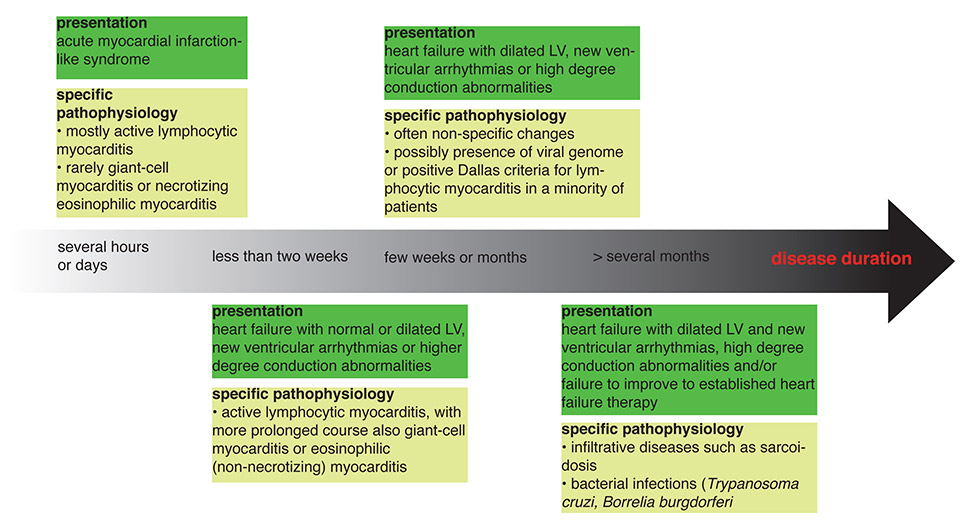
Figure 1
The common clinical presentations of myocarditis and possible specific pathologies. The scheme is adapted from [64].
LV = left ventricle
DOI: https://doi.org/10.4414/smw.2013.13841
Heart failure affects around 1%–2% of the general population in developed countries and prognoses remain poor despite medical advances [1, 2]. Inflammatory dilated cardiomyopathy (iDCM), which most commonly results from infection-triggered myocarditis, accounts for around a tenth of heart failure cases [3]. Epidemiological data further suggest that iDCM is an important cause of heart failure and sudden death in children and young adult patients [4].
Myocarditis usually results from infections with viruses or the protozoan Trypanosoma cruzi (Chagas’ disease), and is often associated with autoimmune responses against heart tissue. In most of these cases, histology reveals a predominantly lymphocytic pattern of infiltrates. In some of the affected patients, myocarditis can evolve into a chronic inflammatory process, which results in dilation of heart chambers, excessive accumulation of fibrillar, mainly type I, collagen, and impaired contractility.
Clinical presentation of myocarditis is highly variable and ranges from an asymptomatic course to fulminant heart failure and sudden cardiac death. The broad clinical picture of myocarditis also includes chest pain and electrocardiogram (ECG) alterations, which cannot be differentiated from the acute coronary syndrome on the basis of symptoms only [5]. Therefore, a diagnosis of myocarditis is usually not straightforward and warrants a variety of examinations. Serum levels of troponins I and T, C-reactive protein, and several cytokines have been suggested as helpful diagnostic biomarkers, but they all lack sensitivity and specificity. Right heart catheterisation and coronary angiography provides haemodynamic data and helps to exclude coronary abnormalities, but is not diagnostic. Echocardiography is critical to assess cardiac function, pericardial effusion and potential valvular abnormalities during the initial diagnostic work-up, but is also not diagnostic for myocarditis. Cardiovascular magnetic resonance imaging (CMR) allows visualisation of even smaller lesions in the myocardium, but its specificity for inflammation is still under investigation. So far, endomyocardial biopsies still represent the diagnostic gold standard despite their low sensitivity. Heart biopsies are usually analysed for the presence of eosinophils, giant cells, macrophages, granulocytes or lymphocytes. Myocarditis was originally defined using the Dallas criteria of an “inflammatory infiltrate of the myocardium with necrosis and/or degeneration of adjacent myocytes not typical of ischaemic damage associated with coronary heart disease” [6]. Of note, the sensitivity and prognostic relevance of myocardial biopsies can be enhanced with immunohistochemistry. Detection of >14 CD3+ T cells or CD68+ macrophages per high power field by means of immunohistochemistry provides a more sensitive and prognosticly relevant diagnostic criterion for inflammatory cardiomyopathy than the meanwhile almost historical Dallas criteria [7]. Additionally, increased intracardiac expression of human leucocyte antigens (e.g. HLA-DR) or adhesion molecules (e.g. intracellular adhesion molecules) have been suggested to reflect inflammatory activity of myocarditis.
Inflammatory dilated cardiomyopathy results from myocarditis. As mentioned above, there is a wide variation in the presentation of myocarditis, which can range from fulminant heart failure to very subtle signs of nonspecific inflammation. Importantly, and as we will discuss later, the extent of initial cardiac inflammation does not at all imply that acute myocarditis will really progress to iDCM and end stage heart failure.
Myocarditis can develop as a result of infectious and noninfectious triggers (table 1). Viruses are the most important causes of myocarditis in the western world and usually give rise to a pattern of lymphocytic infiltration in the myocardium. Enteroviruses, especially Coxsackievirus B, but also hepatitis C virus, cytomegalovirus and adenoviruses were commonly reported in biopsies in the past [8–10]. In more recent studies from Germany, human herpesvirus 6 and parvovirus B-19 were predominant [11, 12]. Whether this represents a geographical observation that might be due to methodological differences or a true change in virus predominance is still controversial [13]. Recent reports associate myocarditis also with H1N1 influenza [14]. In addition bacteria such as Borrelia burgdorferi (Lyme disease) or parasites such as Trypanosoma cruzi (Chages’ disease) are important causative organisms in certain regions of the world.
Noninfectious causes of myocarditis include drug-induced hypersensitivity syndromes that might be triggered by numerous drugs such as diuretics (hydrochlorothiazides, furosemide), antibiotics (ampicillin, tetracycline, azithromycin), neuroreactive drugs (phenytoin, tricyclic antidepressants, benzodiazepines) [15–17] and tumour necrosis factor (TNF) antagonists [18]. Also local trauma caused by irradiation or after cardiotomy can lead to myocarditis. Several autoimmune disorders such as Churg-Strauss syndrome [19, 20], or systemic lupus erythematosus [21] can also lead to an inflammatory response within the myocardium.
Giant cell myocarditis represents a rare and distinct disease entity with a specific histological pattern and poor prognosis. Its pathogenesis, however, is not clear yet, and there is so far no evidence for a causative infectious agent. Autoimmune mechanisms are most likely involved, given the fact that giant cell myocarditis often affects people with a history of autoimmune disorders.
| Table 1:Myocarditis – causative agents. | |
| Infectious: | |
| Viral | Cocksackievirus B3 Parvovirus B-19 Human herpesvirus 6 Cytomegalovirus Hepatitis C virus Adenovirus Influenza virus Ebstein-Barr virus |
| Bacterial | Borrelia burgdorferi Corynebacterium diphteriae Rheumatic fever (group A streptococci) |
| Parasitic | Trypanosoma Babesia |
| Noninfectious: | |
| Toxic | Radiation Alcohol Specific drug toxicity (e.g. doxorubicin, cyclophosphamide) |
| Hypersensitivity | Numerous drugs ( e.g. penicillins, sulphonamides, tricyclic antidepressants) vaccines |
| Autoimmune disorder | Rheumatoid arthritis, Churg-Strauss syndrome, Kawasaki disease, (dermato)myositis, systemic lupus erythematosus, scleroderma |
| Cause not specified: | |
| Giant cell myocarditis Necrotising eosinophilic myocarditis | |
There is no clear consensus on the meanings of the terms “myocarditis” and “inflammatory dilated cardiomyopathy (iDCM)”, with some authors speaking of iDCM also in the phase of acute cardiac inflammation. To us, iDCM refers to a functionally impaired and morphologically altered heart, with histological/immunohistochemical evidence of both inflammation and tissue remodelling (e.g. Masson’s trichrom staining for fibrosis or CD3+/CD45+ staining for inflammation). It is difficult to assess the absolute risk of developing iDCM after myocarditis, mainly because there is no noninvasive gold standard for early and sensitive diagnosis of myocarditis. Therefore, a substantial number of subclinical cases of myocarditis might go undiagnosed, eventually leading to an underestimation of the absolute risk for iDCM development.
