Minimal residual disease monitoring: the new standard for treatment evaluation of haematological malignancies?
DOI: https://doi.org/10.4414/smw.2014.13907
Mathieu
Hauwel, Thomas
Matthes
Summary
Minimal residual disease (MRD) refers to the small number of malignant cells that remain after therapy when the patient is in remission and shows no symptoms or overt signs of disease. Current treatment protocols for haematological malignancies allow most patients to obtain some form of MRD state, but cure seldom follows and in most cases fatal relapses occur sooner or later, leaving a bitter impression of having won a battle yet lost the war.
MRD detection and quantification are used for evaluation of treatment efficiency, patient risk stratification and long-term outcome prediction. Whereas multicolour flow cytometry (MCFC) and polymerase chain reaction (PCR) based methods constitute the two most commonly used techniques for MRD detection, next generation sequencing will certainly be widely employed in the future.
As MRD reflects the nature of the malignant disease itself, including its sensitivity to the drug regimens applied, it constitutes the ideal method for surveillance and patient follow-up. The morphological examination of peripheral blood or bone marrow smears, although still an indispensable part of routine laboratory testing, is clearly insufficient for patient management, and clinicians should not ask themselves whether to look for MRD or not, but how and when.
Abbreviations
ALL acute lymphoblastic leukaemia
AML acute myeloid leukaemia
ASO-PCR allele-specific oligonucleotide-PCR
BCR B-cell receptor
CLL chronic lymphocytic lymphoma
CML chronic myeloid leukaemia
CR complete remission
cytoCR flow cytometry CR
DNA deoxyribonucleic acid
FISH fluorescent in-situ hybridisation
FL follicular lymphoma
iCR immunofixation CR
IgH immunoglobulin heavy chain
LAIP leukaemia-associated immunophenotype
LSCs leukaemic stem cells
MCFC multicolour flow cytometry
MRD minimal residual disease
mRNA messenger RNA
NGS new generation sequencing
NMR nuclear magnetic resonance
PCR polymerase chain-reaction
PET positron emission tomography
PFS progression-free survival
Ph Philadelphia chromosome
RNA ribonucleic acid
RT-PCR reverse transcriptase-PCR
sCR serum free light chain ratio CR
TCR T-cell receptor
Introduction
Minmal residual disease (MRD) is defined as the small number of cancer cells that persist in a patient during or after treatment, even though clinical and microscopic examinations confirmed complete remission (CR) and the patient shows no signs or symptoms of disease. Because MRD is seen as the major cause of disease relapse, its detection is at the crossroads of past and future concerns regarding haematological malignancy management. It gives important feedback about conventional treatment success and helps in selecting therapeutic alternatives. The concept and clinical significance of MRD have been particularly well investigated in acute leukaemias, where patients have approximately 1012 malignant cells at diagnosis and may, after therapy, with a microscopically normal bone marrow (cytological CR or haematological CR), still harbour up to 1010 cancer cells. Although achieving this type of response certainly means good news for the patient, invariably disease relapses occur sooner or later, originating from residual malignant cells that have not been eliminated despite the treatments used.
Several highly sensitive methods are available for MRD detection, including molecular biology – based on the detection of cancer-specific deoxyribonucleic acid (DNA), ribonucleic acid (RNA) or proteins – to measure minute levels of cancer cells in blood or tissue samples, sometimes as low as one cancer cell in a million normal cells. Flow cytometry is a fast and cost-effective method widely used for tumour immunophenotyping at diagnosis. Recent advances in multicolour (>8) panel development are bringing the sensitivity level of cytometric analysis close to that achieved so far by molecular biology techniques. However, all these techniques require great technical expertise and are still mainly used in the context of clinical trials. Slowly they are beginning to be introduced into routine clinical practice.
Techniques for MRD detection
DNA-based tests
These are based on detecting a leukaemia-specific DNA sequence. Generally, this is achieved through the use of the polymerase chain-reaction (PCR), since cytogenetic techniques such as caryotyping, fluorescence in-situ hybridisation (FISH), or comparative genome hybridisation (CGH) lack the sensitivity required for MRD monitoring. The DNA sequence chosen may contribute to the genesis of the leukaemia, or may simply be linked to it. The markers used for DNA-based testing are often chromosomal translocations such as t(8;21) and t(15;17) in acute myeloid leukaemia (AML), or t(14;18) and t(11;14) in lymphomas (table 1). Other genes used for MRD detection include microsatellites, immunoglobulin and T-cell receptor (TCR) genes. The allele-specific oligonucleotide PCR (ASO-PCR) is based on the fact that B- and T-cell leukaemias exhibit a distinct immunoglobulin and TCR gene rearrangement at the V(D)J junctional region that can be used as a specific marker for that particular leukaemic clone.
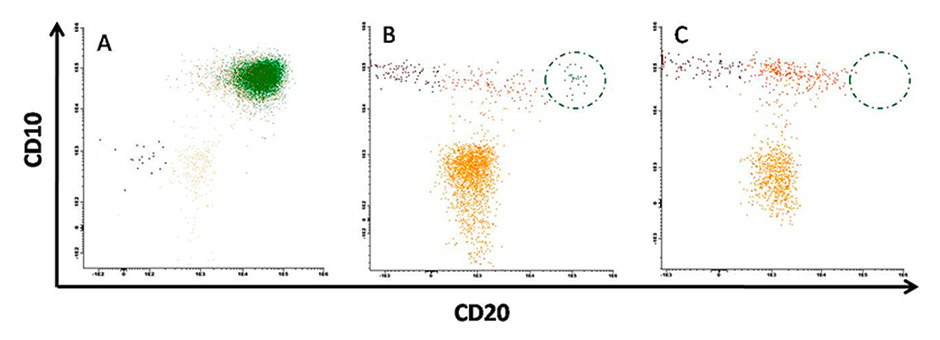
Figure 1
Minimal residual disease detection using flow cytometry.
