Targeting DNA double-strand break signalling and repair: recent advances in cancer therapy
DOI: https://doi.org/10.4414/smw.2013.13837
Daniela
Hühn, Hella A
Bolck, Alessandro A
Sartori
Abstract
Genomic instability, a hallmark of almost all human cancers, drives both carcinogenesis and resistance to therapeutic interventions. Pivotal to the ability of a cell to maintain genome integrity are mechanisms that signal and repair deoxyribonucleic acid (DNA) double-strand breaks (DSBs), one of the most deleterious lesions induced by ionising radiation and various DNA-damaging chemicals. On the other hand, many current therapeutic regimens that effectively kill cancer cells are based on the induction of excessive DSBs. However, these drugs often lack selectivity for tumour cells, which results in severe side effects for the patients, thus compromising their therapeutic potential. Therefore, the development of novel tumour-specific treatment strategies is required.
Unlike normal cells, however, cancer cells are often characterised by abnormalities in the DNA damage response including defects in cell cycle checkpoints and/or DNA repair, rendering them particularly sensitive to the induction of DSBs. Therefore, new anticancer agents designed to exploit these vulnerabilities are becoming promising drugs for enhancing the specificity and efficacy of future cancer therapies. Here, we summarise the latest preclinical and clinical developments in cancer therapy based on the current knowledge of DSB signalling and repair, with a special focus on the combination of small molecule inhibitors with synthetic lethality approaches.
Introduction
Cancer is the major cause of death in Switzerland among people aged 45–84 years [1]. The latest Swiss cancer statistics indicate that prostate cancer in men and breast cancer in women are the most common types, with 6,000 and 5,500 incidences per year, respectively. Notably, lung cancer is still the leading cause of cancer-related death in the Swiss population, accounting for approximately 3,000 deaths each year [2].
Almost all human cancers are characterised by genomic instability, which is considered to play a key role in the conversion of a normal cell into a premalignant cell [3]. Mechanisms contributing to genomic instability include aberrant repair of deoxyribonucleic acid (DNA) lesions as well as defective signalling to cell-cycle checkpoints and induction of apoptosis. Damaging agents, emanating from endogenous and environmental sources such as oxidative stress and ultraviolet (UV) radiation, constantly challenge the integrity of DNA. Remarkably, spontaneous DNA damage, mostly hydrolytic cytosine deamination and oxidative DNA base damage, occurs at a rate of up to 105 lesions per cell per day [4, 5].
In order to counteract these insults and preserve genome stability, cells activate a coordinated signal-transduction network, which is collectively known as the DNA damage response (DDR). Generally, this response consists of a series of events such as detection of the DNA damage by sensors, accumulation of repair factors by mediators and repair of the lesion by effectors [6]. Cells are equipped with a variety of distinct, but partially compensatory, DNA repair mechanisms, each addressing a specific type of lesion [5]. DNA double-strand breaks (DSBs) are considered to be the most hazardous lesions, since a single unrepaired DSB may trigger cell death whereas a misrepaired DSB potentially results in mutations such as chromosomal rearrangements, which can promote carcinogenesis. Therefore, activation of cell-cycle checkpoints and faithful repair in response to DSBs are a primary barrier to malignant transformation.
The fact that DSBs are highly cytotoxic is exploited in conventional cancer treatment with radiation therapy and certain chemotherapeutic drugs such as DNA crosslinkers and topoisomerase inhibitors. Although those agents induce DSBs in all cells, hyperproliferating cancer cells are much more susceptible to killing than normal cells. However, most of these well-established treatments cause a number of adverse effects, mainly by affecting the fast-dividing cells of the patient, such as haematopoietic stem cells, hair follicles and cells lining the stomach and intestines. Therefore, novel strategies to treat cancer are eagerly anticipated and the subject of extensive research.
In this review, we summarise how DSB repair and its genetic interactions have emerged as targets for improved cancer treatment strategies in the recent past. We also highlight the current knowledge of small molecule inhibitors (SMIs) of DSB signalling and repair factors, which are promising candidates for clinical use. For a comprehensive overview of targeting DDR pathways for cancer therapy, we direct the reader to recently published reviews [7–9].
DNA damage response
The DDR is a multifaceted signalling network, which is elicited upon detection of DNA lesions in order to coordinate the cell cycle, DNA repair and possibly senescence or apoptosis (fig. 1). Three members of the phosphatidylinositol-3-kinase (PI3K) related kinases (PIKKs) ‒ ATM (ataxia telangiectasia mutated), ATR (ATM- and Rad3-related) and DNA-PKcs (DNA-dependent protein kinase catalytic subunit) ‒ become activated upon DNA damage to trigger the DDR [10]. Through a cascade of phosphorylation events, ATM and ATR activate multiple proteins, most notably p53 and the downstream checkpoint kinases CHK1 and CHK2, which in turn phosphorylate WEE1 kinase and CDC25 phosphatases. Consequently, through regulating the activity of cyclin-dependent kinases (CDKs), the progression from one cell cycle phase to another is delayed [11]. Depending on which CDK is inhibited; the cell cycle is arrested either at the G1/S or the G2/M transition. The resulting cell-cycle arrest allows time for repair, thereby preventing genome duplication or cell division in the presence of damaged DNA. Thus, cells with an abrogated DDR generally display an increased sensitivity towards DNA-damaging agents.