Several observational trials aimed at identifying clinical factors that could predict the development of iDCM after a diagnosis of viral myocarditis. Goldberg et al. reported in a series of 109 biopsy-proven cases of active myocarditis that left bundle-branch block (relative risk [RR] 2.9), impaired left ventricular function of <40% (RR 2.9) and syncope (RR 8.5) were associated with a fatal course or need for transplantation [22]. Pulmonary hypertension is another risk factor in iDCM. Elevated pulmonary pressures are in general associated with adverse outcomes in cardiomyopathies [23], but this is even more important in iDCM [24]. More recently, elevated blood levels of interferon-β were reported to be predictive of better clearance of the myocardial virus load and improved survival on follow-up [25].
So far, we still cannot adequately explain why myocarditis progresses to iDCM in some patients while others recover completely.
The induction of major histocompatibility and intercellular adhesion molecules on cardiac myocytes indicate an autoimmune inflammatory response in patients with myocarditis or inflammatory dilated cardiomyopathy [26]. HLA class II genes are highly polymorphic, but nevertheless the majority of autoimmune diseases, including myocarditis with dilated cardiomyopathy, are linked to a limited set of class II‐DR or ‐DQ alleles. This may partially explain the increased risk of cardiovascular disease in patients with chronic inflammatory conditions [27]. A strong initial inflammatory reaction seems to be beneficial once the patient survives the acute phase. In fact, McCarthy et al. demonstrated in 147 patients with biopsy-proven myocarditis that those presenting with fulminant myocarditis had a significantly higher survival at 11-year follow-up than those with acute or borderline myocarditis [28]. The importance of the initial inflammatory response was also evident from the Myocarditis Treatment Trial: patients with stronger cellular (higher white cell counts and levels of natural killer cells and macrophages) and humoral (higher levels of cardiac immunoglobulin G [IgG], general IgG or antiskeletal-muscle IgG) immune responses developed less severe disease [29]. This beneficial effect of a strong inflammatory response might be due to a better clearance of the viral genome from the heart. In a study of Kühl et al., involving 172 patients with viral myocarditis that were followed for 7 months, those patients with successful clearance of the virus as evidenced by myocardial biopsy had significantly improved left ventricular function on follow up [12].
Autoimmune mechanisms play an important role in myocarditis and its progression to iDCM. Caforio et al. demonstrated that autoreactive antibodies against cardiac α-myosin are frequently present in patients with myocarditis [30]. Furthermore, they showed that the presence of antimyosin antibodies correlated with worsening of left ventricular function in patients with biopsy-proven myocarditis [31]. Besides antimyosin antibodies, several other autoantibodies against cardiac proteins, such as antibodies against β1-adrenoreceptors and cardiac laminin, were reported to be highly expressed in iDCM [32, 33].
It has been recently reported that heart-specific human and mouse T cells recognising cardiac myosin are not eliminated during negative selection in the thymus [34]. Activation of self-antigen specific T cells, however, does not necessarily lead to a pathogenic response. Development of autoimmune-mediated inflammation mostly depends on the cytokine profile produced by autoreactive T cells. So far, three major subsets of CD4+ T helper cells, Th1, Th2 and Th17 cells, were reported to be involved in autoimmune processes in animal models and in humans [35–39]. However the ultimate Th subset or cytokine profile mediating myocarditis remains elusive. Whereas effector Th cells promote inflammation, another subset of regulatory T cells serves to control it. Depletion of regulatory T cells results in multiorgan inflammation including fatal autoimmune myocarditis [40]. Furthermore, activation of autoreactive Th cells requires presentation of the self-antigen by antigen-presenting cells such as dendritic cells. In animal models, activation of pattern-recognition receptors on dendritic cells is critical for development of autoimmune myocarditis [41, 42].
Genetic factors including major histocompatibility complex genes have been shown to be involved in determining susceptibility or tolerance to autoimmunity. The human histocompatibility leucocyte antigen HLA-D region and its products, the HLA-DR antigens, are essential for the generation of specific T-cell mediated autoimmune responses. They control the mixed lymphocyte reaction (MLR), the graft versus host reaction in vivo and cell-cell interactions involved in immune responses. HLA-DR expression on T cells is a marker of T-cell activation [43]. It has been demonstrated that HLA-DR expressing T cells were markedly increased in the peripheral blood of patients with dilated cardiomyopathy [43]. HLA class II molecules including HLA-DR present antigenic peptides to CD4+ T cells, which is a prerequisite for T-cell activation. Ueno et al. concluded that HLA-DR expressed on the surface of activated T cells promotes immunoreactions that promote the proliferation of immune cells or cytokine production in iDCM patients [44]. Furthermore, there is evidence that several human HLA expression patterns predispose to the development of dilated cardiomyopathy and/or inflammatory cardiomyopathy [45, 46].
A major problem in viral myocarditis and iDCM seems to be the inability of certain patients to successfully eradicate viruses from the myocardium [12]. Interferon-β treatment in entero- or adeno-virus positive patients with longstanding symptoms of iDCM resulted in clearance of the virus in 22 of 22 patients and significant improvement in heart function after 6 months [47]. Long-term follow up at 120 months demonstrated increased survival with interferon-β treatment as compared with patients that failed to clear the virus [25]. Interestingly, high endogenous levels of interferon-β correlated with higher spontaneous virus clearance and improved outcome [25].
Another approach aims at autoimmune responses involved in iDCM development. This approach is based on the observation that, in some patients, inflammation persists even after clearance of the virus. However, in the Myocarditis Treatment Trial, immunosuppression with ciclosporin or azathioprine was not superior to supportive therapy in patients with acute myocarditis [29]. This is different in subsets of iDCM patients: Wojnicz et al. treated patients with iDCM for more than 6 months and increased immunohistochemical activation markers in cardiac biopsies in a placebo-controlled, nonblinded study with steroids plus azathioprine [48]. Although immunosuppressive therapy failed to improve death, transplantation or hospital readmission rates, it improved both left ventricular function and New York Heart Association (NYHA) class. Frustaci et al. selected, in a randomised, double-blinded study, iDCM patients without evidence of virus persistence in heart biopsies for immunosuppression, and showed marked functional improvement [49]. Intravenous immunoglobulin (IVIG) was also considered as a potential therapeutic candidate because of its immunomodulatory effects. Unfortunately, robust data are lacking, with only one larger randomised and appropriately blinded trial investigating the effect of IVIG in a cohort of 67 patients with recent-onset iDCM [50]. In this trial, left ventricular function increased in all patients over the study period of 1 year, with no additional benefit with IVIG. This is in contrast to a series of case reports that speak of dramatic improvements in some cases. In a more recent study, 17 patients with chronic iDCM and high myocardial parvovirus B-19 virusload were treated with IVIG, which improved left ventricular function and functional capacity within 6 months, but this study lacks both randomisation and a control group. We believe that more data from high-powered studies is needed in order to recommend routine use of IVIG in patients with myocarditis or iDCM. However, the use of IVIG might be reasonable in children [51].
