Practical issues in the management of the long QT syndrome: focus on diagnosis and therapy
DOI: https://doi.org/10.4414/smw.2013.13843
Summary
The long QT syndrome (LQTS) is a leading cause of sudden death in the young. It is not as rare as previously assumed, given its established prevalence of 1:2,000 live births. It is characterised by prolongation of the QT interval and by the occurrence of syncope, due to torsades-des-pointes ventricular tachycardia, cardiac arrest and sudden death; these life-threatening cardiac events are usually, but not always, associated with physical or emotional stress. It is a genetic disorder, and knowledge of the genotype impacts significantly on management. Extremely effective therapies are available, which makes the existence of undiagnosed affected and symptomatic patients inexcusable. Indeed, mortality for properly treated patients has now declined to around 1% over a 10-year period.
This review, aimed at the clinical cardiologist, discusses briefly the essential genetic information and focuses primarily on the main issues of diagnosis and therapy. One special point of interest is in the impact of genetics on clinical management and the potential medicolegal consequences of not pursuing genetic screening in the proband and hence in the family members.
Introduction
The types of patient that nowadays come to be seen by cardiologists and internists are progressively changing. It is no longer exceptional to see a patient either suspected to be affected by the long QT syndrome (LQTS) or found at a precompetitive sport examination to have a prolonged QT interval. With an established prevalence of 1 in 2,000 live births [1], LQTS can no longer be regarded as a rare disease and physicians are no longer justified for not knowing how to diagnose it and manage it properly.
LQTS, a genetic disorder, is one of the leading causes of sudden cardiac death below the age of 20 and the availability of very effective therapies makes inexcusable the existence of symptomatic and undiagnosed patients. There are several features that make LQTS a disease easy to suspect under the proper circumstances. Typically, patients present with a prolongation of the QT interval on the ECG and often report episodes of syncope or cardiac arrest, especially under conditions of physical or emotional stress.
This review, which capitalises on a personal experience with LQTS that dates back to 1971 [2, 3] and on a life largely spent studying this intriguing disease, will touch briefly on the main genetic data and will focus primarily on diagnosis and therapy. For the readers who might also be interested in specific aspects of LQTS, such as more detailed genetics, modifier genes and additional issues two very recent reviews may offer adequate information [4, 5].
Essential genetics
Sixteen genes have so far been identified as either responsible for or associated with LQTS [4]. The three main genes, KCNQ1 (LQT1), KCNH2(LQT2) and SCN5A(LQT3), account for approximately 75% of clinically definite cases of LQTS, whereas the minor genes contribute an additional 5% collectively. An estimated 20% of LQTS remains genetically elusive.
KCNQ1encodes the α-subunit of the K+ channel Kv7.1 which generates IKs and, being physiologically increased by sympathetic activation, is essential for QT adaptation when heart rate increases. When IKs is defective, the QT interval fails to shorten appropriately during tachycardia, thus creating a highly arrhythmogenic condition. Heterozygous KCNQ1mutations cause the dominant Romano-Ward LQT1 syndrome and account for the majority of disease-causing variants. Homozygous mutations in KCNQ1, or compound heterozygous mutations, cause the recessive Jervell and Lange-Nielsen variant, characterised by deafness because of the reduced IKs in the inner ear [6].
The second most common gene harbouring LQTS mutations is KCNH2, which encodes the α-subunit of the K+ channel conducting the IKr current. The rapid IKr (KCNH2) and the slow IKs (KCNQ1) are two independent components of the delayed rectifier IK current, the major determinant of phase three of the cardiac action potential. Mutations in KCNH2cause a reduction in IKr current.
The third major LQTS gene is SCN5A, which encodes the α-subunit of the cardiac sodium channel conducting the depolarising sodium inward current. SCN5Amutations produce the LQTS phenotype by increasing the delayed Na+ inward current and, therefore, prolonging the transmembrane portion of the K+ channel. In December 1995, a few months after this gene discovery, we reported the first evidence that the Na+ channel blocker mexiletine reduces the late Na+ current [7]. This finding paved the way to the search for gene-specific therapies.
Given the large and growing number of genetic variants identified so far, to distinguish pathogenic mutations from rare variants is critically important. The probability for a missense mutation to be pathogenic appears to depend largely on its location. In general, genetic variants located in the pore and transmembrane regions are much more likely to be pathogenic. Whenever a functional study of the specific mutation has been performed, the results may help in assessing its clinical relevance. When these data are missing, as is often the case, it is important to establish whether within the family the mutation cosegregates with either symptoms or QT prolongation. An important take-home message is that the laboratory finding of an amino acid substitution should not be automatically taken as an indication of a disease-causing mutation.
An example of how advanced genetic methodology can help in solving complex cases and in identifying new genes comes from an extremely recent publication [8]. We performed exome sequencing in two unrelated infants with very early occurrence of recurrent cardiac arrests due to ventricular fibrillation, major QT prolongation and T wave alternans, and who were genotype-negative. By comparing the deoxyribonucleic acid (DNA) of the patients and of their parents we identified de-novo mutations in CALM1 and in CALM2. This prompted candidate-gene screening in a cohort of 82 LQTS genotype-negative patients. Two more patients were carriers of either CALM1 or CALM2 mutations. Significantly, all four patients share strikingly similar clinical manifestations: major QT prolongation (all >600 ms), T wave alternans, onset of cardiac arrest in infancy, multiple episodes of ventricular fibrillation terminated by an implantable cardioverter defibrillator (ICD) and mostly triggered by sympathetic activation, and poor response to any therapeutic attempt. CALM1 and CALM2 are two of the three human genes encoding calmodulin, a ubiquitous multifunctional Ca2+ binding protein essential for many intracellular signalling processes that transduces Ca2+ signals to influence activity of ion channels, kinases and other target proteins regulating physiological functions in heart and brain [9]. Overexpression of calmodulin mutants with defective Ca2+binding produces major prolongation of ventricular action potentials [10], thus representing a plausible mechanistic link with QT interval prolongation and arrhythmia risk.
