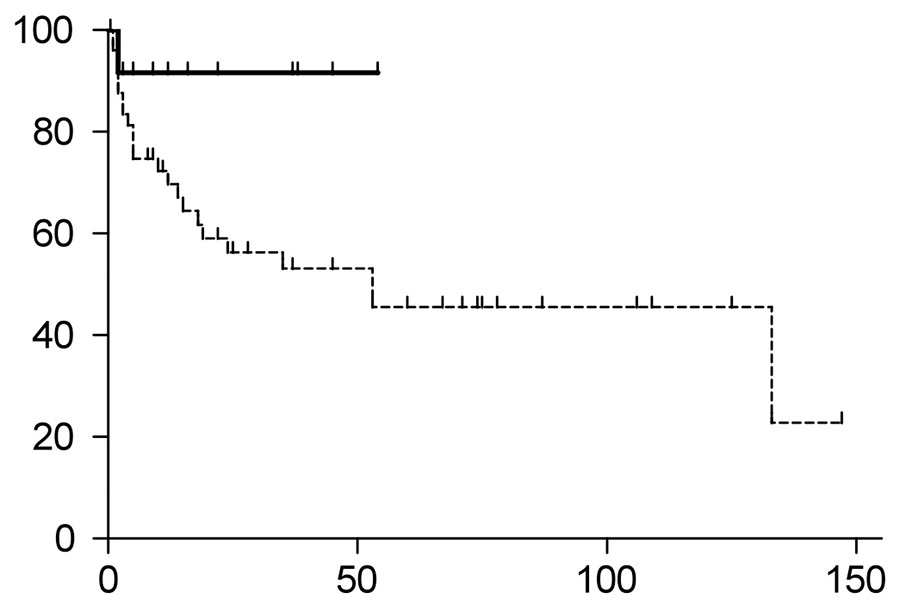
Figure 1B
Overall survival of patients treated with HDCT/ASCT (continuous line) versus conventional chemotherapy (dotted line). The HDCT/ASCT showed a trend toward better overall survival (p = 0.0651). The HDCT/ASCT group had not yet reached median overall survival, whereas the median overall survival of the conventional chemotherapy group was 53 months.
ASCT = autologous stem cell transplantation; HDCT = high-dose chemotherapy
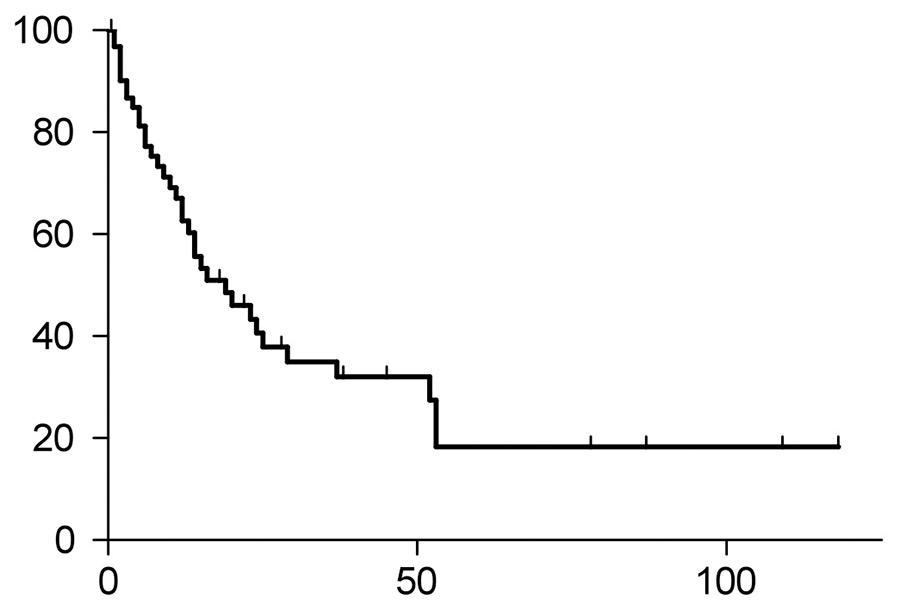
Figure 1C
Time to progression (TTP) of the entire study group. Median TTP was 39 months, and 36 patients had progression of their disease.