The importance of heart-specific autoantibodies for the development and maintenance of myocarditis and iDCM [52, 53] prompted the evaluation of IgG adsorption via plasmapharesis as a therapeutic tool. In a study of 34 patients with iDCM and autoantibodies against β-adrenoreceptors, plasmapharesis of IgG resulted in improved functional capacity and left ventricular function. A later study further demonstrated that the beneficial effect is mostly due to reduction of a subgroup of IgG, IgG3 [54].
All these studies, therefore, demonstrate possible therapeutic options for two groups of patients: those with failure to clear the virus from the myocardium, who might benefit from antiviral treatments; and those with persistent inflammation due to autoimmune mechanisms, who might profit from immunosuppressive therapies. To date, unfortunately, none of the above-described treatments is widely established in clinical practice and therefore remain to be administered in the setting of clinical studies.
Treatment concepts for iDCM mainly follow the general therapy for systolic heart failure as described in the current guidelines of the European Society of Cardiology (ESC; available online for free at http://www.escardio.org [55]). Mainstays of medical therapy are angiotensin converting enzyme inhibitors, β-blockers, aldosterone antagonists and diuretics. Digoxin should be used with caution in the iDCM population as animal models point towards a possible adverse effect in myocarditis [56]. Indications for implantation of intracardiac defibrillators and/or cardiac resynchronisation therapy (CRT) devices follow the same guidelines as for the general heart failure population. Nevertheless, patients with active myocarditis are particularly prone to unexpected changes in lead threshold and/or sensing owing to the ongoing inflammatory remodelling processes. Severe cases of myocarditis with acute deterioration or deterioration despite optimal medical therapy qualify for implantation of a temporary external ventricular assist device, which can stabilise the patient and bridge the time to transplantation or recovery [57].
Every patient with a diagnosis of active myocarditis should avoid competitive physical activity for at least 6 months, even if full recovery occurs before this [58]. This is especially important in patients with heart failure, or if an iDCM patient develops fever or laboratory evidence of systemic inflammation of any cause. Patients with myocarditis can develop potentially fatal arrhythmias, but they often resolve spontaneously after the acute phase and therapy is therefore often supportive. Continuous ECG-monitoring of hospitalised patients with acute myocarditis allows potentially life-threatening arrhythmias such as heart block or severe sinus bradycardia to be detected early. Owing to the high rate of spontaneous remission of arrhythmia after resolution of the acute inflammatory phase, devices such as permanent pacemakers or cardioverters/defibrillators are usually only recommended if myocarditis progresses, that is, iDCM develops or arrhythmias do not resolve. The current guidelines on ventricular arrhythmias and sudden death feature a detailed section on arrhythmia in myocarditis [59].
The clinical manifestations of myocarditis can vary to a great extent. Acute myocarditis can remain subclinical and manifest only as transient ECG abnormalities or increases in troponin following a viral illness or vaccination. As an example, 4 out of 501 study participants (0.8%) showed increased troponin levels following vaccination for smallpox; however, the reported incidence of symptomatic myocarditis following smallpox vaccination is much lower at only 5.5 cases per 10,000 vaccinations [60].
Figure 1
The common clinical presentations of myocarditis and possible specific pathologies. The scheme is adapted from [64].
LV = left ventricle
On the other end of the spectrum, patients with the acute myocardial infarction-like syndrome present with chest pain suggestive of acute coronary syndrome, but no evidence of coronary stenosis on angiography. The majority of these patients were found to have evidence of myocarditis [61, 62]. The differentiation from acute ischaemia can be difficult in these cases as inflammatory patches in the myocardium during myocarditis can result in wall-motion abnormalities on echocardiography like those seen during ischaemia.
If symptomatic, acute myocarditis often presents with the symptoms of a new-onset heart failure such as dyspnoea, exercise intolerance and fatigue. Echocardiography may show a normal or dilated left ventricle with left ventricular function ranging from low-normal to severely impaired, eventually requiring mechanical circulatory support. Heart failure in acute myocarditis often improves with standard heart failure therapy, and failure to do so should prompt further investigations such as endomyocardial biopsy (EMB; see next section).
Chronic disease can manifest with slow deterioration of heart function over several months or persistent deterioration of heart function despite established medical heart failure therapy. Figure 1 outlines the time course of myocarditis and underlying pathologies.
As outlined above, the main problem for clinicians in diagnosing myocarditis is the high variability of symptoms. It is therefore important to be aware of clues in the history of a patient that should prompt further investigations. In general, every new onset and otherwise unexplained cardiac abnormality, including clinical signs of heart failure, arrhythmias and conduction abnormalities, is suspicious. This is especially true in young patients, as well as in patients with a history of recent upper respiratory tract infection or enteritis. New-onset skin abnormalities such as exanthema and rashes may point to acute viral childhood infections such as parvovirus B-19 or drug reactions (e.g. following vaccination, change in medication, etc.).
We recommend a two-step approach to the patient with suspected myocarditis: initial workup should include a thorough clinical examination with an emphasis on signs of heart failure and abnormal cardiac bruits or murmurs, as well as signs of associated diseases such as exanthema, organomegaly or lymphadenopathy. Electrocardiography is indicated to quantify specific or nonspecific repolarisation abnormalities, arrhythmias or conduction blocks that might prompt immediate intervention. Cardiac enzymes, especially troponins, are elevated in most cases of relevant myocarditis. However, the real sensitivity in the setting of myocarditis is unknown. Cardiac enzymes also lack specificity for myocarditis as they can be elevated in cases of pericarditis.
Echocardiography is used to check left ventricular function and search for other causes of heart failure such as regional wall-motion abnormalities suggestive of coronary artery disease or valvular disease. We appreciate algorhithms, which allow us to exclude ischaemia with reasonable probability, but these methods are still not broadly available, validated and reliable. Coronary angiography should therefore be performed at a low level of suspicion of ischaemia.
The second step includes more specific tests for myocarditis: endomyocardial biopsy (EMB) and cardiac magnetic resonance imaging (CMR). Unfortunately, the sensitivity of EMB is in general fairly low, even with five to ten samples per intervention, and the method carries a certain risk for complications (around 6%, of which 2.7% are due to sheath insertion and 3.3% due to the biopsy procedure [64]). However, EMB remains the sole intervention that can with certainty confirm the general diagnosis of myocarditis and the specific subset of the disease. It is important that biopsies be taken from the left and right ventricles (septal region of the right and left ventricles plus the free wall of the left ventricle) to increase sensitivity. EMB is generally recommended in patients with haemodynamically relevant new onset of heart failure of less than 2 weeks duration, as well as in patients with new onset of heart failure for 2 weeks to 3 months duration that presents with a dilated left ventricle, arrhythmias and conduction blocks of type Mobitz II and above. In addition, EMB should be performed in patients with new onset of heart failure who do not improve within 2 weeks following initiation of medical heart failure therapy. Other scenarios that might prompt EMB include, for example, longer standing heart failure of more than 3 months. However, the evidence for EMB in these settings often lacks controlled studies and is based mostly on expert consensus. An excellent overview of the possible clinical scenarios that might benefit from EMB was published by Cooper and colleagues [64]. In real life, the most important factors limiting the usefulness of EMB are the hands of the interventional cardiologist and the examining pathologist, who both need to have profound experience of investigating myocarditis. These conditions are mostly met in big centres attached to the respective registries. We suggest that the indication for EMB should in general be discussed with a specialist cardiologist familiar with the method.