The cases reported above are a good example of how a clinical observation of a very severe form of LQTS, not explained by the current knowledge, has prompted the successful use of advanced genetic methodologies leading to the identification of a novel and important disease-gene for LQTS, thus opening the way for better understanding of the underlying mechanisms. Should any reader have knowledge of similar cases, my laboratory will gladly perform advanced genetic testing at no cost.
The success rate of genotyping has greatly increased. In patients with a clinically definite diagnosis of LQTS the success rate of my cardiovascular genetics laboratory is 80–85%. A very similar rate is currently observed by the Mayo Clinic group (Rochester, MN, USA), headed by Dr Ackerman.
Diagnosis
Typical cases present no diagnostic difficulty for physicians aware of the disease, as the association between stress-induced syncope and QT interval prolongation is too striking to be missed. Special attention has to be paid to specific aspect of the electrocardiogram (ECG) besides the sheer prolongation of the QT interval, such as presence of notches on the T waves which is typical for LQT2 [11] (fig. 1A) and the occasional appearance of T wave alternans [3] (fig. 1B). There are rather specific T-wave patterns associated with the three main genotypes (figs 1A and 2), but exceptions do exist and are not infrequent. Accordingly, the ECG cannot be used to assume the presence of a given genotype; at best, it may provide a “reasonable suspicion”, which needs to be confirmed or dismissed by actual genetic screening.
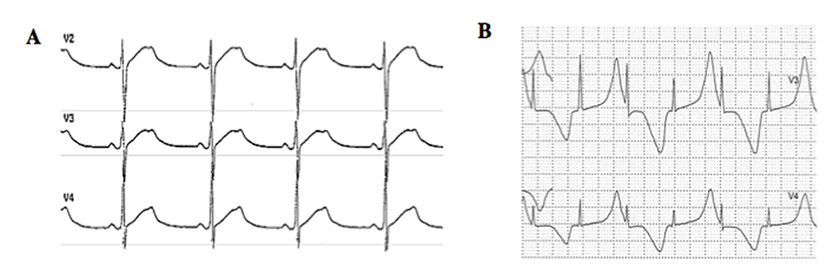
Figure 1
(A) Example of notched T waves from a 37-year old man affected by long QT syndrome (LQTS) who suffered two syncopal episodes. (B) Example of T wave alternans from a 2-year old LQTS patient with multiple episodes of cardiac arrest. Both tracings are from a 24-hour Holter recording. (Reproduced from reference [39].)
However, borderline cases are more complex and require the evaluation of multiple variables besides clinical history and ECG. The diagnosis of LQTS is facilitated by the use of a diagnostic score, initially published in 1993 [12] and subsequently updated [13], which has become known as the “Schwartz score” (table 1). This score tabulates the elements of the personal history, family history and ECG QTc values to qualitatively frame an index of suspicion for LQTS, and is helpful for the assessment of the index case and of family members. From the standpoint of actionability, a diagnostic score ≥3.5 points makes the diagnosis very likely. This score is also dynamic. As an example, serial ECG evaluations in the patient and in the patient’s first-degree relatives can impact on the probability of LQTS: the presence of a family member already diagnosed as affected by LQTS adds 0.5 points, which moves the total points to the threshold of 3.5 points in the case of an index case with 3 points provided by a QTc ≥480 ms.
The importance of a correct diagnosis has assumed a new dimension in the molecular era. A new responsibility for the clinician lies in the identification of the most logical candidates for molecular screening, and relates to the availability and cost of genetic testing. The study by Taggart et al. [14] is relevant. In a group of 176 consecutive patients diagnosed as affected by LQTS and sent to the Mayo Clinic for management and genetic testing, 41% were regarded as unaffected, 32% as probably affected and only 27% as definite cases of LQTS. Genetic testing confirmed the clinical assessment because disease-causing mutations were found in none of the unaffected, in 34% of the probably affected and in 78% of the definitely affected. It follows that an exceedingly large number of patients incorrectly received the clinical diagnosis of LQTS from their own cardiologists.
It is indeed in the selection of patients with a suspicion of LQTS that the Schwartz score becomes especially useful. As the score gives importance to the degree of QT prolongation, it should be obvious that it cannot help in the identification of silent mutation carriers. The smart approach consists of the use of the Schwartz score for the selection of those patients who should undergo molecular screening (everyone with a score ≥3.0) and of the use of cascade screening [15, 16] for the identification of all affected family members, including the silent mutation carriers. Indeed, it is important to know that, whereas the search for the disease-causing mutation in the proband is expensive and time consuming, once it has been identified screening all the family members for the presence or absence of the same mutation is inexpensive and very rapid.
The effectiveness of cascade screening for the early identification of affected family members also carries medicolegal implications, as we have recently discussed [5]. Cascade screening can be performed only after positive genotyping of the proband. It follows that the physician who does not attempt to genotype the proband clinically affected by LQTS has wilfully decided to ignore the possibility that some of his or her family members are carriers of the disease and thereby exposed to the risk of life-threatening arrhythmias. Similarly, when the disease-causing mutation is identified in the proband, the physician who does not propose initiating cascade screening has also wilfully decided to leave the affected family members – approximately 50% of first-degree relatives – uninformed about their status and unprotected.