A. Patient diagnosed with a massive B-lineage acute lymphoblastic leukaemia clone characterised by CD10/CD20 double positivity (green) and bone marrow dysplasia. B. Same patient after chemotherapy. Bone marrow regeneration is confirmed by a high number of progenitors (yellow) and normal B cell differentiation is resumed (brown to orange). However residual disease was also detected (green). C. Normal bone marrow from healthy donor. 106 total events were acquired in each case.
Although sensitivity of these different PCR-techniques is high (up to 1 in 105 cells in some studies), they are only applicable in patients with known translocations or other known and specific DNA markers. ASO-PCR requires the development of reagents (patient-specific probes) and assay conditions for each individual patient, which is laborious, expensive and time consuming.
RNA-based tests
These are based on detecting a leukaemia-specific messenger RNA (mRNA) sequence. Generally, this is achieved through the use of reverse transcription of the mRNA followed by reverse transcriptase polymerase chain reaction (RT-PCR). These mRNA-based tests are normally used when a DNA test is impractical. For example, the t(9;22) BCR-Abl translocation in chronic myeloid leukaemia (CML) may occur over a large length of chromosomes 9 and 22, which makes DNA-based testing difficult and inefficient. However, RNA is a much less stable target for diagnostics than DNA and requires careful handling and processing.
The markers used for RNA-based testing are almost exclusively chromosomal translocations, such as t(9;22) BCR-Abl, t(15;17) PML-RARA, and t(12;21) ETV6-RUNX1 (TEL-AML1); these translocations are not specific to an individual and remain stable throughout the course of the disease (see table 1).
Next generation sequencing
With the development of new sequencing methods it has become possible to search not only for known mutations/translocations, but also for all clonal gene mutations and rearrangements present in diagnostic samples, and to track their evolution during therapy. In a recent study, consensus primers and high-throughput sequencing were employed to amplify and sequence all rearranged immunoglobulin heavy chain (IgH) and TCR gene segments present in bone marrow samples collected throughout treatment in patients with B-lineage acute lymphoblastic leukaemia (B-ALL). The assay was shown to be quantitative at frequencies down to 105 with a lower limit of detection of 106 [1]. This was at least one to two orders of magnitude higher than standard ASO-PCR and flow-cytometric methods, respectively. Although the costs for this assay are still rather prohibitive and only few data are already published, it can be assumed that, with the technology being rapidly improved, this assay will gain widespread use in the clinics.
Flow cytometry tests
These tests are based on the detection of specific proteins on the surface of cells, in most cases white blood cells. Leukaemic white blood cells cells often show quite unusual and unique combinations of these cell surface proteins (LAIP, leukemia-associated immunophenotype) compared with normal cells. The proteins can be stained with fluorescent dye-labelled antibodies and detected using MCFC. The absence or abnormal expression of tens of different proteins can thus be determined for a single cell. In several minutes, millions of cells from a patient sample can be analysed.
Flow cytometry is the most commonly used technique for the diagnosis and characterisation of haematological malignancies. Although the method is widely used, a high level of expertise is required to interpret the data proficiently when it comes to rare event detection such as MRD. The sensitivity for the detection of malignant cells varies according to the type of leukaemia, the panel of antibodies used, the number of cells analysed and the expertise of the laboratory. Currently, sensitivities of 1 in 104 up to 1 in 105 cells can routinely be reached, and it is expected that with modern flow cytometers and software, and panels of 10 antibodies or more, sensitivity even up to 1 in 106 cells could be possible (fig. 1).
Choice of the test
Which method, among those described above, is the best to measure MRD is still a matter of intense debate. Numerous clinical trials are ongoing to determine for each specific disease the method to employ, the optimal time and frequency of MRD assessment, as well as the thresholds that allow the best stratification of patients and show the most predictive power. The various methods are thereby compared on the basis of their sensitivity, specificity and applicability to the largest number of patients, as well as their costs. International efforts are being undertaken to standardise MRD detection and to facilitate its introduction into routine clinical practice.
Whether other exciting new methods, like the analysis of the proteome or the metabolome with chromatography, mass spectrometry or NMR spectroscopy (nuclear magnetic resonance spectroscopy) are sufficiently sensitive for MRD detection in haematology has still to be investigated [2]. A first promising study in multiple myeloma has been published recently, which showed the potential of metabolic profiles obtained by NMR in identifying new biomarkers that might be used to monitor response to treatment in multiple myeloma patients [3].
|
Table 1:Most common genes and translocations used for detection of minimal residual disease. |
|
Disease
|
Targets
|
|
Acute lymphoblastic leukaemia
|
t(9;22) BCR-Abl |
| |
t(12;21) ETV6-RUNX1 (TEL-AML1) |
| |
patient-specific Ig or TCR genes |
|
Acute myeloid leukaemia
|
t(15;17) PML-RARa |
| |
t(8;21) AML1-RUNX1T1 (AML-ETO) |
| |
inv(16) CBFb/MYH11 |
| |
FLT3-ITD |
| |
NPM1 |
|
Chronic lymphocytic leukaemia
|
patient-specific Ig genes |
|
Chronic myeloid leukaemia
|
t(9;22) BCR-Abl |
|
Follicular lymphoma
|
t(14;18) IgH/BCL2 |
| |
patient-specific Ig genes |
|
Mantle cell lymphoma
|
t(11;14) IgH/CCND1 (IgH/BCL1) |
| |
patient-specific Ig genes |
|
Multiple myeloma
|
M-protein |
|
|
t(4;14) |
|
|
patient-specific Ig genes |
| Ig = immunoglobulin; IgH = immunoglobulin heavy chain; TCR = T-cell receptor |
The clinical significance of MRD
In the last century, physicians relied mainly on clinical examination and microscopic examination of blood or bone marrow smears to evaluate the response to the chemotherapy they had given to their patients and to categorise them into progressive or stable disease and partial or complete response. With the development of more sensitive techniques, such as those explained above, the original definition of CR became unsatisfactory and we now distinguish between six different types of CR, depending on the technique used for MRD detection and the sensitivity achieved: clinical examination (clinical CR), the microscope (haematological CR), caryotype and FISH analyses (cytogenetic CR), RT-PCR or PCR (molecular CR), MCFC (cytometric CR), or high-throughput sequencing (HTPS CR). In the treatment strategies currently in use for haematological malignancies such as acute leukaemias and lymphoid neoplasms, MRD testing has several important roles.