CMR is another valuable tool in the diagnosis of suspected myocarditis. The advantages of CMR are its noninvasive nature and the good correlation with the diagnosis of myocarditis using EMB [65]. Furthermore, late gadolinium enhancement in CMR was demonstrated to be a strong predictor for mortality following myocarditis [66]. The importance of CMR is also reflected in the 2012 ESC guidelines for the diagnosis of heart failure, which recommend both CMR and EMB for the diagnosis of myocarditis [67]. However, one needs to understand that, so far, CMR cannot visualise the inflammation itself, only indirect signs such as oedema during the acute phase and fibrosis in the chronic phase. There are ongoing efforts to visualise the inflammatory infiltrate, but this is still used in animals and under experimental conditions only [68].
In clinical practice, the use of CMR might be limited by availability, patients carrying noncompatible implanted devices and the general lack of information derived from tissue biopsies, such as the presence of lymphocyte subsets or virus load.
The molecular mechanisms of the development of inflammatory heart diseases are poorly understood. Insight from animal models is required to better characterise the pathophysiology of inflammatory cardiomyopathy and to develop tools to specifically modulate disease progression. Mouse models are very useful in generating and testing hypotheses, as they mirror many mechanistic aspects of human diseases, mainly because of the possibility to over express or knock out specific target genes. In contrast, the ability to study disease in humans is limited as a result of ethical and technical concerns.
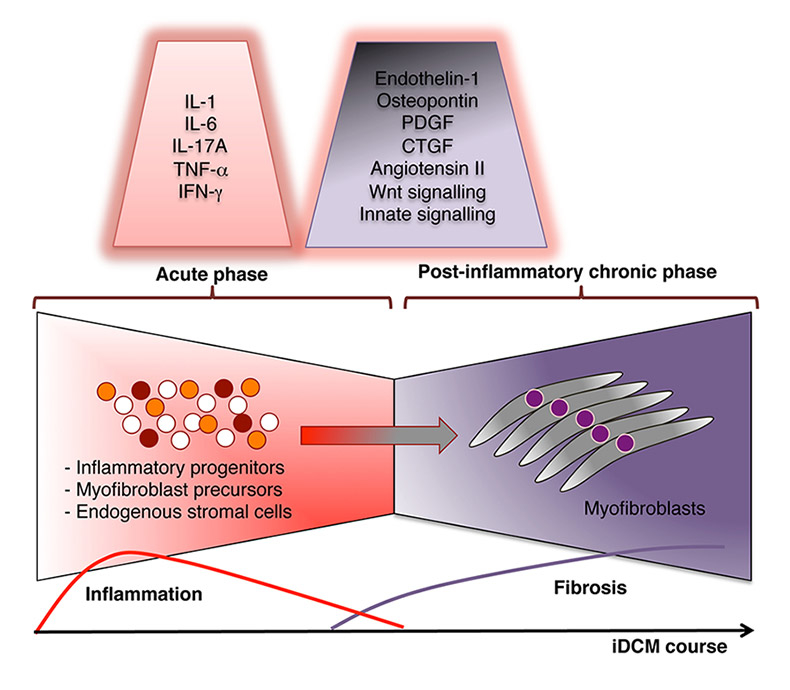
Figure 2
Factors mediating myocardial fibrosis formation in inflammatory dilated cardiomyopathy (iDCM).
Active heart inflammation (myocarditis) is characterised by the massive infiltration of mononuclear cells, including monocytes/macrophages, granulocytes, T cells, dendritic cells, mast cells and cell progenitors. The inflammation slowly resolves and is replaced by fibrotic tissue. Postinflammatory tissue remodelling is a direct cause of heart failure in iDCM. Signalling orchestra modulates the transition from active myocarditis to iDCM. Tissue remodelling is associated with disruption of the highly organised myocardial extracellular matrix and excessive accumulation of collagen and collagen-producing myofibroblasts that leads to heart dysfunction. Mainly inflammatory progenitors and, to some extent, endogenous stromal cells or circulating myofibroblast precursors represent the cellular sources of myocardial myofibroblasts. The most potent factors mediating this specific transition from active myocarditis into iDCM are listed.
CTGF = connective tissue growth factor; IFN = interferon; IL = interleukin; PDGF = platelet-derived growth factor; TNF = tumour necrosis factor
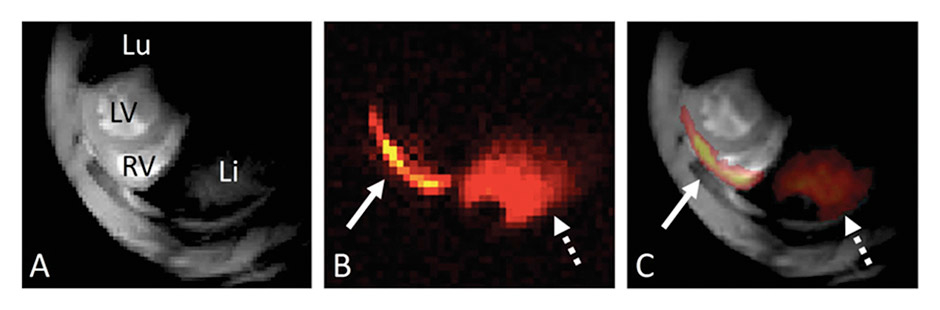
Figure 3
Fluorine-19 magnetic resonance imaging of myocarditis.
(A) 1H magnetic resonance imaging (MRI) slice in the short-axis orientation at the base of the heart in the experimental autoimmune myocarditis mouse model; 1H-MRI depicts the right and left ventricles (RV and LV) as well as the lung (Lu) and liver (Li). (B) 19F-MRI of the same anatomical location. Two regions with a 19F signal can be observed: a thin line at the level of the myocardium (whole arrow) and a larger region at the level of the liver (dotted arrow). (C) Fusion of the 1H and thresholded 19F images: the 19F signal collocalises with the RV free wall and the liver (reproduced from [68]).
Animal models allow the cellular and molecular mechanisms of iDCM to be studied. Understanding of these mechanisms can lead to the development of more efficient and more precise treatment strategies. Animal models of experimental autoimmune myocarditis (EAM) offer an attractive option for the study of the critical process of transition from active inflammation to the end-stage heart failure phenotype in the absence of infectious agents. In the classical EAM model, susceptible rats or mice are either immunised with cardiac self-antigens together with a nonspecific adjuvant or injected with activated dendritic cells loaded with cardiac-specific peptides (nonclassical model) [41]. EAM is mediated by cardiac-specific CD4+ T helper cells that invade the myocardium and induce acute myocarditis. Later, inflammation largely resolves, but the process of pathological remodelling continues and many animals develop myocardial fibrosis, ventricular dilation and heart failure on follow-up.