This is an extremely important aspect because there are still cardiologists who believe that once they have convincingly diagnosed a patient as affected by LQTS there is no need to perform genetic screening. The impact of genetic screening on clinical cardiology is exemplified by the fact that not performing cascade screening could lead to a number of otherwise avoidable deaths among those genotype positive / phenotype negative family members of affected patients. Cascade screening forcefully demonstrates that molecular biology and genetics can no longer be regarded as tools for researchers and that they represent an essential component of good medical care. One additional and related issue concerns the importance of performing a molecular autopsy in all cases of unexplained sudden death in the young. In almost 30% of these cases a channelopathy may be identified [17] and cascade screening may start. There is a widespread agreement on this approach [18, 19].
Genetic testing also has practical aspects that need to be considered. The time necessary to screen a proband is often long, not uncommonly 4 to 6–7 months, partly because approximately 5% of patients are found to carry two independent mutations [20, 21] and are thereby at higher risk for cardiac events, which mandates genetic screening of at least the five more common genes, not stopping with the identification of the first disease-causing mutation. Once the culprit mutation in the proband is identified everything changes, and cascade screening among family members to verify who carries the same mutation takes approximately 7–10 days.
The cost of genetic testing can also be a problem for the unfortunate LQTS patients who live in countries without a fully functional and protective national health service. The cost may vary from country to country. In Italy, and specifically in the Lombardy region where I live and work, patients pay a ticket of 66 euros and the hospital receives a reimbursement of 1200 euros for the proband and of 515 euros for each family member. My personal policy has always been that if I am contacted for foreign patients who cannot afford genetic testing I do my best to have it done with my research funds, but this is a personal position.
|
Table 1: 1993–2012 long QT syndrome (LQTS) diagnostic criteria (From reference [13]). |
| |
Points
|
|
Electrocardiographic Findings#
|
|
| A |
QTc ^ |
≥ 480 ms |
3 |
| 460–479 ms |
2 |
| 450–459 (male) ms |
1 |
| B |
QTc ^ 4th minute of recovery from exercise stress test ≥480 ms |
1 |
| C |
Torsade de pointes * |
2 |
| D |
T wave alternans |
1 |
| E |
Notched T wave in three leads |
1 |
| F |
Low heart rate for age @
|
0.5 |
|
Clinical history
|
|
| A |
Syncope * |
With stress |
2 |
| Without stress |
1 |
| B |
Congenital deafness |
0.5 |
|
Family history
|
|
| A |
Family members with definite LQTS $
|
1 |
| B |
Unexplained sudden cardiac death below age 30 among immediate family members $
|
0.5 |
|
# In the absence of medications or disorders known to affect these electrocardiographic features
^ QTc calculated using Bazett's formula where QTc = QT/√RR
* Mutually exclusive
@ Resting heart rate below the 2nd percentile for age
$ The same family member cannot be counted in A and B |
| Score
≤1 point:
low probability of LQTS
1.5 to 3 points:
intermediate probability of LQTS
≥3.5 points
high probability |
Score |
≤1 point: |
low probability of LQTS |
|
1.5 to 3 points: |
intermediate probability of LQTS |
|
≥3.5 points |
high probability |
| Score |
≤1 point: |
low probability of LQTS |
| |
1.5 to 3 points: |
intermediate probability of LQTS |
| |
≥3.5 points |
high probability |
|
Table 2:M-FACT * Risk score (From reference [40]). |
| |
–1 point
|
0 points
|
1 point
|
2 points
|
| Event free on therapy for >10 years |
Yes |
|
|
|
| QTc (ms) |
|
≤500 |
> 500 – ≤ 550 |
>550 |
| Prior aborted cardiac arrest |
|
No |
Yes |
|
| Events on therapy |
|
No |
Yes |
|
| Age at implant |
|
>20 yrs |
≤20 yrs |
|
| *Acronym derived from M (Minus 1 point for being free of cardiac events while on therapy for >10 years); F (“Five Hundred” and “Five hundred and Fifty ms QTc); A (Age ≤20 years atimplant); C (Cardiac arrest); T (events on Therapy). |
Clinical management and therapy
The key elements of management of LQTS patients are β-adrenergic blocking agents, left cardiac sympathetic denervation (LCSD), the ICD, and common sense. They are complemented currently by gene-specific approaches, both pharmacological and behavioural.
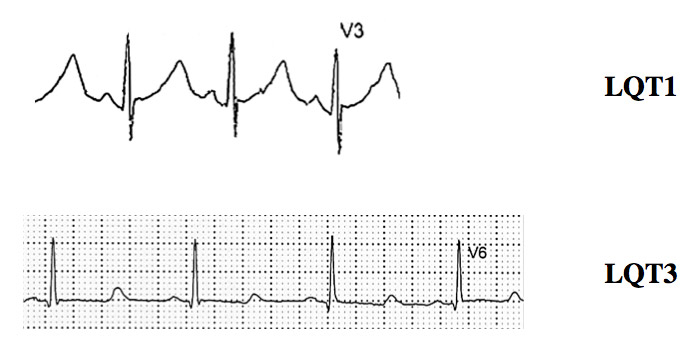
Figure 2
ECG tracings of LQT1 and LQT3 patients.