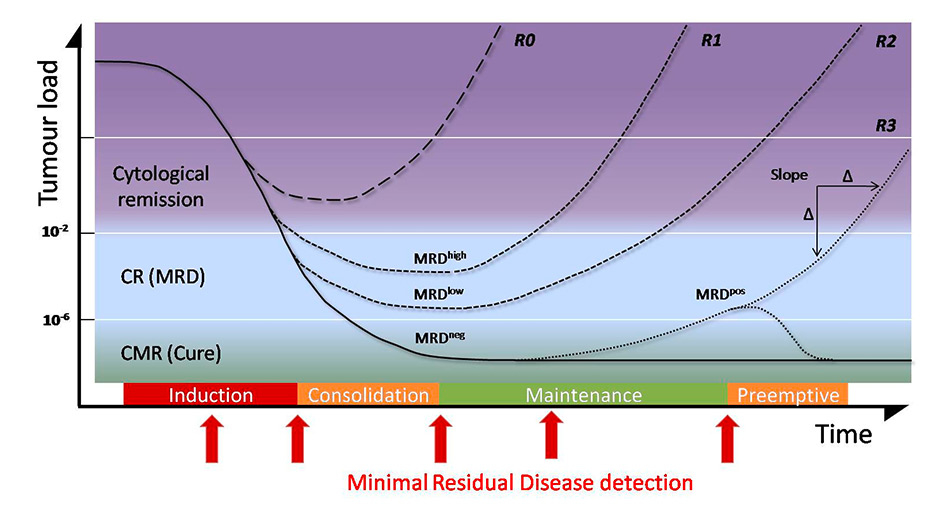
Figure 2
Evolution of tumour load and minimal residual disease (MRD) before, during and after chemotherapy.
After therapy, the level of MRD is one of the best predictive factors of relapse. A patient who did not achieve MRD level will relapse very quickly (R0). Depending of the level of minimal disease present, relapse will occur sooner or later (R1, R2, R3). Patients who achieve MRD negativity are considered to be cured and can be maintained in that state with pre-emptive treatment triggered by MRD positive results. The slope of the tumour growth curve is a further predictive factor.
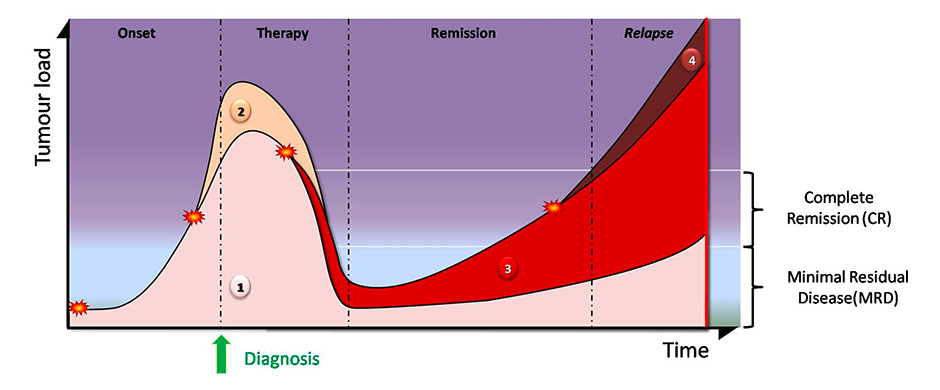
Figure 3
Clonal evolution during chemotherapy.
Tumour clones derive from somatic mutations (stars). The original parent clone (1) can give rise to subclones (2, 4). Additional mutations due to chemotherapy/radiotherapy can also trigger the development of new subclones (3). Therapy eliminates some clones (2) but some others (1, 3) survive and are detected as minimal residual disease. Clonal composition may be very different between the initial diagnosis and after relapse.
Determining treatment efficiency
After the appropriate chemotherapy regimen has been selected and administered, quantitative methods such as the ones described above allow the follow-up of tumour reduction and the determination whether the tumour has been eradicated or whether traces of it remain (fig. 2). This quantification can be considered as an early surrogate marker of treatment efficacy since it constitutes the collective end result of all of the cellular mechanisms that determined the patients’ response to therapy. It gives physicians the opportunity to identify good responders who could benefit from treatment de-escalation and/or from a milder consolidation/maintenance therapy. Thus, unnecessary exposure to toxic agents can be reduced and the treatment personalised. In addition to the accurate measurement of the remaining tumour burden, these techniques also provide information about the kinetics of tumour reduction. In clinical trials, MRD measurements are commonly used as an early endpoint that allows comparison of the efficacy of different treatments.
MRD as prognostic factor of relapse (MRD as post-therapy prognosticator)
Biomarkers and clinical scores established at diagnosis for a large number of haematological malignancies are highly predictive of outcome in a specific therapeutic context, but sometimes have been shown to lose their prognostic value in a different treatment setting [4]. Owing to its a posteriorinature, MRD detection is probably less susceptible to treatment-related variability than biomarkers and constitutes a highly informative factor for estimating the probability of disease relapse. The correlation between the level of MRD and the probability of relapse seems to be stronger than for any other prognostic factor including age, sex, type of mutation and treatment used [5]. However, one has to keep in mind that the presence of leukaemic cells is not necessarily an indication that relapse is imminent. These cells might have been altered by the treatment in such a way that they become incapable of proliferation. They may be able to divide but are held in check by the patient's immune system.