Cardiac dysfunction correlates with the extension of myocardial fibrosis [69, 70]. Pathological tissue remodelling is associated with disruption of a highly organised myocardial extracellular matrix and excessive accumulation of collagen and collagen-producing myofibroblasts. Degradation of extracellular matrix is under control of the family of zinc-dependent matrix metalloproteinases (MMPs) and their inhibitors – tissue inhibitors of MMPs (TIMPs). Inhibition of MMP activity may therefore be a potential therapeutic target [71]. In inflammatory heart diseases, there are two possible major scenarios for accumulation of pathogenic myofibroblasts. In myocarditis, some inflammatory cells can either transform into pathogenic myofibroblasts, or produce inflammatory mediators that activate and change resident cardiac fibroblasts into myofibroblasts. In EAM, inflammatory progenitor cells are identified as the major cellular source for myofibroblasts [72]. Although activation of resident fibroblasts is believed to represent a major mechanism of fibrogenesis in ischaemic heart failure, there are no direct data available to quantify their contribution to inflammation-triggered myocardial fibrosis.
Data from animal models demonstrate that certain cytokines and growth factors produced by inflammatory cells control or modulate the transition from myocarditis to the iDCM phenotype. Transforming growth factor-β (TGF-β) is a primary profibrotic cytokine that regulates cell growth, apoptosis, differentiation and migration. In particular, TGF-β induces conversion of fibroblasts or progenitor cells into pathological myofibroblasts [72, 73]. Inhibition of TGF-β activity with blocking antibodies prevents development of postinflammatory fibrosis in EAM. Interleukin-1 (IL-1) is another inflammatory cytokine, which stimulates cardiac fibroblast migration, and controls collagen synthesis and MMP and TIMP expression [70]. IL-1 receptor signalling controls fibrogenic pathways not only in EAM [74], but also in healing infarcts in ischaemic heart models [75]. In EAM, IL-17A is one of the major cytokines produced by autoreactive T cells. Recent data demonstrated that IL-17A is not required for myocarditis induction, as initially assumed, but controls disease progression and postinflammatory fibrosis formation by affecting TGF-β and IL-1β production and MMP activity. On the other hand, Fairweather and colleagues suggest that Th2 cytokines such as IL-4 and IL-13 also promote iDCM phenotype development [76]. Osteopontin is a cytokine that specifically regulates myofibroblast differentiation [77]. In EAM, osteopontin is dispensable for myocarditis development [78] and its role in fibrosis remains to be determined. However, insight from virus-triggered myocarditis models clearly associates osteopontin with cardiac fibrosis [79]. Another group of bioactive molecules contributing to the postinflammatory fibrotic processes in the heart are growth factors. In viral model of myocarditis, platelet-derived growth factor, as well as connective tissue growth factor, has also been suggested to mediate cardiac fibrosis [80].
All these results underscore the critical role of several specific inflammatory cytokines and growth factors in myocardial fibrosis, and therefore make the signalling pathways activated by these cytokines potential therapeutic targets. However, before translating experimental findings into clinical application, in-depth understanding of the mechanisms and biological function of the targeted molecule in physiological and in pathophysiological conditions is required. For example, although in animal models TNF-α has been recognised as an attractive target with clear proinflammatory and profibrotic activities, large-scale, randomised clinical trials evaluating TNF-α antagonists for the treatment of dilated cardiomyopathy had to be stopped early because of excessive mortality in the treatment group [81].
The mechanism responsible for the transition from active myocarditis to iDCM is a complicated process that involves several linked signalling pathways. One of the most important pathways requires activation of angiotensin II [82]. Angiotensin II, a crucial regulator of the cardiovascular system, is not only required for physiological functions such as maintaining the blood pressure, but is also involved in the pathophysiolgy of the heart, for example, in hypertension and atherosclerosis [82]. Angiotensin II contributes to fibrogenesis through the production of profibrotic factors and has been implicated in the pathogenesis of diverse cardiovascular diseases. Elevated intracardiac angiotensin II levels have been found in overloaded hearts with fibrosis [83]. Angiotensin II, together with TGF-β, endothelin-1, connective tissue growth factor and platelet-derived growth factor work synergistically, which results in their important contribution to myofibroblast differentiation and fibrosis development.
Moreover, under the pathological stress the heart reactivates several signalling pathways that traditionally were thought to be operational only in the developing heart. On one hand, Wnt signalling controls heart development, but on the other hand, it also plays a pivotal role in adult cardiac remodelling, mainly in the organ fibrogenesis during adult cardiac remodelling following heart injury of any cause. The canonical Wnt pathway is a series of events that occur when Wnt proteins bind to frizzled family receptors, ultimately resulting in translocation of β-catenin from the cytoplasm into the nucleus [84]. Consequently, inhibition of nuclear β-catenin signalling significantly reduced postinfarct mortality and the functional decline of left ventricular function following chronic left anterior descending coronary artery ligation [84]. Natural Wnt inhibitors – soluble frizzled-related proteins (sFRPs) – block Wnt-dependent activation of the canonical Wnt pathway. For example, sFRPs injected into the heart attenuated left ventricular remodelling [85]. Our unpublished observations also point to a critical role of Wnt signalling in postinflammatory remodelling in the heart also.
Innate mechanisms also interfere with tissue fibrosis in inflammatory heart disease. We recently found that Toll-like receptor/MyD88 signalling on inflammatory cells controls fibrosis and iDCM progression in the EAM [74]. Moreover, Fairweather and colleagues [86] showed that in viral (Coxsackievirus B induced) myocarditis, Toll-like receptor-4 deficient mice show reduced levels of myocardial inflammation and of fibrosis development, indicating the important role of innate immunity in iDCM progression.
Taking together, all mediators mentioned above might represent potential targets for therapeutic strategies aiming to reduce or inhibit pathological myocardial fibrosis in iDCM (fig. 2).
Prevention of myofibroblast accumulation in the postinflammatory heart represents a novel potential therapeutical approach. So far, it has been believed that resident cardiac fibroblasts convert into myofibroblasts in response to inflammation and promote tissue fibrosis. Recent findings from the mouse model of EAM point to another concept of fibrogenesis in iDCM. Using the EAM model, we recently demonstrated that infiltrates in the acutely inflamed heart contain a pool of cells that differentiate into myofibroblasts in response to TGF-β signalling. Our data suggest that at least 60% of the myofibroblasts in end-stage remodelled hearts originate from extracardiac inflammatory cells [72]. We identified inflammatory CD133+ progenitor cells as a direct source of myofibroblasts in EAM [72].
Interestingly, CD133+ progenitor cells are not only precursors of myofibroblasts, but rather represent multilineage progenitors, which under the specific conditions can also differentiate into macrophages, dendritic cells or even cardiomyocyte-like cells [87–89]. Thus, prevention of fibrosis can be achieved either by preventing fibrogenic lineage commitment or by inducing nonfibrogenic lineage differentiation of CD133+ progenitor cells. Accordingly, we showed that blocking TGF-β signalling completely prevents myofibroblast formation and fibrosis in EAM. Macrophage colony-stimulating factor (M-CSF) effectively stimulates macrophage lineage from CD133+ progenitor cells [89]. We showed that injection of M-CSF during acute myocarditis results in macrophage commitment of CD133+ cells, which show impaired capacity to form myofibroblasts. This treatment prevents also fibrosis formation in EAM [89]. These results allow us to propose progenitor-targeted approaches as a novel category of potential treatments for iDCM patients in the future.