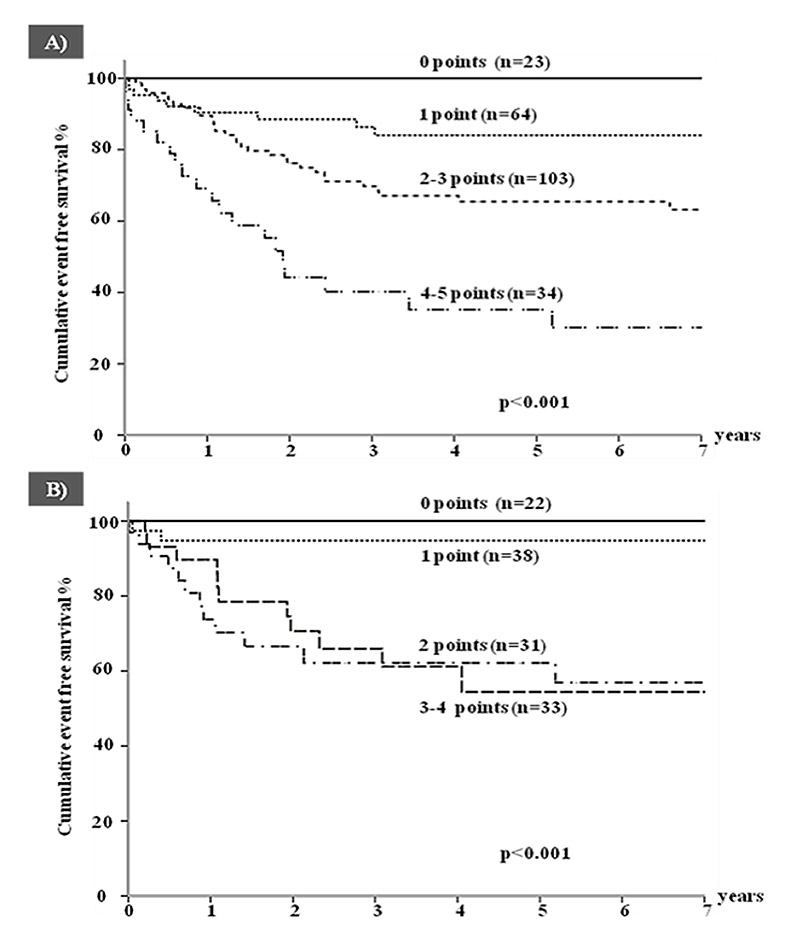
Figure 3
Cumulative event-free survival for a first appropriate implantable cardioverter defibrillator shock according to increasing risk score (M-FACT) in (A) all patients and (B) in patients with no prior aborted cardiac arrest. (Reproduced from reference [40]).
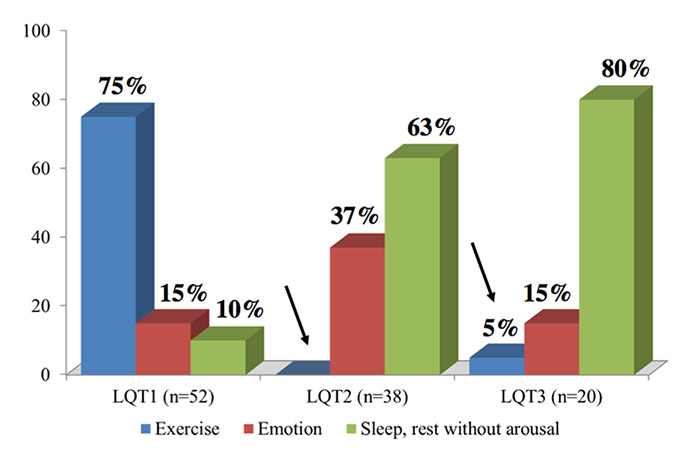
Figure 4
Triggers for lethal cardiac events in LQT1, LQT2 and LQT3 patients. The arrows indicate the rare occurrence of these events during sympathetic activation in patients without mutations affecting the IKs current. (Modified from reference [27].)
Βeta-blockers
Beta-adrenergic blocking agents represent the first-choice therapy in symptomatic LQTS patients. They seldom produce excessive bradycardia, especially if the dosage is gradually increased over several weeks. Contrary to commonly held views, β-blockers are not all equally effective. The most effective are propranolol and nadolol. Propranolol remains the most widely used drug, at 2 to 3 mg/kg/day; sometimes, the dosage is increased further. Nadolol is also used often because its longer half-life allows twice-a-day administration, usually at 1 to 1.5 mg/kg/day, and for this reason is preferred for teenagers. Unfortunately, as a result of limited commercial interest, nadolol is no longer available, or is available only with difficulty, in many countries. Metoprolol is definitely less effective [22], and a switch from propranolol or nadolol to metoprolol has been associated with tragic recurrences. Metoprolol should not be used in the management of symptomatic LQTS. Also, atenolol seems somewhat less effective, but the data available are limited [23]. No data exist on the other β-blockers.
In a study of 869 LQTS patients of unknown genotype, overall mortality on β-blocker therapy was 2%, and was 1.6% when limited to patients with syncope (no cardiac arrest) and without events in the first year of life [24]. Beta-blockers are extremely effective in LQT1 patients. Data from two large studies [25, 26] indicate that mortality is 0.5% and sudden death combined with cardiac arrest reaches 1%. Beta-Blocker noncompliance and use of QT-prolonging drugs are responsible for almost all life-threatening β-blocker failures in LQT1 patients [26]; conversely, compliance and the avoidance of QT-prolonging drugs are associated with 97% reduction in the risk for cardiac events. Compared with LQT1, LQT2 patients have more life-threatening events despite β-blockers, but most of these are resuscitated cardiac arrests (6–7%) [25].