MRD as remission control and trigger for pre-emptive treatment
The treatment of relapsing leukaemias or lymphomas is associated with high mortality/morbidity and remains in general unsatisfactory. Therefore, preventing relapse constitutes an important goal in disease management, with MRD monitoring providing an ideal tool to predict imminent relapse and to trigger the appropriate therapeutic response while the patient is clinically still asymptomatic and the tumour burden is low.
In a study of mantle cell lymphoma, pre-emptive treatment of molecular relapses successfully converted 92% of the patients back to MRD negativity for a median progression-free survival (PFS) of 3.7 years [6]. Although no long-term remissions were obtained, some patients underwent successive molecular relapse / pre-emptive treatment cycles, preserving them from clinical relapse. In another study of AML patients monitored after allogeneic transplant, azacytidine treatment was used on the basis of the results of MRD detection: if CD34-positive donor chimerism fell below 80%, treatment was given [7]. No long-term remission could be obtained, but PFS was increased by several months. This strategy thus led to a significant improvement in the quality of life of the patients and also provided an extended time-window for donor lymphocyte infusions and for the search for a second transplant. In a study with patients suffering from promyelocytic leukaemia, pre-emptive therapy with arsenic trioxide based on positive PCR results for the t(15:17) was able to decrease the probability of 3-year relapse from 12% down to 3% [8].
Analysis of genetic drift prior to relapse
Next generation sequencing (NGS) has led to the discovery of until-recently unsuspected tumour heterogeneity in and between patients. It was found that tumours not only show important clonal diversity and evolution during treatment, but that even at diagnosis several clones exhibiting different mutations can be detected in the patients. These clonal populations are then subject to intense selective pressures under the various chemotherapies, leading to the selection and expansion of the fittest clones, similar to the principle of Darwinian evolution [9–12] (fig. 3).
MRD detection in acute leukaemias and lymphomas
MRD in ALL
MRD analyses are typically performed on bone marrow specimens, although it has been shown that for T-ALL concordant results can be obtained by the analysis of peripheral blood samples [13]. Difficulties in evaluating haematological MRD in ALL lie in the fact that morphological analysis is unable to distinguish between ALL blast cells and lymphoid precursors (haematogones) or activated mature lymphocytes. This distinction is particularly difficult in samples of bone marrow recovering from chemotherapy or transplantation, where haematogones may account for 10% of the lymphoid cells.
MRD is currently one of the most powerful prognostic indicators for disease-free and overall survival in childhood ALL, and there is strong evidence supporting its prognostic significance also in adult ALL. Depending upon the method used, up to 70% of children with ALL (80% of adults) will have MRD detectable immediately after the completion of induction therapy. Multiple studies have shown that children with detectable MRD have significantly decreased disease-free and overall survival and show higher relapse rates than children without detectable residual tumour cells. A multicentre study compared the ability of real-time quantitative PCR and MCFC to predict bone marrow relapse using 726 surveillance samples from 228 children with ALL [14]. Concordant results were obtained with these two techniques in 84% of day 29 samples using an MRD threshold of 0.1%. Thus, MRD monitoring has been introduced into many treatment protocols for risk assignment and selection of therapeutic regimens.
MRD studies have also demonstrated prognostic value when measured before or after allogeneic haematopoietic cell transplantation. Patients with MRD are more likely to relapse than those without detectable MRD [15–18].
Leukaemic clones can establish a prolonged “dormancy.” In one study, bone marrow samples obtained at relapse in 8 of 12 children, who relapsed more than 10 years after the diagnosis of ALL, demonstrated an IgH or TCR gene rearrangement identical to that present in the diagnostic bone marrow specimen [19]. This suggests that the leukaemic cells, or clonal precursors, survived for more than a decade, and raises the question when, if ever, we can truly be confident of cure.
While it is currently unclear whether the identification of MRD should lead to subsequent therapy, the presence of a persistently positive MRD result should trigger preparation for potential disease relapse in the future. For example, a search for potential donors may be undertaken for patients who have not yet undergone allogeneic haematopoietic cell transplantation. Ongoing trials are evaluating the escalation of therapy intensity in MRD positive cases and the reduction of therapy intensity in MRD negative cases [20].
MRD in AML
The achievement of haematological CR is a prerequisite for cure in AML and can be achieved in 50%–80% of cases (depending on age), but does not provide sufficient insight into the quality of the response, since the majority of patients with a haematological CR relapse within 3–5 years after diagnosis. It is still not clear which is the best method to measure MRD in AML, and numerous trials have been performed using either PCR or RT-PCR techniques, or assays based on flow cytometry. The advantage of MCFC-based assays is that they determine accurately the number of leukaemic cells and are applicable to the majority of AML patients, whereas PCR- and RT-PCR-based methods only allow for MRD detection in the approximately 50% of patients with a suitable molecular target.
The analysis of the AML blast phenotype with MCFC often detects multiple LAIPs on subsets of blasts already at diagnosis [21, 22]. Interestingly, comparisons of paired samples from presentation and relapse often show immunophenotypic shifts and LAIP changes, thus rendering MRD detection challenging. One approach, which became possible with the development of 10- and 12-colour cytometers, is to monitor as many independent LAIPs as possible per patient. This reduces the likelihood of false-positive MRD results due to the potential presence of LAIP antigen combinations at low frequencies in normal bone marrow, bone marrow after chemotherapy, or after growth factor administration. In experienced laboratories, a positive MRD result with MCFC rests on the identification of only 20–50 clustered abnormal events. Because leukaemic cells are quantified in relation to other cells in the specimen, the smallest abnormal cell cluster that can be reliably called MRD depends on the total number of cells analysed. With 2–5x105 cells acquired, a 20–50 cell cluster represents a sensitivity of 1 in 104 (0.01%). The lower the number of abnormal cells present, the higher the number of cells required to be analysed. As a result, if the sample quantity is limited, the sensitivity of the MRD assay will be lowered. This stresses the importance of sample quality for accurate MRD evaluation.