Within the entire cohort of 63 patients, the two groups showed no differences with regards to sex, blood cell counts, BNP, β2–microglobulin, albumin, type and level of gammopathy (typically with intact immunoglobulin and associated light chain disease), proteinuria, creatinine and level of malignant plasma cell infiltration in the bone marrow at initial diagnosis. However, patient characteristics differed for age (p = 0.0006) and troponin-T levels (p = 0.0279), two parameters well known to be associated with outcome of AL-amyloidosis patients. The mean level of free light chains at diagnosis was 740 mg/l for the entire study group, 1652 mg/l in the HDCT/ASCT group and 355 mg/l in the conventional chemotherapy group (p = 0.1039). Five patients in the group with HDCT/ASCT had a first-line treatment before HDCT/ASCT, whereas the other eight patients directly proceeded to the high-dose treatment. For the conventional chemotherapy group, the various regimens are listed in table 1. Clinical characteristics at diagnosis, laboratory values at initial presentation, types of therapy, and the course of the disease of the entire study population are also summarised in table 1.
At diagnosis, a total of 38 patients (60%) had cardiac involvement as assessed with echocardiography, 35 patients (56%) had renal involvement, 17 patients (27%) had nerve involvement and 5 patients (8%) had liver involvement. Other organs involved included lung, intestine, spleen, larynx or tongue. Forty-eight percent had one organ involved, 30% had two-organs disease, 19% had three organs involved and 3% had four- (or more) organ disease. The conventional chemotherapy group had a higher percentage of cardiac involvement than the HDCT/ASCT group (66% vs 38%).
After a mean follow-up of 31 months for the entire cohort at the cut-off day on 31 December 2012, one death (7.7%) occurred in the HDCT/ASCT group, and 23 deaths (46%) in the conventional chemotherapy group (p = 0.0116). The single death in the HDCT/ASCT group was transplant related, and it occurred at day 10 after ASCT; thus the transplant-related mortality of the HDCT/ASCT cohort was 7.7%.
The median overall survival of the entire cohort was 133 months (fig. 1A). Patients undergoing HDCT/ASCT had a trend towards better overall survival compared with the conventional chemotherapy group (p= 0.0651, fig. 1B, hazard ratio 0.3694, 95% confidential interval 0.1282‒1.064). The p-value did not reach significance, most likely because of the small cohort size. The HDCT/ASCT group had not yet reached the median survival, whereas the conventional chemotherapy group had a median survival of 53 months.
Of the entire study population, 36 patients had disease progression (fig. 1C), with four patients (31%) in the HDCT/ASCT group and 32 patients (64%) in the conventional chemotherapy group (p = 0.0566, fig. 1D). The median time to progression (TTP) of the entire study population was 19 months, with the median TTP not yet reached in the HDCT/ASCT group, and 15 months in the group with conventional chemotherapy (p = 0.248, hazard ratio 0.6012, 95% confidential interval 0.2536‑1.425).
| Table 1: Characteristics of the study population. | |||||
| HDCT/ASCT | Conventional chemotherapy | All | p-value | ||
| n | 13 | 50 | 63 | ||
| Age | mean ± SEM range | 59.2 ± 1.7 62–69 | 69.6 ± 1.4 45–93 | 67.4 ± 1.3 45–93 | <0.0001 |
| Sex | male/female | 5/8 | 33/17 | 38/25 | 0.1107 |
| Troponin-T (µg/l)(×) | mean ± SEM range | 0.015 ± 0.004 0.003–0.034 | 0.08 ± 0.03 0.001–0.369 | 0.081 ± 0.024 0.001–0.369 | 0.0279 |
| BNP (ng/l)(°) | mean ± SEM range | 240 ± 102 19–978 | 5252 124–40000 | 3104 19–40000 | 0.1983 |
| β2-microglobulin (mg/l) (§) | mean ± SEM range | 2.9 ± 0.8 1.4–11.2 | 6 ± 1.3 1.8–28 | 4.9 ± 0.95 1.4–28 | 0.1182 |