Among LQT3 patients, major events have been reported to occur more frequently (10%–15%) despite β-blockers [25, 27] and have contributed to the incorrect notion that β-blockers are of limited or no value for LQT3 patients. This misconception is the consequence of including LQT3 patients who present with events in the first year of life with those who present with events at a later time [28]. Indeed, the presence of a cardiac event in the first year of life is associated with an extremely poor prognosis, independent of treatment. In patients presenting at an older age, mortality on β-blocker therapy is approximately 3% and is highest in those with QTc values close to 600 ms. This information comes from the largest study ever performed in LQT3, with data on 400 patients [29].
Other pharmacological interventions
Besides sodium channel blockers (see below) there is no place at this time for other drugs in the management of LQTS,. Digitalis shortens the QT interval and for this reason was initially used in some patients, but it was a complete failure [2]. There are a few reports, mostly from Japan, on potassium channel openers, but the data are too scanty to draw any conclusion. The efficacy of β-blockers, and of the additional therapies discussed below, is such to discourage any pharmacological experimentation in LQTS; the only exceptions are represented by the few desperate cases where all the traditional therapies, at full dosage, have failed.
Left cardiac sympathetic denervation
Left cardiac sympathetic denervation, performed either via an extrapleural approach [30] or by thoracoscopy [31], requires removal of the first four thoracic ganglia (T1–T4). The cephalic portion of the left stellate ganglion should be left intact to avoid Horner's syndrome. Whenever the local surgeons do not have adequate personal experience with either extrapleural or videoscopic LCSD, the traditional and easy approach represented by opening the second left intercostal space allows clear visualisation of the stellate ganglion and the sympathetic chain. The rationale for LCSD, largely based on its rather striking antifibrillatory effect [32], has been reviewed recently [33], and includes a major reduction in noradrenaline release at the ventricular level without post-denervation supersensitivity [34] and with no reduction in heart rate [35].
The largest series of LCSD cases was published in 2004 and included 147 LQTS patients who underwent sympathectomy during the previous 35 years [36]. They represented a group at very high risk (99% symptomatic, with an extremely long mean QTc (563 ± 65 ms), previous cardiac arrest in 48%, and recurrent syncope despite full-dose β-blockers in 75%. During a mean follow-up of 8 years, there was a 91% reduction in cardiac events. In five patients who underwent LCSD owing to multiple ICD shocks and electrical storms, over a 4-year follow-up there was a 95% decrease in the number of shocks with a dramatic improvement in the quality of life of the patients and of their families. LCSD produced a mean QTc shortening of 39 ms, pointing to an action on the substrate as well as on the trigger (i.e. reduction in local noradrenaline release). The major protective effect of LCSD has been fully confirmed in the large series reported by the Mayo Clinic [31, 37]. It should be clear that whenever syncopal episodes recur despite full-dose β-blocker therapy, LCSD should be considered and implemented without hesitation. Also, failure by the attending physician to provide the family with adequate information of the pros and cons of LCSD as compared with ICD implantation may carry medicolegal consequences [38]. Lack of a surgeon familiar with LCSD in a nearby hospital is not a valid reason for not mentioning this important therapeutic option to patients or their families.
Our current indications for denervation therapy include: (1) patients with appropriate ventricular fibrillation terminating ICD shocks; (2) patients with LQTS-triggered breakthrough cardiac events while on adequate drug therapy; (3) patients with failure to tolerate β-blocker therapy because of unacceptable side effects or because of asthma; and (4) high-risk, very young patients where primary drug therapy may not be sufficiently protective and where there are hopes of LCSD serving as a “bridge to an ICD” [31].
The untoward effects of LCSD are very limited and should never deprive a patient of a very effective means of making ventricular fibrillation less likely. The left hand becomes slightly warmer and dryer; very seldom this happens also on the left side of the forehead. The possible side effect that generates largely unwarranted concerns is the possible occurrence of Horner’s syndrome (left ptosis); this happens if the nerves passing through the stellate ganglion and directed to the left eye are cut. As these nerves pass through the upper half of the stellate ganglion, correct surgery leaves intact the upper half of the stellate ganglion. In this way Horner’s syndrome is avoided. In our 40-year experience with LCSD in hundreds of patients, Horner’s syndrome has occurred in 1.5%. The possibility that, in an individual patient, these nerves pass at a lower level within the stellate ganglion can never be excluded, and in this case Horner’s syndrome will occur. We always inform the patients about this possibility with this percentage. Finally, during surgery the stellate ganglion may be compressed and a modest lowering of the left eyelid may occur; it is almost always a transient phenomenon lasting a few weeks. We can never exclude the possibility that the left eyelid may remain 1–2 mm lower but this, while potentially evident on a close-up photograph, is not noticeable in normal social life.
Implantable cardioverter defibrillator
Clinical cardiologists find the decision to implant an ICD relatively easy. In the case of appropriate shocks, the cardiologist will have saved the life of the patient; in the case of no shocks and the possibility of complications, the cardiologist will have done the best for the patient’s protection. Conversely, the decision to not implant an ICD could, in the case of a tragic outcome, lead to serious medicolegal consequences for the physician if such a decision was not supported by a valid rationale. Even though these considerations should play no or a minimal role in medical decisions, in the era of “defensive medicine” they actually do.