Independent of the initial LAIP is the “different-from-normal” approach, which uses a standard antibody panel for every patient and recognises leukaemic cells on the basis of their specific antigenic profile, which differs sufficiently from the profile of the normal haematopoietic elements in the various myeloid cell lineages to allow them to be distinguished even when present at very low levels [21–23].
MCFC also provides a means to detect “leukaemic stem cells” (LSCs). These cells are contained within the CD34+CD38neg cell compartment and are supposed to constitute the stem cells of the leukaemic clone, in analogy to normal stem cells, which are at the origin of normal haematopoiesis [24]. LSCs in AML differ from normal haematopoietic stem cells in their high expression of several antigens, including CD25, CD123 and C-type lectin-like molecule-1 (known as CLL-1) [25]. Their frequency at diagnosis predicts the occurrence of MRD and their presence before and after therapy is associated with relapse (“stem cell MRD”) [22, 24, 25]. The persistence of LSCs after therapy and outgrowth at relapse may explain treatment failure in cytoMRD-negative patients, because routine MRD antibody panels fail to detect LSCs.
Standard prognostic features at diagnosis (i.e. cytogenetics and gene mutation status) predict the occurrence of MRD after therapy, as well as PFS and overall survival. MRD measured post-induction can improve on this risk-assessment, because it reflects physiological mechanisms not evident in disease features present at diagnosis, including multidrug resistance drug-efflux pumps and apoptosis regulation.
The practical implication of this information is that accurate risk allocation in AML has to wait at least until the MRD status of a patient after induction is known [26, 27].
MRD in chronic myeloid leukaemia
The Philadelphia chromosome (Ph), resulting from the balanced translocation between the c-Abl gene on chromosome 9q34 and the B-cell receptor (BCR) gene on chromosome 22, denoted as t(9;22), was the first genetic abnormality to be associated with a human cancer. Current World Health Orgainisation diagnostic criteria for CML require detection of the Ph chromosome or its products, the BCR-Abl fusion mRNA and the BCR-Abl protein. For MRD detection, RT-PCR techniques have been developed, which consist of a double, sequential amplification of the fusion mRNA based on the use of “nested” primers [28]. This technique is extremely sensitive, capable of routine detection of one Ph positive cell in 105 to 106 normal cells. There are many variables in this RT-PCR assay, including which internal standard to use, and how to compare results obtained in different laboratories. International standards have been developed and consensus guidelines have been established by the National Comprehensive Cancer Network and the European LeukemiaNet, which describe in detail the practical aspects of the assay, the time-points during treatment with kinase inhibitors when the assay should be performed, as well as the interpretation of the results obtained [29, 30]. Importantly, an international scale has been developed for the measurement of BCR-Abl transcripts, in which a major molecular response is defined as a BCR-Abl transcript level of 0.1% or less (this represents a 3-log reduction from a standardised baseline) and a complete molecular response as a BCR-Abl transcript level that is undetectable by quantitative RT-PCR in an assay with adequate sensitivity (e.g., 4.5-logs) [31]. Several studies have focused on the quantification of BCR-Abl transcripts after the start of a treatment with tyrosine kinase inhibitors. A major or complete molecular response at 3, 6, 8, 12 or 18 months after the start of treatment constitutes in each case an important milestone for predicting a favourable long-term outcome, including PFS and overall survival [32, 33].
Interestingly, BCR-Abl fusion transcripts can also be detected at a very low level (one cell in 108 to 109) in haematopoietic cells from some normal individuals [34]; this defines the limits of useful sensitivity of RT-PCR for MRD detection in CML.
MRD in lymphoid neoplasms
The three most widely used clonal markers in patients with lymphoid neoplasms are the t(14;18), the t(11;14) and the IgH gene rearrangement. The t(14;18) was the first lesion routinely employed for PCR-based MRD analysis in lymphoid neoplasms and it is detectable in approximately 60%–80% of follicular lymphoma patients and in 20% of diffuse large cell lymphomas.
Together with positron emission tomography (PET) scan imaging techniques, it constitutes a powerful post-treatment outcome predictor in follicular lymphoma. Patients with PCR-negative bone marrow after chemotherapy, after autologous or allogeneic BM transplantation have a superior outcome compared with those with persistently detectable residual lymphoma cells [35]. As in follicular lymphoma, some CLL patients who achieve a complete remission after chemotherapy or a transplant still have a detectable malignant clone, and, as in follicular lymphoma, the outcome of patients is worse if such a MRD is detected [36, 37]. Several studies have compared MCFC and PCR techniques [36, 38] and showed similar sensitivities of 1 in 104 for both techniques, and a major international effort has led to the establishment of guidelines for MRD detection by flow cytometry [39].
Recently, a method based on NGS of a pool of amplimers generated by the use of degenerate consensus primers to amplify all IgH genes in a mixture of polyclonal lymphoid cells was described [1]. This technique was shown to be extremely sensitive (detection limit 1 in 106) and to predict relapses in patients after a reduced-intensity allotransplant with high accuracy [40]. It remains to be seen whether in the future this approach can be implemented for large multicentre clinical trials.
MRD in multiple myeloma
Disease evolution in multiple myeloma can be followed by measuring either paraprotein levels or serum free light chains. Additionally, bone marrow plasma cells can be quantified in order to assess response if a patient lacks measurable paraprotein levels in serum or urine, or has unmeasurable levels of serum free light chains.