| Albumin (g/l) (¨) | mean ± SEM range | 31.5 ± 2.3 14–43 | 31.3 ± 1.3 5–43 | 31.4 ± 1.1 5–43 | 0.9380 |
| Light chain (*) | kappa/lambda | 5/7 | 15/34 | 20/41 | 0.5050 |
| Light chain value (mg/L) (Y) | mean ± SEM range | 1652 11.8–13631 | 355 ± 67 2.7–1098 | 740 2.7–13631 | 0.1039 |
| Subtypes | IgG IgA IgM light chain only | 3 3 1 6 | 14 4 5 27 | 17 7 6 33 | |
| Paraprotein value (g/l) ($) | mean ± SEM range | 17.3 ± 5.4 5.6–36.9 | 15.1 ± 1.6 2.7–22.3 | 15.6 ± 1.8 2.7–36.9 | 0.5892 |
| Proteinuria (g/l) | yes/no mean ± SEM range | 8/5 2.9 ± 1 0.3–6.8 | 25/25 2.9 ± 0.4 0.14–6 | 33/30 2.9 ± 0.4 0.14–6.8 | 0.5423 0.9765 |
| Haemoglobin (g/l) | mean ± SEM range | 136 ± 4.2 96–153 | 122 ± 3.4 82–181 | 125 ± 2.9 82–181 | 0.0529 |
| Leucocytes (G/l) | mean ± SEM range | 7.6 ± 0.4 5.2–10.8 | 8 ± 0.4 4.1–17 | 7.9 ± 0.3 4.1–17 | 0.5540 |
| Platelets (G/l) | mean ± SEM range | 279 ± 16.8 210–387 | 307 ± 19.9 57–886 | 301 ± 16 57–886 | 0.4668 |
| Creatinin (µmol/l) (&) | mean ± SEM median range | 98.6 ± 21.9 64 51–290 | 153 ± 22.6 89 53–968 | 142 ± 18.5 85.5 51–968 | 0.2258 |
| Plasma cell infiltration of the bone marrow (%) (c) | mean ± SEM range | 26 ± 6.8 10–80 | 24 ± 2.9 0–90 | 25 ± 2.7 0–90 | 0.7711 |
| Mean follow-up (months) | 20.4 | 31.4 | 30.9 | ||
| Progression | yes / no | 4/9 | 32/18 | 36/27 | 0.0566 |
| Dead | yes / no | 1/12 | 23/27 | 24/39 | 0.0116 |
| First line treatment + | VAD bortezomib/dex./cycloph. bortezomib/dex. thalidomide/dex. melphalan/pred. lenalidomide/dex. | 1 2 2 0 0 0 | 6 3 15 2 15 2 | 7 5 17 2 15 2 | |
| Number of involved organs | 1 2 3 ≥4 | 8 (61%) 4 (31%) 1 (8%) 0 | 22 (44%) 15 (30%) 11 (22%) 2 (4%) | 30 (48%) 19 (30%) 12 (19%) 2 (3%) | |
| Involved organs | heart kidney liver nerve various (X) | 5 (38%) 7 (54%) 1 (8%) 4 (31%) 4 (31%) | 33 (66%) 28 (56%) 4 (8%) 13 (26%) 15 (30%) | 38 (60%) 35 (56%) 5 (8%) 17 (27%) 19 (30%) | |
| Response to induction (") | complete remission partial remission stable disease/progression disease | 0 4 0 | 5 17 3 | 5 21 3 | |
| Revised Mayo stage (^) | I II III IV | 2 5 1 0 | 0 3 2 4 | 2 8 3 4 | |
| ×Troponin-T data were available from 23 patients, and °BNP data from 25 patients; §Information on β2microglobulin was available from 31 patients, and on albumin from 53 patients,;*Information on the light chain subtype was not available from 2 patients, yand on the light chain level from 26 patients; $information on the paraprotein value was available from 29 patients, &on creatinine from 60 patients, and con the plasma cell infiltration in the bone marrow from 53 patients; "data on the response to induction treatment was available from 34 patients, and ^sufficient information allowing classification according to the revised Mayo stage criteria was available from 17 patients; Xincluded lung, intestine, spleen, larynx, or tongue. +VAD: vincristine, adriamycin, dexamethasone; dex: dexamethasone; pred: prednisone; cycloph: cyclophos-phamide. | |||||
Only few data are available so far on treatment and outcome of AL-amyloidosis patients in Switzerland. In particular, one single-centre study reported experience with 16 patients with AL-amyloidosis treated with HDCT and ASCT [27]. In this study, we report a single-centre cohort of 63 patients in the period from January 1995 to December 2012, diagnosed and treated at the Department of Clinical Oncology and Institute of Pathology at the University Hospital and University of Bern, Switzerland. The study population was divided into a group of patients treated with HDCT/ASCT and a second group comprising patients undergoing conventional chemotherapy. We found that amyloidosis patients receiving HDCT and ASCT tended to have a more favourable outcome than patients treated with conventional chemotherapy (p = 0.0651).