It is important to analyse the available knowledge before jumping to erroneous conclusions. There is a stunning mismatch in ICD utilisation for patients with LQTS: some programmes in the United States implant an ICD in approximately 80% of their patients with LQTS, whereas among two of the largest LQTS Clinics in the world, my own in Pavia and that of Michael Ackerman at the Mayo Clinic, the ICD utilisation rate is approximately 3% and 15%, respectively [39]. I have to add here that since 2000, among the very large number of patients I follow up with my associates, there has been only one case of sudden death, and this was in a 5-year-old child with cardiac arrest in the first year of life for whom we had strongly recommended ICD implantation but whose parents had refused. My interpretation of this good outcome is that it depends largely on the fact that our patients come to our centre for a full check-up (ECG, 12-lead 24-hour Holter and exercise stress test) every 6–12 months. My very first patient has come every year since 1971. This allows us to adjust therapy as necessary, reassess risk, and realise if there are signs of cardiac electrical instability that require significant changes in the therapeutic strategy.
There is an overall consensus in favour of immediately implanting an ICD in the case of a documented cardiac arrest, either on or off therapy, even though there are some exceptions, such as a clearly drug-induced event in an otherwise asymptomatic patient with modest QT prolongation. By contrast, opinions differ strongly regarding the use of ICDs in patients without cardiac arrest.
The current knowledge is, and should be, based on the largest ICD study published so far, which provided information on 233 LQTS patients [40]. It was disquieting to realise that the majority of implanted patients had not suffered a cardiac arrest and, moreover, that many had not even failed β-blocker therapy. Asymptomatic patients, almost absent among the LQT1 and LQT2 groups with an ICD, represented 45% of LQT3 patients, indicating that the mere presence of a SCN5Amutation, even in an asymptomatic individual, was deemed sufficient for ICD implantation. During a mean follow-up of 4.6 years, at least one appropriate shock was received by 28% of patients and adverse events occurred in 25% of them. Given the practical importance of identifying in advance those patients with the highest and lowest probability of receiving appropriate shocks, which represents the justification for the ICD implant, a score (M-FACT) was developed, based on simple clinical variables available in a doctor’s office during a first visit [40] (table 2, fig. 3). M-FACT includes QTc duration, age at implant and cardiac events despite therapy. Appropriate ICD therapies were predicted by age <20 years at implantation, a QTc above 500 ms, prior cardiac arrest and cardiac events while on drug therapy; within 7 years, appropriate shocks occurred in no patients with none of these factors and in 70% of those with all factors. Similar observations have been drawn from the largest single centre study of ICD use in patients with LQTS [41].
I suggest implantation of an ICD in: (1) all patients who survived a cardiac arrest while compliant on adequate drug therapy; (2) most of those who survived a cardiac arrest except those with a reversible/preventable cause, and possibly some of those with previously undiagnosed and therefore untreated LQT1; (3) those with LQTS-triggered syncope despite a full dose of β-blocker, whenever the option of LCSD is either not available or discarded after discussion with the patient; (4) all patients with syncope despite a full dose of β-blocker and LCSD; (5) exceptionally, asymptomatic postpubertal LQT2 women with a QTc ≥550 ms and asymptomatic patients with a QTc >550 ms who also manifest signs of high electrical instability (e.g. T-wave alternans) or other evidence of being at high risk despite β-blockade and LCSD (e.g. long sinus pauses followed by abnormal T-wave morphologies) [40, 41]. For patients with Jervell and Lange-Nielsen or Timothy syndrome, who appear incompletely protected by antiadrenergic therapies, we recommend to consider on a case-by-case basis, the possibility of triple therapy, namely β-blockers plus LCSD plus ICD.
Gene-specific therapy and management
The amazing unravelling of the genotype-phenotype correlation [42] has made of LQTS the first disease for which the initial steps of gene-specific management have become possible.
LQT1 patients are at higher risk during sympathetic activation, such as during exercise and emotional stress [27] (fig. 4). They should not participate in competitive sports and, probably, they should also avoid intense exercise training in order to avoid the potentiation of potentially dangerous vagal reflexes [43]. Swimming is particularly dangerous for LQT1 patients, as 99% of the arrhythmic episodes associated with swimming occur in this group [22]. LQT2 patients are very sensitive to serum potassium levels, which should not be allowed to fall. When reasonable levels are not maintained by diet or with oral K+ supplements, a combination with K+-sparing agents such as spironolactone should be considered. As these patients are at especially high risk when aroused from sleep or rest by a sudden noise [27], we recommend that telephones and alarm clocks are removed from their bedrooms. Also, when parents have to wake up their children in the morning, they should do it gently and without yelling. Women with LQT2 [44], but not LQT1 [45], are at increased risk during the postpartum period and compliance with LQT2-directed therapy, adequate rest and avoidance of QT prolonging medications is particularly important during this time. If a mother chooses not to breastfeed, we recommend that the male partners of LQT2 mothers take care of night-time feeding during the first 3‑4 months post-partum.
The demonstration that LQT3-causing SCN5Amutations have a “gain-of-function” effect [46] suggested the testing of sodium-channel blockers, particularly mexiletine, as possible adjuvants in the management of LQT3 patients [3]. The effect of mexiletine is mutation-specific [47] and that is why the efficacy of mexiletine should be tested in all LQT3 patients under continuous ECG monitoring by the acute oral drug test technique, using half of the daily dose. Within 90 minutes the peak plasma concentration is reached and if the QTc is shortened by more than 40 ms, without evidence of PR prolongation, QRS widening and eliciting a Brugada ECG pattern, then mexiletine could/should be added to β-blocker therapy. Flecainide cannot be advised for the treatment of LQTS patients. Even though there is no conclusive evidence for a beneficial effect of mexiletine and definite failures have occurred, there is also growing evidence of significant benefit in a number of individual cases. Highly malignant forms manifesting in infancy owing to mutations causing extremely severe electrophysiological dysfunctions and which were corrected by the combination of mexiletine and propranolol have been reported [48]. Despite very limited information [49], there are hopes for the potential benefit of ranolazine, a sodium-channel blocker especially specific to the late sodium current. With respect to β-blocker therapy, given its direct late sodium-current blocking properties, propranolol is probably the LQT3-preferred β-blocker.