Underdiagnosis of active myocarditis, mostly due to a mild or even asymptomatic disease course, represents a major limitation for most preventive treatment approaches. In fact, most patients present in the later stages of disease progression, when the typical end-stage heart failure phenotype has already developed. The mechanisms of the development of inflammatory heart diseases development are poorly understood. Insight from animal models is required to better characterise the pathophysiology of inflammatory cardiomyopathy and to develop tools specifically to modulate disease progression.
In animal models, however, it is also difficult to follow clearly disease courses, as cardiac infiltrates or cellular components are routinely determined by postmortem analysis. Convectional approaches to scoring inflammation in animal tissues are limited and do not allow analysis of disease progression in the same animal. In order to gain insight into disease mechanisms in vivo, the development of reliable and reproducible imaging tools for visualising myocardial inflammation and tissue fibrosis are necessary. A noninvasive, quantitative measurement of disease severity would dramatically improve the quality of inflammation-related datasets, and significantly reduce the costs. A novel magnetic resonance-based imaging technique offers such an option. We have just reported the successful use of 19F magnetic resonance imaging in picturing 19F signal in the inflammatory cells recruited into inflamed heart during the peak of the EAM (fig. 3).
1 Redfield MM, Jacobsen SJ, Burnett JC, Jr., Mahoney DW, Bailey KR, Rodeheffer RJ. Burden of systolic and diastolic ventricular dysfunction in the community: appreciating the scope of the heart failure epidemic. JAMA. 2003;289:194–202.
2 Mosterd A, Hoes AW, de Bruyne MC, Deckers JW, Linker DT, Hofman A, et al. Prevalence of heart failure and left ventricular dysfunction in the general population; The Rotterdam Study. Eur Heart J. 1999;20:447–55.
3 Felker GM, Thompson RE, Hare JM, Hruban RH, Clemetson DE, Howard DL, et al. Underlying causes and long-term survival in patients with initially unexplained cardiomyopathy. N Engl J Med. 2000;342:1077–84.
4 Kallwellis-Opara A, Dorner A, Poller WC, Noutsias M, Kuhl U, Schultheiss HP, et al. Autoimmunological features in inflammatory cardiomyopathy. Clin Res Cardiol. 2007;96:469–80.
5 Morgera T, Di Lenarda A, Dreas L, Pinamonti B, Humar F, Bussani R, et al. Electrocardiography of myocarditis revisited: clinical and prognostic significance of electrocardiographic changes. Am Heart J. 1992;124:455–67.
6 Aretz HT, Billingham ME, Edwards WD, Factor SM, Fallon JT, Fenoglio JJ, Jr. et al. Myocarditis. A histopathologic definition and classification. Am J Cardiovasc Pathol. 1987;1:3–14.
7 Sagar S, Liu PP, Cooper LT, Jr. Myocarditis. Lancet. 2012;379:738–47.
8 Matsumori A, Yutani C, Ikeda Y, Kawai S, Sasayama S. Hepatitis C virus from the hearts of patients with myocarditis and cardiomyopathy. Lab Invest. 2000;80:1137–42.
9 Rose NR, Neumann DA, Herskowitz A. Coxsackievirus myocarditis. Adv Intern Med. 1992;37:411–29.
10 Schonian U, Crombach M, Maser S, Maisch B. Cytomegalovirus-associated heart muscle disease. Eur Heart J. 1995;16(Suppl O):46–9.
11 Kindermann I, Kindermann M, Kandolf R, Klingel K, Bultmann B, Muller T, et al. Predictors of outcome in patients with suspected myocarditis. Circulation. 2008;118:639–48.
12 Kuhl U, Pauschinger M, Seeberg B, Lassner D, Noutsias M, Poller W, et al. Viral persistence in the myocardium is associated with progressive cardiac dysfunction. Circulation. 2005;112:1965–70.
13 Yilmaz A, Klingel K, Kandolf R, Sechtem U. A geographical mystery: do cardiotropic viruses respect national borders? Author reply. J Am Coll Cardiol. 2008;52:82;82–83.
14 Ukimura A, Satomi H, Ooi Y, Kanzaki Y. Myocarditis Associated with Influenza A H1N1pdm2009. Influenza Res Treat. 2012;2012:351979.
15 Ben m’rad M, Leclerc-Mercier S, Blanche P, Franck N, Rozenberg F, Fulla Y, et al. Drug-induced hypersensitivity syndrome: clinical and biologic disease patterns in 24 patients. Medicine (Baltimore). 2009;88:131–40.
16 Pursnani A, Yee H, Slater W, Sarswat N. Hypersensitivity myocarditis associated with azithromycin exposure. Ann Intern Med. 2009;150:225–6.
17 Taliercio CP, Olney BA, Lie JT. Myocarditis related to drug hypersensitivity. Mayo Clin Proc. 1985;60:463–8.
18 Slattery E, Ismail N, Sheridan J, Eustace K, Harewood G, Patchett S. Myocarditis associated with infliximab: a case report and review of the literature. Inflamm Bowel Dis. 2011;17:1633–4.
19 Vinit J, Bielefeld P, Muller G, Pfitzenmeyer P, Bonniaud P, Lorcerie B, et al. Heart involvement in Churg-Strauss syndrome: retrospective study in French Burgundy population in past 10 years. Eur J Intern Med. 2010;21:341–6.
20 Rigamonti F, De Benedetti E, Letovanec I, Rosset A, Chizzolini C. Cardiac involvement in Churg-Strauss syndrome mimicking acute coronary syndrome. Swiss Med Wkly. 2012;142:w13543.
21 Wijetunga M, Rockson S. Myocarditis in systemic lupus erythematosus. Am J Med. 2002;113:419–23.
22 Goldberg L. Predictors of adverse outcome in biopsy-proven myocarditis. J Am Coll Cardiol. 1999;33:A850.
23 Abramson SV, Burke JF, Kelly JJ, Jr., Kitchen JG, 3rd, Dougherty MJ, Yih DF, et al. Pulmonary hypertension predicts mortality and morbidity in patients with dilated cardiomyopathy. Ann Intern Med. 1992;116:888–95.
24 Cappola TP, Felker GM, Kao WH, Hare JM, Baughman KL, Kasper EK. Pulmonary hypertension and risk of death in cardiomyopathy: patients with myocarditis are at higher risk. Circulation. 2002;105:1663–8.
25 Kuhl U, Lassner D, von Schlippenbach J, Poller W, Schultheiss HP. Interferon-Beta improves survival in enterovirus-associated cardiomyopathy. J Am Coll Cardiol. 2012;60:1295–6.
26 Wojnicz R, Nowalany-Kozielska E, Wodniecki J, Szczurek-Katanski K, Nozynski J, Zembala M, et al. Immunohistological diagnosis of myocarditis. Potential role of sarcolemmal induction of the MHC and ICAM-1 in the detection of autoimmune mediated myocyte injury. Eur Heart J. 1998;19:1564–72.
27 Mangalam AK, Rajagopalan G, Taneja V, David CS. HLA class II transgenic mice mimic human inflammatory diseases. Adv Immunol. 2008;97:65–147.
28 McCarthy RE, 3rd, Boehmer JP, Hruban RH, Hutchins GM, Kasper EK, Hare JM, et al. Long-term outcome of fulminant myocarditis as compared with acute (nonfulminant) myocarditis. N Engl J Med. 2000;342:690–5.
29 Mason JW, O'Connell JB, Herskowitz A, Rose NR, McManus BM, Billingham ME, et al. A clinical trial of immunosuppressive therapy for myocarditis. The Myocarditis Treatment Trial Investigators. N Engl J Med. 1995;333:269–75.