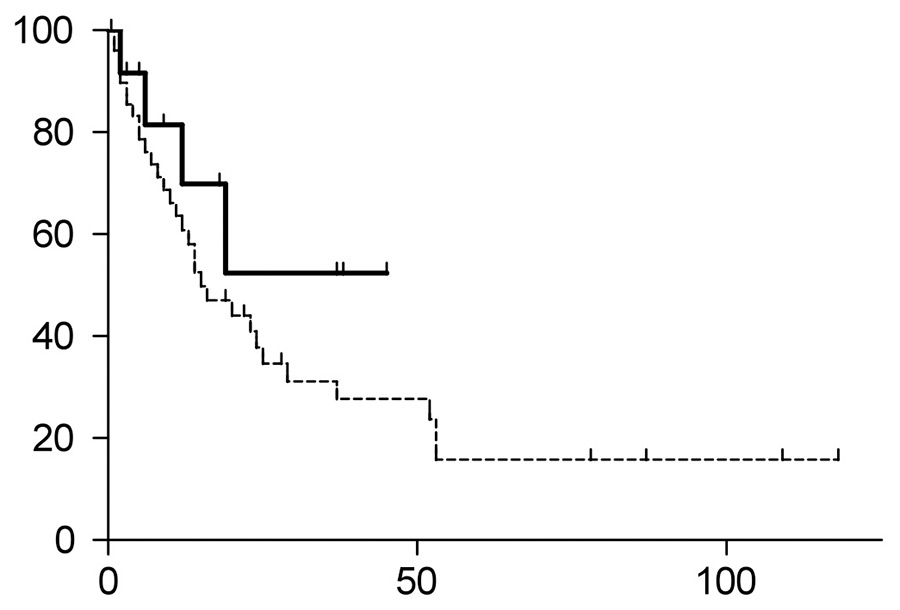
Figure 1D
Time to progression (TTP of) patients treated with HDCT/ASCT (continuous line) versus conventional chemotherapy regimens (dotted line). The HDCT/ASCT group had not yet reached the median TTP; the TTP of the conventional chemotherapy group was 15 months (p = 0.248).ASCT = autologous stem cell transplantation; HDCT = high-dose chemotherapy
Previous reports have suggested that response rates and overall survival are better in systemic AL-amyloidosis patients treated with HDCT/ASCT as compared with conventional chemotherapy regimens [9–13]. However, only a minority of amyloidosis patients, usually those with limited organ disease and/or those lacking significant cardiac involvement, are eligible for HDCT/ASCT [8]. In contrast, a single randomised study failed to show a benefit for HDCT/ASCT compared with oral melphalan and dexamethasone [32]; however, 26% of the patients in the transplantation arm of this multicentre study ultimately did not receive HDCT/ASCT, and the ASCT treatment-related mortality was excessive (24%). The baseline patient characteristics in this randomised trial were equally balanced between the two groups, with a similar organ involvement and age as the HDCT/ASCT group in our study [32].
Our data from systemic AL-amyloidosis patients indicate a trend toward better overall survival for patients undergoing HDCT/ASCT: the median survival was not reached in the HDCT/ASCT group versus 53 months in the conventional chemotherapy group (p = 0.0651). The two groups of this cohort differed significantly in two clinical characteristics: patients undergoing HDCT/ASCT were younger (59 vs 70 years, p = 0.0006), and their troponin-T values were lower (0.015 μg/l vs 0.08 μg/l, p = 0.0279). Thus, a possible factor contributing to the improved outcome, in addition to the more intensive treatment, of patients receiving HDCT/ASCT may be their better performance status and superior cardiac function. Ultimately, patient cohorts with matched characteristics within a sufficiently powered randomised clinical trial will be necessary to define the impact of the better performance status on outcome.