Independent of genotype, all LQTS patients should avoid any cardiac or noncardiac drug that blocks the IKr current. A list of such drugs is available at http://www.qtdrugs.org and should be given to every patient because their family physician may not be aware of this potentially lethal, unwanted side effect. This is a precise responsibility of the cardiologist who follows these patients.
References
1 Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, et al. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120:1761–7.
2 Schwartz PJ, Periti M, Malliani A. The long Q-T syndrome. Am Heart J. 1975;89:378–90.
3 Schwartz PJ, Malliani A. Electrical alternation of the T wave. Clinical and experimental evidence of its relationship with the sympathetic nervous system and with the long QT syndrome. Am Heart J. 1975;89:45–50.
4 Schwartz PJ, Crotti L, Insolia R. Long QT syndrome: from genetics to management. Circ Arrhythm Electrophysiol. 2012;5:868–77.
5 Schwartz PJ, Ackerman MJ, George AL Jr, Wilde AM. Impact of genetics on the clinical management of channelopathies. J Am Coll Cardiol. 2013;62:169–80.
6 Schwartz PJ, Spazzolini C, Crotti L, Bathen J, Amlie JP, Timothy K, et al. The Jervell and Lange-Nielsen Syndrome. Natural history, molecular basis, and clinical outcome. Circulation. 2006;113:783–90.
7 Schwartz PJ, Priori SG, Locati EH, Napolitano C, Cantù F, Towbin JA, et al. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene-specific therapy. Circulation. 1995;92:3381–6.
8 Crotti L, Johnson CN, Graf E, De Ferrari GM, Cuneo BF, Ovadia M, et al. Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation. 2013;127:1009–17.
9 Maier LS, Bers DM, Brown JH. Calmodulin and Ca2+/calmodulin kinases in the heart physiology and pathophysiology. Cardiovasc Res. 2007;73:629–30.
10 Alseikhan BA, DeMaria CD, Colecraft HM, Yue DT. Engineered calmodulins reveal the unexpected eminence of Ca2+ channel inactivation in controlling heart excitation. Proc Natl Acad Sci U S A. 2002;99:17185–90.
11 Malfatto G, Beria G, Sala S, Bonazzi O, Schwartz PJ. Quantitative analysis of T wave abnormalities and their prognostic implications in the idiopathic long QT syndrome. J Am Coll Cardiol. 1994;23:296–301.
12 Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome: an update. Circulation. 1993;88:782–4.
13 Schwartz PJ, Crotti L. QTc behavior during exercise and genetic testing for the long-QT syndrome. Circulation. 2011;124:2181–4.
14 Taggart NW, Haglund CM, Tester DJ. Diagnostic miscues in congenital long-QT syndrome. Circulation. 2007;115:2613–20.
15 Schwartz PJ. Cascades or waterfalls, the cataracts of genetic screening are being opened on clinical cardiology. J Am Coll Cardiol. 2010;55:2577–9.
16 Hofman N, Tan HL, Alders M, van Langen IM, Wilde AA. Active cascade screening in primary inherited arrhythmia syndromes: does it lead to prophylactic treatment? J Am Coll Cardiol. 2010;55:2570–6.
17 Tester DJ, Ackerman MJ. Postmortem long QT syndrome genetic testing for sudden unexplained death in the young. J Am Coll Cardiol. 2007;49:240–6.
18 Schwartz PJ, Crotti L. Can a message from the dead save lives? J Am Coll Cardiol. 2007;49:247–9.
19 Michaud K, Fellmann F, Abriel H, Beckmann JS, Mangin P, Elger BS. Molecular autopsy in sudden cardiac death and its implication for families: discussion of the practical, legal and ethical aspects of the multidisciplinary collaboration. Swiss Med Wkly. 2009;139:712–8.
20 Schwartz PJ, Priori SG, Napolitano C. How really rare are rare diseases?: the intriguing case of independent compound mutations in the long QT syndrome. J Cardiovasc Electrophysiol. 2003;14:1120–1.
21 Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC. Compound mutations: a common cause of severe long-QT syndrome. Circulation. 2004;109:1834–41.
22 Chockalingam P, Crotti L, Girardengo G, Johnson JN, Harris KM, van der Heijden JF, et al. Not all beta-blockers are equal in the management of long QT syndrome types 1 and 2: Higher recurrence of events under metoprolol. J Am Coll Cardiol. 2012;60:2092–9.
23 Chatrath R, Bell CM, Porter CJ, Ackerman MJ. β-Blocker therapy failures in probands with genotyped long QT syndrome. Pediatric Cardiol. 2004;25:459–65.
24 Moss AJ, Zareba W, Hall WJ, Schwartz PJ, Crampton RS, Benhorin J, et al. Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation. 2000;101:616–23.
25 Priori SG, Napolitano C, Schwartz PJ, Grillo M, Bloise R, Ronchetti E, et al. Association of long QT syndrome loci and cardiac events among patients treated with beta-blockers. JAMA. 2004;292:1341–4.