A comparison of CR detection by negative immunofixation (iCR), normal serum free light chain ratio (sCR) and undetectable myeloma cells by MCFC (cytoCR) in 102 patients with multiple myeloma treated with novel agents (bortezomib and lenalidomide) showed that 43% patients achieved iCR, 30% achieved sCR and 30% achieved cytoCR. There was no significant survival difference between patients with sCR versus iCR; importantly, however, patients in cytoCR showed significantly increased PFS and time to progression compared with those in sCR or iCR, suggesting increased sensitivity of MCFC to detect MRD [41]. Although a head-to-head comparison between cytoCR assessed by MCFC and molecular CR measured by ASO-PCR showed that ASO-PCR was slightly more sensitive and specific than cytometry, it was applicable in a lower proportion of multiple myeloma patients (75% vs 90%, respectively) and was more time-consuming than cytometry [42].
In a study of 241 patients with multiple myeloma who had attained a morphological complete response following high-dose chemotherapy and autologous haematopoietic cell transplantation, the presence of MRD detected by MCFC was associated with a greater chance of early relapse [43].
Rawstron et al assessed MRD using MCFC in a large cohort of 378 uniformly treated patients following induction therapy and at 100 days after autologous stem cell transplantation, as well as after induction therapy in a nontransplantation group [44]. This study demonstrated the applicability and clinical utility of assessing MRD. It confirmed the feasibility of carrying out MRD analysis in a majority of patients enrolled in a multicentre study and demonstrated clinical utility of this assay, given that patients with MRD had an inferior outcome compared with those without detectable MRD. Importantly, this study also showed that maintenance therapy provided benefit to those patients who were MRD negative; moreover, maintenance therapy could convert patients to MRD negative status, which correlated with improved outcome.
Concluding remarks
MRD-positive patients are going to relapse sooner or later. From this abrupt statement, one can logically conceive MRD negativity as the ultimate goal of any treatment, but should it be reached at all cost and is it reachable at all, in every single case? Although the quality of antitumour drugs is continuously improving, aggressive first-line therapies (such as highly intensive chemotherapy or pretransplant whole body irradiation) have potentially lethal side effects, the most demoralising of which is secondary malignancy. Treatment must be adapted individually, if not personalised. Thus, achieving MRD-negative status using the most sensitive, cost-effective and practical technique available should become a goal for future strategies in the treatment of haematological malignancies, since achievement of MRD negativity is even more important than the regimen used, according to Varghese [45].
However, several still unresolved questions remain: (1.) some patients, despite having achieved a MRD-negative status unfortunately relapse, suggesting that the currently achieved sensitivities are for some disease entities still not sufficient; (2.) the biologically relevant MRD levels may vary for the different types of leukaemia or lymphoma, and thus thresholds have to be defined separately for each disease. This means numerous time-consuming and costly clinical studies.
Whereas in Switzerland PCR- and RT-PCR-based methods are routinely used and standardised according to international guidelines (i.e. BCR-Abl), MCFC tests for MRD are not performed on a routine basis in most laboratories and no standardisation has yet been reached. The cost of comprehensive studies is obviously a deterrent for many small laboratories; therefore, local consortiums should be created to pool efforts and resources in order to build local expertise, not to mention that experience gained in MRD detection will also improve the way diagnosis is established in the first place. The Swiss Cytometry Society and the Swiss Flow Cytometry School*, which has recently been founded at the Geneva University Hospital, could be some of the partners actively helping to establish common protocols and standardized assays in Switzerland.
References
1 Faham M, Zheng J, Moorhead M, Carlton VE, Stow P, Coustan-Smith E, et al. Deep-sequencing approach for minimal residual disease detection in acute lymphoblastic leukemia. Blood. 2012;120(26):5173–80. PubMed PMID: 23074282. Pubmed Central PMCID: 3537310.
2 Leichtle AB, Dufour JF, Fiedler GM. Potentials and pitfalls of clinical peptidomics and metabolomics. Swiss Med Wkly. 2013;143:w13801. PubMed PMID: 23771768.
3 Puchades-Carrasco L, Lecumberri R, Martinez-Lopez J, Lahuerta JJ, Mateos MV, Prosper F, et al. Multiple myeloma patients have a specific serum metabolomic profile that changes after achieving complete remission. Clin Cancer Res. 2013;19(17):4770–9. PubMed PMID: 23873687.
4 Sehn LH. Optimal use of prognostic factors in non-Hodgkin lymphoma. Hematology Am Soc Hematol Educ Program. 2006:295–302. PubMed PMID: 17124075.
5 Campana D. Should minimal residual disease monitoring in acute lymphoblastic leukemia be standard of care? Curr Hematol Malig Rep. 2012;7(2):170–7. PubMed PMID: 22373809.
6 Andersen NS, Pedersen LB, Laurell A, Elonen E, Kolstad A, Boesen AM, et al. Pre-emptive treatment with rituximab of molecular relapse after autologous stem cell transplantation in mantle cell lymphoma. J Clin Oncol. 2009;27(26):4365–70. PubMed PMID: 19652064.
7 Platzbecker U, Wermke M, Radke J, Oelschlaegel U, Seltmann F, Kiani A, et al. Azacitidine for treatment of imminent relapse in MDS or AML patients after allogeneic HSCT: results of the RELAZA trial. Leukemia. 2012;26(3):381–9. PubMed PMID: 21886171. Pubmed Central PMCID: 3306138.
8 Grimwade D, Jovanovic JV, Hills RK, Nugent EA, Patel Y, Flora R, et al. Prospective minimal residual disease monitoring to predict relapse of acute promyelocytic leukemia and to direct pre-emptive arsenic trioxide therapy. J Clin Oncol. 2009;27(22):3650–8. PubMed PMID: 19506161.