30 Caforio AL, Goldman JH, Haven AJ, Baig KM, Libera LD, McKenna WJ. Circulating cardiac-specific autoantibodies as markers of autoimmunity in clinical and biopsy-proven myocarditis. The Myocarditis Treatment Trial Investigators. Eur Heart J. 1997;18:270–5.
31 Lauer B, Schannwell M, Kuhl U, Strauer BE, Schultheiss HP. Antimyosin autoantibodies are associated with deterioration of systolic and diastolic left ventricular function in patients with chronic myocarditis. J Am Coll Cardiol. 2000;35:11–8.
32 Muller J, Wallukat G, Dandel M, Bieda H, Brandes K, Spiegelsberger S, et al. Immunoglobulin adsorption in patients with idiopathic dilated cardiomyopathy. Circulation. 2000;101:385–91.
33 Wolff PG, Kuhl U, Schultheiss HP. Laminin distribution and autoantibodies to laminin in dilated cardiomyopathy and myocarditis. Am Heart J. 1989;117:1303–9.
34 Lv H, Havari E, Pinto S, Gottumukkala RV, Cornivelli L, Raddassi K, et al. Impaired thymic tolerance to alpha-myosin directs autoimmunity to the heart in mice and humans. J Clin Invest. 2011;121:1561–73.
35 Rangachari M, Mauermann N, Marty RR, Dirnhofer S, Kurrer MO, Komnenovic V, et al. T-bet negatively regulates autoimmune myocarditis by suppressing local production of interleukin 17. J Exp Med. 2006;203:2009–19.
36 Valaperti A, Marty RR, Kania G, Germano D, Mauermann N, Dirnhofer S, et al. CD11b+ monocytes abrogate Th17 CD4+ T cell-mediated experimental autoimmune myocarditis. J Immunol. 2008;180:2686–95.
37 Nindl V, Maier R, Ratering D, De Giuli R, Zust R, Thiel V, et al. Cooperation of Th1 and Th17 cells determines transition from autoimmune myocarditis to dilated cardiomyopathy. Eur J Immunol. 2012;42:2311–21.
38 Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2012;12:560–7.
39 Luger D, Silver PB, Tang J, Cua D, Chen Z, Iwakura Y, et al. Either a Th17 or a Th1 effector response can drive autoimmunity: conditions of disease induction affect dominant effector category. J Exp Med. 2008;205:799–810.
40 Ono M, Shimizu J, Miyachi Y, Sakaguchi S. Control of autoimmune myocarditis and multiorgan inflammation by glucocorticoid-induced TNF receptor family-related protein(high), Foxp3-expressing CD25+ and CD25- regulatory T cells. J Immunol. 2006;176:4748–56.
41 Eriksson U, Ricci R, Hunziker L, Kurrer MO, Oudit GY, Watts TH, et al. Dendritic cell-induced autoimmune heart failure requires cooperation between adaptive and innate immunity. Nat Med. 2003;9:1484–90.
42 Marty RR, Dirnhofer S, Mauermann N, Schweikert S, Akira S, Hunziker L, et al. MyD88 signaling controls autoimmune myocarditis induction. Circulation. 2006;113:258–65.
43 Ueno A, Murasaki K, Hagiwara N, Kasanuki H. Increases in circulating T lymphocytes expressing HLA-DR and CD40 ligand in patients with dilated cardiomyopathy. Heart Vessels. 2007;22:316–21.
44 Caforio AL, Stewart JT, Bonifacio E, Burke M, Davies MJ, McKenna WJ, et al. Inappropriate major histocompatibility complex expression on cardiac tissue in dilated cardiomyopathy. Relevance for autoimmunity? J Autoimmun. 1990;3:187–200.
45 Portig I, Sandmoeller A, Kreilinger S, Maisch B. HLA-DQB1* polymorphism and associations with dilated cardiomyopathy, inflammatory dilated cardiomyopathy and myocarditis. Autoimmunity. 2009;42:33–40.
46 Taylor JA, Havari E, McInerney MF, Bronson R, Wucherpfennig KW, Lipes MA. A spontaneous model for autoimmune myocarditis using the human MHC molecule HLA-DQ8. J Immunol. 2004;172:2651–8.
47 Kuhl U, Pauschinger M, Schwimmbeck PL, Seeberg B, Lober C, Noutsias M, et al. Interferon-beta treatment eliminates cardiotropic viruses and improves left ventricular function in patients with myocardial persistence of viral genomes and left ventricular dysfunction. Circulation. 2003;107:2793–8.
48 Wojnicz R, Nowalany-Kozielska E, Wojciechowska C, Glanowska G, Wilczewski P, Niklewski T, et al. Randomized, placebo-controlled study for immunosuppressive treatment of inflammatory dilated cardiomyopathy: two-year follow-up results. Circulation. 2001;104:39–45.
49 Frustaci A, Russo MA, Chimenti C. Randomized study on the efficacy of immunosuppressive therapy in patients with virus-negative inflammatory cardiomyopathy: the TIMIC study. Eur Heart J. 2009;30:1995–2002.
50 McNamara DM, Holubkov R, Starling RC, Dec GW, Loh E, Torre-Amione G, et al. Controlled trial of intravenous immune globulin in recent-onset dilated cardiomyopathy. Circulation. 2001;103:2254–9.
51 Drucker NA, Colan SD, Lewis AB, Beiser AS, Wessel DL, Takahashi M, et al. Gamma-globulin treatment of acute myocarditis in the pediatric population. Circulation. 1994;89:252–7.
52 Caforio AL, Mahon NJ, Tona F, McKenna WJ. Circulating cardiac autoantibodies in dilated cardiomyopathy and myocarditis: pathogenetic and clinical significance. Eur J Heart Fail. 2002;4:411–7.
53 Lauer B, Schannwell M, Kuhl U, Strauer BE, Schultheiss HP. Antimyosin autoantibodies are associated with deterioration of systolic and diastolic left ventricular function in patients with chronic myocarditis. J Am Coll Cardiol. 2000;35:11–8.
54 Staudt A, Bohm M, Knebel F, Grosse Y, Bischoff C, Hummel A, et al. Potential role of autoantibodies belonging to the immunoglobulin G-3 subclass in cardiac dysfunction among patients with dilated cardiomyopathy. Circulation. 2002;106:2448–53.
55 McMurray JJ, Adamopoulos S, Anker SD, Auricchio A, Bohm M, Dickstein K, et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2012;33:1787–847.
56 Matsumori A, Igata H, Ono K, Iwasaki A, Miyamoto T, Nishio R, et al. High doses of digitalis increase the myocardial production of proinflammatory cytokines and worsen myocardial injury in viral myocarditis: a possible mechanism of digitalis toxicity. Jpn Circ J. 1999;63:934–40.
57 Chen JM, Spanier TB, Gonzalez JJ, Marelli D, Flannery MA, Tector KA, et al. Improved survival in patients with acute myocarditis using external pulsatile mechanical ventricular assistance. J Heart Lung Transplant. 1999;18:351–7.
58 Cakic S, Eriksson U. Sports and inflammatory heart diseases. Schweizerische Zeitschrift für Sportmedizin und Sporttraumatologie. 2011;59:87–9.
59 Zipes DP, Camm AJ, Borggrefe M, Buxton AE, Chaitman B, Fromer M, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death). J Am Coll Cardiol. 2006;48:e247–346.