The retrospective character and the small number of patients in the HDCT/ASCT group limit the power of this study. Nevertheless, these real-life observations may allow some conclusions. Based on the differences in age and troponin-T values in our two subgroups, our study confirms current treatment algorithms recommending intensive treatment to fit amyloidosis patients with limited organ involvement and with clinically insignificant cardiac involvement, whereas amyloidosis patients not meeting these criteria should be treated with conventional chemotherapy regimens. As with many other rare tumour types, further research questions aiming to improve the treatment of amyloidosis patients must be based on sufficiently large numbers of patients. Thus, an obvious step will be to extend this single centre cohort to additional centres, ideally towards the establishment of a Swiss cohort of AL-amyloidosis patients. Such an initiative is currently being initiated, and it will hopefully be accompanied by the initiation of the first clinical trials in Switzerland specifically focusing on amyloidosis patients. This is of particular clinical relevance since amyloidosis patients are usually excluded from trials of multiple myeloma patients and treatment regimens for these two groups of patients are similar.
Acknowledgement:The authors wish to thank Marion Bleckmann, Barbara Muster and Irene Briner for assistance with the ASCT data.
Funding / potential competing interests: No financial support and no other potential conflict of interest relevant to this article was reported.
1 Merlini G, Seldin DC, Gertz MA. Amyloidosis: pathogenesis and new therapeutic options. J Clin Oncol. 2011;29(14):1924–33.
2 Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349(6):583–96.
3 Falk RH, Comenzo RL, Skinner M. The systemic amyloidoses. N Engl J Med. 1997;337(13):898–909.
4 Comenzo RL. Amyloidosis. Curr Treat Options Oncol. 2006;7(3):225–36.
5 Leung N, Nasr SH, Sethi S. How I treat amyloidosis: the importance of accurate diagnosis and amyloid typing. Blood. 2012;120(16):3206–13.
6 Sack FU, Kristen A, Goldschmidt H, Schnabel PA, Dengler T, Koch A, et al. Treatment options for severe cardiac amyloidosis: heart transplantation combined with chemotherapy and stem cell transplantation for patients with AL-amyloidosis and heart and liver transplantation for patients with ATTR-amyloidosis. Eur J Cardiothorac Surg. 2008;33(2):257–62.
7 Merlini G, Wechalekar AD, Palladini G. Systemic light chain amyloidosis: an update for treating physicians. Blood. 2013;121(26):5124–30.
8 Kumar S, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Colby C, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol. 2012;30(9):989–95.
9 Dispenzieri A, Kyle RA, Lacy MQ, Therneau TM, Larson DR, Plevak MF, et al. Superior survival in primary systemic amyloidosis patients undergoing peripheral blood stem cell transplantation: a case-control study. Blood. 2004;103(10):3960–3.
10 Bahlis NJ, Lazarus HM. Multiple myeloma-associated AL amyloidosis: is a distinctive therapeutic approach warranted? Bone Marrow Transplant. 2006;38(1):7–15.
11 Skinner M, Sanchorawala V, Seldin DC, Dember LM, Falk RH, Berk JL, et al. High-dose melphalan and autologous stem-cell transplantation in patients with AL amyloidosis: an 8-year study. Ann Intern Med. 2004;140(2):85–93.
12 Kyle RA, Therneau TM, Rajkumar SV, Offord JR, Larson DR, Plevak MF, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002;346(8):564–9.
13 Schonland SO, Lokhorst H, Buzyn A, Leblond V, Hegenbart U, Bandini G, et al. Allogeneic and syngeneic hematopoietic cell transplantation in patients with amyloid light-chain amyloidosis: a report from the European Group for Blood and Marrow Transplantation. Blood. 2006;107(6):2578–84.
14 Passweg JR, Halter J, Bucher C, Gerull S, Heim D, Rovo A, et al. Hematopoietic stem cell transplantation: a review and recommendations for follow-up care for the general practitioner. Swiss Med Wkly. 2012;142:w13696.
15 Passweg JR, Baldomero H, Bargetzi M, Bucher C, Chalandon Y, Duchosal MA, et al. Haematopoietic stem cell transplantation: activity in Switzerland compared with surrounding European countries. Swiss Med Wkly. 2013;143:w13757.
16 Sanchorawala V, Skinner M, Quillen K, Finn KT, Doros G, Seldin DC. Long-term outcome of patients with AL amyloidosis treated with high-dose melphalan and stem-cell transplantation. Blood. 2007;110(10):3561–3.