9 Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150(2):264–78. PubMed PMID: 22817890. Pubmed Central PMCID: 3407563.
10 Walter MJ, Shen D, Ding L, Shao J, Koboldt DC, Chen K, et al. Clonal architecture of secondary acute myeloid leukemia. N Engl J Med. 2012;366(12):1090–8. PubMed PMID: 22417201. Pubmed Central PMCID: 3320218.
11 Walker BA, Wardell CP, Melchor L, Brioli A, Johnson DC, Kaiser MF, et al. Intraclonal heterogeneity is a critical early event in the development of myeloma and precedes the development of clinical symptoms. Leukemia. 2013 Jul 2. PubMed PMID: 23817176.
12 Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer. 2012;12(5):335–48. PubMed PMID: 22495321.
13 Coustan-Smith E, Sancho J, Hancock ML, Razzouk BI, Ribeiro RC, Rivera GK, et al. Use of peripheral blood instead of bone marrow to monitor residual disease in children with acute lymphoblastic leukemia. Blood. 2002;100(7):2399–402. PubMed PMID: 12239148.
14 Thorn I, Forestier E, Botling J, Thuresson B, Wasslavik C, Bjorklund E, et al. Minimal residual disease assessment in childhood acute lymphoblastic leukaemia: a Swedish multi-centre study comparing real-time polymerase chain reaction and multicolour flow cytometry. Br J Haematol. 2011;152(6):743–53. PubMed PMID: 21250970.
15 Knechtli CJ, Goulden NJ, Hancock JP, Grandage VL, Harris EL, Garland RJ, et al. Minimal residual disease status before allogeneic bone marrow transplantation is an important determinant of successful outcome for children and adolescents with acute lymphoblastic leukemia. Blood. 1998;92(11):4072–9. PubMed PMID: 9834212.
16 Knechtli CJ, Goulden NJ, Hancock JP, Harris EL, Garland RJ, Jones CG, et al. Minimal residual disease status as a predictor of relapse after allogeneic bone marrow transplantation for children with acute lymphoblastic leukaemia. Br J Haematol. 1998;102(3):860–71. PubMed PMID: 9722317.
17 Uzunel M, Mattsson J, Jaksch M, Remberger M, Ringden O. The significance of graft-versus-host disease and pretransplantation minimal residual disease status to outcome after allogeneic stem cell transplantation in patients with acute lymphoblastic leukemia. Blood. 2001;98(6):1982–4. PubMed PMID: 11535539.
18 Elorza I, Palacio C, Dapena JL, Gallur L, Sanchez de Toledo J, Diaz de Heredia C. Relationship between minimal residual disease measured by multiparametric flow cytometry prior to allogeneic hematopoietic stem cell transplantation and outcome in children with acute lymphoblastic leukemia. Haematologica. 2010;95(6):936-41. PubMed PMID: 20179088. Pubmed Central PMCID: 2878791.
19 Vora A, Frost L, Goodeve A, Wilson G, Ireland RM, Lilleyman J, et al. Late relapsing childhood lymphoblastic leukemia. Blood. 1998;92(7):2334–7. PubMed PMID: 9746771.
20 Bassan R, Spinelli O, Oldani E, Intermesoli T, Tosi M, Peruta B, et al. Improved risk classification for risk-specific therapy based on the molecular study of minimal residual disease (MRD) in adult acute lymphoblastic leukemia (ALL). Blood. 2009;113(18):4153–62. PubMed PMID: 19141862.
21 Freeman SD, Jovanovic JV, Grimwade D. Development of minimal residual disease-directed therapy in acute myeloid leukemia. Semin Oncol. 2008;35(4):388–400. PubMed PMID: 18692689.
22 Ossenkoppele GJ, van de Loosdrecht AA, Schuurhuis GJ. Review of the relevance of aberrant antigen expression by flow cytometry in myeloid neoplasms. Br J Haematol. 201100;153(4):421–36. PubMed PMID: 21385170.
23 Loken MR, Alonzo TA, Pardo L, Gerbing RB, Raimondi SC, Hirsch BA, et al. Residual disease detected by multidimensional flow cytometry signifies high relapse risk in patients with de novo acute myeloid leukemia: a report from Children’s Oncology Group. Blood. 2012;120(8):1581–8. PubMed PMID: 22649108. Pubmed Central PMCID: 3429302.
24 Gerber JM, Smith BD, Ngwang B, Zhang H, Vala MS, Morsberger L, et al. A clinically relevant population of leukemic CD34(+)CD38(-) cells in acute myeloid leukemia. Blood. 2012;119(15):3571–7. PubMed PMID: 22262762. Pubmed Central PMCID: 3325044.
25 Will B, Steidl U. Multi-parameter fluorescence-activated cell sorting and analysis of stem and progenitor cells in myeloid malignancies. Best Pract Res Clin Haematol. 2010;23(3):391–401. PubMed PMID: 21112038. Pubmed Central PMCID: 3052971.
26 Buccisano F, Maurillo L, Del Principe MI, Del Poeta G, Sconocchia G, Lo-Coco F, et al. Prognostic and therapeutic implications of minimal residual disease detection in acute myeloid leukemia. Blood. 2012;119(2):332–41. PubMed PMID: 22039260.
27 San Miguel JF, Vidriales MB, Lopez-Berges C, Diaz-Mediavilla J, Gutierrez N, Canizo C, et al. Early immunophenotypical evaluation of minimal residual disease in acute myeloid leukemia identifies different patient risk groups and may contribute to postinduction treatment stratification. Blood. 2001;98(6):1746–51. PubMed PMID: 11535507.