26 Vincent GM, Schwartz PJ, Denjoy I, Swan H, Bithell C, Spazzolini C, et al. High efficacy of beta-blockers in long-QT syndrome type 1: contribution of noncompliance and QT-prolonging drugs to the occurrence of beta-blocker treatment “failures”. Circulation. 2009;119:215–21.
27 Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, et al. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89–95.
28 Schwartz PJ, Spazzolini C, Crotti L. All LQT3 patients need an ICD. True or false? Heart Rhythm. 2009;6:113–20.
29 Wilde AA, Kaufman ES, Shimizu W, Moss AJ, Benhorin J, Lopes CM, et al. Sodium channel mutations, risk of cardiac events, and efficacy of beta-blocker therapy in type 3 long QT syndrome. (abstr) Heart Rhythm. 2012;9(May Suppl):S321.
30 Odero A, Bozzani A, De Ferrari GM, Schwartz PJ. Left cardiac sympathetic denervation for the prevention of life-threatening arrhythmias. the surgical supraclavicular approach to cervico-thoracic sympathectomy. Heart Rhythm. 2010;7:1161–5.
31 Collura CA, Johnson JN, Moir C, Ackerman MJ. Left cardiac sympathetic denervation for the treatment of long QT syndrome and catecholaminergic polymorphic ventricular tachycardia using video-assisted thoracic surgery. Heart Rhythm. 2009;6:752–9.
32 Schwartz PJ, Snebold NG, Brown AM. Effects of unilateral cardiac sympathetic denervation on the ventricular fibrillation threshold. Am J Cardiol. 1976;37:1034–40.
33 Schwartz PJ. Cutting nerves and saving lives. Heart Rhythm. 2009;6:760–3.
34 Schwartz PJ, Stone HL. Left stellectomy and denervation supersensitivity in conscious dogs. Am J Cardiol. 1982;49:1185–90.
35 Schwartz PJ, Stone HL. Effects of unilateral stellectomy upon cardiac performance during exercise in dogs. Circ Res. 1979;44:637–45.
36 Schwartz PJ, Priori SG, Cerrone M, Spazzolini C, Odero A, Napolitano C, et al. Left cardiac sympathetic denervation in the management of high-risk patients affected by the long QT syndrome. Circulation. 2004;109:1826–33.
37 Coleman MA, Bos JM, Johnson JN, Owen HJ, Deschamps C, Moir C, et al. Videoscopic left cardiac sympathetic denervation for patients with recurrent ventricular fibrillation/malignant ventricular arrhythmia syndromes besides congenital long QT syndrome. Circ Arrhyth Electrophysiol. 2012;5:782–8.
38 Schwartz PJ. Efficacy of left cardiac sympathetic denervation has an unforeseen side effect: medicolegal complications. Heart Rhythm. 2010;7:1330–2.
39 Schwartz PJ, Ackerman MJ. The long QT syndrome A transatlantic clinical approach to diagnosis and therapy. Eur Heart J. 2013 (In press) doi:10.1093/eurheartj/eht089
40 Schwartz PJ, Spazzolini C, Priori SG, Crotti L, Vicentini A, Landolina M, et al. Who are the long-QT syndrome patients who receive an implantable cardioverter defibrillator and what happens to them? Data from the European long-QT syndrome implantable cardioverter-defibrillator (LQTS ICD) Registry. Circulation. 2010;122:1272–82.
41 Horner JM, Kinoshita M, Webster TL, Haglund CM, Friedman PA, Ackerman MJ. Implantable cardioverter defibrillator therapy for congenital long QT syndrome: A single-center experience. Heart Rhythm. 2010;7:1616–22.
42 Schwartz PJ, Priori SG. Long QT syndrome: genotype-phenotype correlations. In: Cardiac Electrophysiology. From Cell To Bedside. IV Edition (Zipes DP and Jalife J, Eds.) WB Saunders Co., Philadelphia, pp. 651–659, 2004.
43 Crotti L, Spazzolini C, Porretta AP, Dagradi F, Taravelli E, Petracci B, et al. Vagal reflexes following an exercise stress test: a simple clinical tool for gene-specific risk stratification in the long QT syndrome. J Am Coll Cardiol. 2012;60:2515–24.
44 Khositseth A, Tester DJ, Will ML, Bell CM, Ackerman MJ. Identification of a common genetic substrate underlying postpartum cardiac events in congenital long QT syndrome. Heart Rhythm. 2004;1:60–4.
45 Heradien MJ, Goosen A, Crotti L, Durrheim G, Corfield V, Brink PA, et al. Does pregnancy increase cardiac risk for LQT1 patients? J Am Coll Cardiol. 2006;48:1410–5.
46 Bennett PB, Yazawa K, Makita N, George AL Jr. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–5.
47 Ruan Y, Liu N, Bloise R, Napolitano C, Priori SG. Gating properties of SCN5A mutations and the response to mexiletine in long-QT syndrome type 3 patients. Circulation. 2007;116:1137–44.
48 Wang DW, Crotti L, Shimizu W, Pedrazzini M, Cantù F, De Filippo P, et al. Malignant perinatal variant of long QT syndrome caused by a profoundly dysfunctional cardiac sodium channel. Circ Arrhythm Electrophysiol. 2008;1:370–8.
49 Moss AJ, Zareba W, Schwarz KQ, Rosero S, McNitt S, Robinson JL. Ranolazine shortens repolarization in patients with sustained inward sodium current due to type-3 long-QT syndrome. J Cardiovasc Electrophysiol. 2008;19:1289–93.