17 Dispenzieri A, Seenithamby K, Lacy MQ, Kumar SK, Buadi FK, Hayman SR, et al. Patients with immunoglobulin light chain amyloidosis undergoing autologous stem cell transplantation have superior outcomes compared with patients with multiple myeloma: a retrospective review from a tertiary referral center. Bone Marrow Transplant. Apr 22 2013.
18 Gertz MA, Lacy MQ, Dispenzieri A, Hayman SR, Kumar SK, Leung N, et al. Effect of hematologic response on outcome of patients undergoing transplantation for primary amyloidosis: importance of achieving a complete response. Haematologica. 2007;92(10):1415–8.
19 Gertz MA, Lacy MQ, Dispenzieri A, Hayman SR, Kumar SK, Dingli D, et al. Autologous stem cell transplant for immunoglobulin light chain amyloidosis: a status report. Leuk Lymphoma. 2010;51(12):2181–7.
20 Seldin DC, Anderson JJ, Sanchorawala V, Malek K, Wright DG, Quillen K, et al. Improvement in quality of life of patients with AL amyloidosis treated with high-dose melphalan and autologous stem cell transplantation. Blood. 2004;104(6):1888–93.
21 Gertz MA, Lacy MQ, Dispenzieri A, Gastineau DA, Chen MG, Ansell SM, et al. Stem cell transplantation for the management of primary systemic amyloidosis. Am J Med. 2002;113(7):549–55.
22 Gertz MA, Lacy MQ, Dispenzieri A, Hayman SR, Kumar S. Transplantation for amyloidosis. Curr Opin Oncol. 2007;19(2):136–41.
23 Kumar S, Dispenzieri A, Gertz MA. High-dose melphalan versus melphalan plus dexamethasone for AL amyloidosis. N Engl J Med. 2008;358(1):91; author reply 92–93.
24 Sanchorawala V, Wright DG, Seldin DC, Dember LM, Finn K, Falk RH, et al. An overview of the use of high-dose melphalan with autologous stem cell transplantation for the treatment of AL amyloidosis. Bone Marrow Transplant. 2001;28(7):637–642.
25 Cibeira MT, Sanchorawala V, Seldin DC, Quillen K, Berk JL, Dember LM, et al. Outcome of AL amyloidosis after high-dose melphalan and autologous stem cell transplantation: long-term results in a series of 421 patients. Blood. 2011;118(16):4346–52.
26 Gertz MA, Lacy MQ, Dispenzieri A, Kumar SK, Dingli D, Leung N, et al. Refinement in patient selection to reduce treatment-related mortality from autologous stem cell transplantation in amyloidosis. Bone Marrow Transplant. 2013;48(4):557–61.
27 Frossard V, Ketterer N, Rosselet A, Meier P, Cairoli A, Duchosal MA, et al. Early intensification and autologous stem cell transplantation in patients with systemic AL amyloidosis: a single-centre experience. Ann Hematol. 2009;88(7):681–5.
28 Gertz MA, Greipp PR, Kyle RA. Classification of amyloidosis by the detection of clonal excess of plasma cells in the bone marrow. J Lab Clin Med. 1991;118(1):33–39.
29 Perfetti V, Garini P, Vignarelli MC, Marinone MG, Zorzoli I, Merlini G. Diagnostic approach to and follow-up of difficult cases of AL amyloidosis. Haematologica. 1995;80(5):409–15.
30 Bradwell AR, Carr-Smith HD, Mead GP, Tang LX, Showell PJ, Drayson MT, et al. Highly sensitive, automated immunoassay for immunoglobulin free light chains in serum and urine. Clin Chem. 2001;47(4):673–80.
31 Gertz MA, Comenzo R, Falk RH, Fermand JP, Hazenberg BP, Hawkins PN, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, 18–22 April 2004. Am J Hematol. 2005;79(4):319–28.
32 Jaccard A, Moreau P, Leblond V, Leleu X, Benboubker L, Hermine O, et al. High-dose melphalan versus melphalan plus dexamethasone for AL amyloidosis. N Engl J Med. 2007;357(11):1083–93.
Contributors:J.R. performed research, analysed data and wrote the paper; Y.B. contributed vital material and analysed data; T.S. analysed data, and T.P. designed research, analysed data and wrote the paper. All authors have read and approved the final version of the manuscript.