28 Wasserman LM. A nested RT-PCR assay to detect BCR/abl. Methods Mol Med. 2004;97:181–9. PubMed PMID: 15064493.
29 Baccarani M, Cortes J, Pane F, Niederwieser D, Saglio G, Apperley J, et al. Chronic myeloid leukemia: an update of concepts and management recommendations of European LeukemiaNet. J Clin Oncol. 2009;27(35):6041–51. PubMed PMID: 19884523.
30 Cortes J, Goldman JM, Hughes T. Current issues in chronic myeloid leukemia: monitoring, resistance, and functional cure. J Natl Compr Canc Netw. 2012;10(Suppl 3):S1–S13. PubMed PMID: 23055247.
31 Hughes T, Deininger M, Hochhaus A, Branford S, Radich J, Kaeda J, et al. Monitoring CML patients responding to treatment with tyrosine kinase inhibitors: review and recommendations for harmonizing current methodology for detecting BCR-ABL transcripts and kinase domain mutations and for expressing results. Blood. 2006;108(1):28–37. PubMed PMID: 16522812. Pubmed Central PMCID: 1895821.
32 Hughes TP, Hochhaus A, Branford S, Muller MC, Kaeda JS, Foroni L, et al. Long-term prognostic significance of early molecular response to imatinib in newly diagnosed chronic myeloid leukemia: an analysis from the International Randomized Study of Interferon and STI571 (IRIS). Blood. 2010;116(19):3758–65. PubMed PMID: 20679528. Pubmed Central PMCID: 3266053.
33 Marin D, Ibrahim AR, Lucas C, Gerrard G, Wang L, Szydlo RM, et al. Assessment of BCR-ABL1 transcript levels at 3 months is the only requirement for predicting outcome for patients with chronic myeloid leukemia treated with tyrosine kinase inhibitors. J Clin Oncol. 2012;30(3):232–8. PubMed PMID: 22067393.
34 Bose S, Deininger M, Gora-Tybor J, Goldman JM, Melo JV. The presence of typical and atypical BCR-ABL fusion genes in leukocytes of normal individuals: biologic significance and implications for the assessment of minimal residual disease. Blood. 1998;92(9):3362–7. PubMed PMID: 9787174.
35 Lobetti-Bodoni C, Mantoan B, Monitillo L, Genuardi E, Drandi D, Barbero D, et al. Clinical implications and prognostic role of minimal residual disease detection in follicular lymphoma. Ther Adv Hematol. 2013;4(3):189–98. PubMed PMID: 23730496. Pubmed Central PMCID: 3666448.
36 Moreno C, Villamor N, Colomer D, Esteve J, Gine E, Muntanola A, et al. Clinical significance of minimal residual disease, as assessed by different techniques, after stem cell transplantation for chronic lymphocytic leukemia. Blood. 2006;107(11):4563–9. PubMed PMID: 16449533.
37 Moreton P, Kennedy B, Lucas G, Leach M, Rassam SM, Haynes A, et al. Eradication of minimal residual disease in B-cell chronic lymphocytic leukemia after alemtuzumab therapy is associated with prolonged survival. J clin oncol. 2005;23(13):2971–9. PubMed PMID: 15738539.
38 Bottcher S, Stilgenbauer S, Busch R, Bruggemann M, Raff T, Pott C, et al. Standardized MRD flow and ASO IGH RQ-PCR for MRD quantification in CLL patients after rituximab-containing immunochemotherapy: a comparative analysis. Leukemia. 2009;23(11):2007–17. PubMed PMID: 19641522.
39 Rawstron AC, Bottcher S, Letestu R, Villamor N, Fazi C, Kartsios H, et al. Improving efficiency and sensitivity: European Research Initiative in CLL (ERIC) update on the international harmonised approach for flow cytometric residual disease monitoring in CLL. Leukemia. 2013;27(1):142–9. PubMed PMID: 23041722.
40 Logan AC, Zhang B, Narasimhan B, Carlton V, Zheng J, Moorhead M, et al. Minimal residual disease quantification using consensus primers and high-throughput IGH sequencing predicts post-transplant relapse in chronic lymphocytic leukemia. Leukemia. 2013;27(8):1659‒65. PubMed PMID: 23419792.
41 Paiva B, Martinez-Lopez J, Vidriales MB, Mateos MV, Montalban MA, Fernandez-Redondo E, et al. Comparison of immunofixation, serum free light chain, and immunophenotyping for response evaluation and prognostication in multiple myeloma. J Clin Oncol. 2011;29(12):1627–33. PubMed PMID: 21402611.
42 Sarasquete ME, Garcia-Sanz R, Gonzalez D, Martinez J, Mateo G, Martinez P, et al. Minimal residual disease monitoring in multiple myeloma: a comparison between allelic-specific oligonucleotide real-time quantitative polymerase chain reaction and flow cytometry. Haematologica. 2005;90(10):1365–72. PubMed PMID: 16219573.
43 Paiva B, Gutierrez NC, Rosinol L, Vidriales MB, Montalban MA, Martinez-Lopez J, et al. High-risk cytogenetics and persistent minimal residual disease by multiparameter flow cytometry predict unsustained complete response after autologous stem cell transplantation in multiple myeloma. Blood. 2012;119(3):687–91. PubMed PMID: 22128143.
44 Rawstron AC, Child JA, de Tute RM, Davies FE, Gregory WM, Bell SE, et al. Minimal Residual Disease Assessed by Multiparameter Flow Cytometry in Multiple Myeloma: Impact on Outcome in the Medical Research Council Myeloma IX Study. J Clinic Oncol. 2013;31(20):2540–7. PubMed PMID: 23733781.
45 Varghese AM, Rawstron AC, Hillmen P. Eradicating minimal residual disease in chronic lymphocytic leukemia: should this be the goal of treatment? Curr Hematol Malig Rep. 2010;5(1):35–44. PubMed PMID: 20425395.