60 Morgan J, Roper MH, Sperling L, Schieber RA, Heffelfinger JD, Casey CG, et al. Myocarditis, pericarditis, and dilated cardiomyopathy after smallpox vaccination among civilians in the United States, January–October 2003. Clin Infect Dis. 2008;46(Suppl 3):S242–250.
61 Kuhl U, Pauschinger M, Bock T, Klingel K, Schwimmbeck CP, Seeberg B, et al. Parvovirus B19 infection mimicking acute myocardial infarction. Circulation. 2003;108:945–50.
62 Sarda L, Colin P, Boccara F, Daou D, Lebtahi R, Faraggi M, et al. Myocarditis in patients with clinical presentation of myocardial infarction and normal coronary angiograms. J Am Coll Cardiol. 2001;37:786–92.
63 Cooper LT, Jr. Myocarditis. N Engl J Med. 2009;360:1526–38.
64 Cooper LT, Baughman KL, Feldman AM, Frustaci A, Jessup M, Kuhl U, et al. The role of endomyocardial biopsy in the management of cardiovascular disease: a scientific statement from the American Heart Association, the American College of Cardiology, and the European Society of Cardiology. Endorsed by the Heart Failure Society of America and the Heart Failure Association of the European Society of Cardiology. J Am Coll Cardiol. 2007;50:1914–31.
65 Baccouche H, Mahrholdt H, Meinhardt G, Merher R, Voehringer M, Hill S, et al. Diagnostic synergy of non-invasive cardiovascular magnetic resonance and invasive endomyocardial biopsy in troponin-positive patients without coronary artery disease. Eur Heart J. 2009;30:2869–79.
66 Grun S, Schumm J, Greulich S, Wagner A, Schneider S, Bruder O, et al. Long-term follow-up of biopsy-proven viral myocarditis: predictors of mortality and incomplete recovery. J Am Coll Cardiol. 2012;59:1604–15.
67 McMurray JJ, Adamopoulos S, Anker SD, Auricchio A, Bohm M, Dickstein K, et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2012;33:1787–847.
68 van Heeswijk RB, De Blois J, Kania G, Gonzales C, Blyszczuk P, Stuber M, et al. Selective In-Vivo Visualization of Immune-cell Infiltration in a Mouse Model of Autoimmune Myocarditis by Fluorine-19 Cardiac Magnetic Resonance. Circ Cardiovasc Imaging. 2013.
69 Pohlers D, Brenmoehl J, Loffler I, Muller CK, Leipner C, Schultze-Mosgau S, et al. TGF-beta and fibrosis in different organs – molecular pathway imprints. Biochim Biophys Acta. 2009;1792:746–56.
70 Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther. 2009;123:255–78.
71 Heymans S, Pauschinger M, De Palma A, Kallwellis-Opara A, Rutschow S, Swinnen M, et al. Inhibition of urokinase-type plasminogen activator or matrix metalloproteinases prevents cardiac injury and dysfunction during viral myocarditis. Circulation. 2006;114:565–73.
72 Kania G, Blyszczuk P, Stein S, Valaperti A, Germano D, Dirnhofer S et al. Heart-infiltrating prominin-1+/CD133+ progenitor cells represent the cellular source of transforming growth factor beta-mediated cardiac fibrosis in experimental autoimmune myocarditis. Circ Res. 2009;105:462–70.
73 Phan SH. The myofibroblast in pulmonary fibrosis. Chest. 2002;122:286S–289S.
74 Blyszczuk P, Kania G, Dieterle T, Marty RR, Valaperti A, Berthonneche C, et al. Myeloid differentiation factor-88/interleukin-1 signaling controls cardiac fibrosis and heart failure progression in inflammatory dilated cardiomyopathy. Circ Res. 2009;105:912–20.
75 Bujak M, Dobaczewski M, Chatila K, Mendoza LH, Li N, Reddy A, et al. Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am J Pathol. 2008;173:57–67.
76 Abston ED, Coronado MJ, Bucek A, Bedja D, Shin J, Kim JB, et al. Th2 regulation of viral myocarditis in mice: different roles for TLR3 versus TRIF in progression to chronic disease. Clin Dev Immunol. 2012;2012:129486.
77 Lenga Y, Koh A, Perera AS, McCulloch CA, Sodek J, Zohar R. Osteopontin expression is required for myofibroblast differentiation. Circ Res. 2008;102:319–27.
78 Abel B, Kurrer M, Shamshiev A, Marty RR, Eriksson U, Gunthert U, et al. The osteopontin – CD44 pathway is superfluous for the development of autoimmune myocarditis. Eur J Immunol. 2006;36:494–9.
79 Szalay G, Sauter M, Haberland M, Zuegel U, Steinmeyer A, Kandolf R, et al. Osteopontin: a fibrosis-related marker molecule in cardiac remodeling of enterovirus myocarditis in the susceptible host. Circ Res. 2009;104:851–9.
80 Leipner C, Grun K, Muller A, Buchdunger E, Borsi L, Kosmehl H, et al. Imatinib mesylate attenuates fibrosis in coxsackievirus b3-induced chronic myocarditis. Cardiovasc Res. 2008;79:118–26.
81 Kwon HJ, Cote TR, Cuffe MS, Kramer JM, Braun MM. Case reports of heart failure after therapy with a tumor necrosis factor antagonist. Ann Intern Med. 2003;138:807–11.
82 Kimura K, Eguchi S. Angiotensin II type-1 receptor regulates RhoA and Rho-kinase/ROCK activation via multiple mechanisms. Focus on “Angiotensin II induces RhoA activation through SHP2-dependent dephosphorylation of the RhoGAP p190A in vascular smooth muscle cells”. Am J Physiol Cell Physiol. 2009;297:C1059–1061.
83 Leask A. Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ Res. 2010;106:1675–80.
84 Bergmann MW. WNT signaling in adult cardiac hypertrophy and remodeling: lessons learned from cardiac development. Circ Res. 2010;107:1198–208.
85 He W, Zhang L, Ni A, Zhang Z, Mirotsou M, Mao L, et al. Exogenously administered secreted frizzled related protein 2 (Sfrp2) reduces fibrosis and improves cardiac function in a rat model of myocardial infarction. Proc Natl Acad Sci U S A. 2010;107:21110–5.
86 Fairweather D, Yusung S, Frisancho S, Barrett M, Gatewood S, Steele R, et al. IL-12 receptor beta 1 and Toll-like receptor 4 increase IL-1 beta- and IL-18-associated myocarditis and coxsackievirus replication. J Immunol. 2003;170:4731–7.
87 Kania G, Blyszczuk P, Valaperti A, Dieterle T, Leimenstoll B, Dirnhofer S, et al. Prominin-1+/CD133+ bone marrow-derived heart-resident cells suppress experimental autoimmune myocarditis. Cardiovasc Res. 2008;80:236–45.
88 Blyszczuk P, Behnke S, Luscher TF, Eriksson U, Kania G. GM-CSF promotes inflammatory dendritic cell formation but does not contribute to disease progression in experimental autoimmune myocarditis. Biochim Biophys Acta. 2012.
89 Blyszczuk P, Berthonneche C, Behnke S, Glonkler M, Moch H, Pedrazzini T, et al. Nitric oxide synthase 2 is required for conversion of pro-fibrogenic inflammatory CD133+ progenitors into F4/80+ macrophages in experimental autoimmune myocarditis. Cardiovasc Res. 2013;97:219–29.
Funding / potential competing interests: No financial support and no other potential conflict of interest relevant to this article was reported.