DNA repair pathways
Depending on the type of DNA damage, cells invoke specific DNA repair pathways in order to restore the genetic information (see fig. 1). Briefly, minor changes to DNA such as oxidised or alkylated bases, small base adducts and single-strand breaks (SSBs) are restored by the base excision repair (BER) pathway [4]. A key player in this process is poly-adenosine-diphosphate-ribose (PAR) polymerase (PARP). Upon detection of SSBs, PARP covalently transfers PAR chains to itself and to acceptor proteins in the vicinity of the lesion, thereby facilitating the repair of SSBs. More complex, DNA helix-distorting base lesions, such as those induced by UV light, are repaired by nucleotide excision repair (NER) [5]. Another kind of damage disturbing the helical structure of DNA is represented by base mismatches. Mismatch repair factors recognise and process misincorporated nucleotides as well as insertion or deletion loops that arise during recombination or from errors of DNA polymerases [12]. Covalent links between the two strands of the double helix represent a type of DNA damage referred to as interstrand crosslinks (ICLs). ICLs represent the most deleterious lesions produced by chemotherapeutic agents such as mitomycin C (MMC), cisplatin and cyclophosphamide. ICL repair is complex and involves the collaboration of several repair pathways, namely Fanconi anaemia, NER, translesion synthesis and homologous recombination (HR) [13].
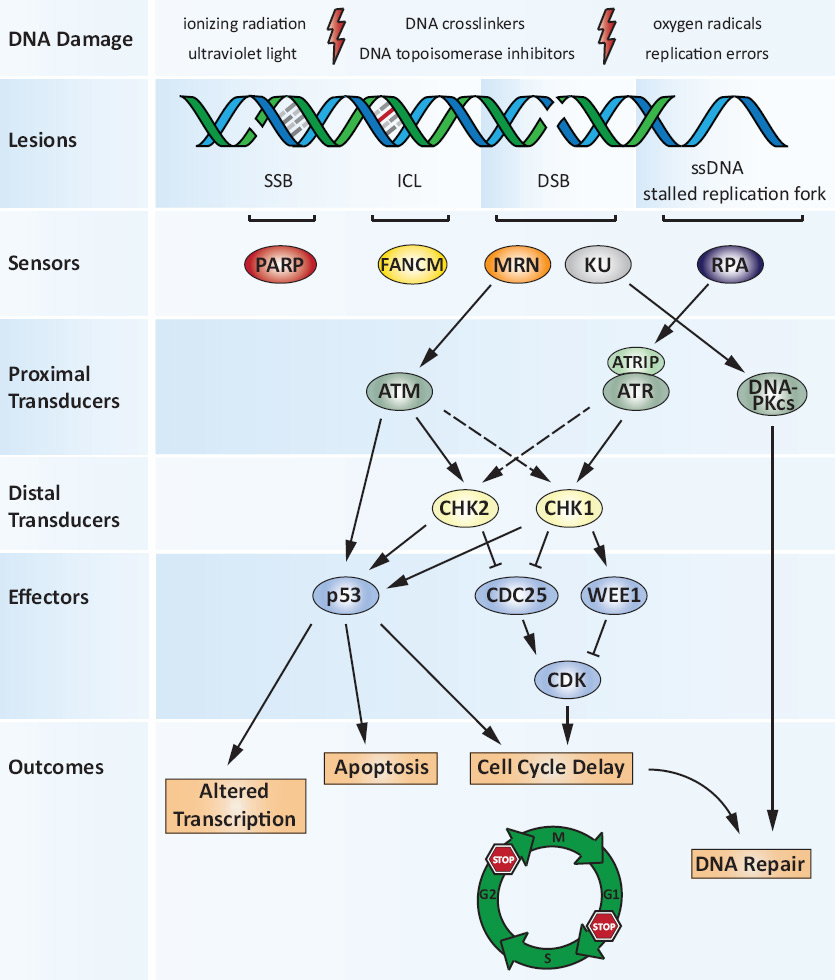
Figure 1
The DNA damage response.
Exogenous and endogenous DNA damaging agents generate various types of lesions including DNA single- and double-strand breaks (SSBs and DSBs). The multifunctional MRN complex detects DSBs, while FANCM is required for the DNA interstrand crosslink (ICL)-induced checkpoint response. PARP predominantly acts as a SSB sensor protein. RPA binds to regions of single-stranded DNA (ssDNA) that are exposed at stalled replication forks or after DSB resection. MRN and RPA mediate the recruitment of ATM and ATR-ATRIP, respectively, and the subsequent activation of the respective pathways, coordinating cell-cycle checkpoints, DNA repair and apoptotic responses to DNA damage. The Ku70/Ku80 heterodimer (KU) competes with MRN for binding to DSBs. KU recruits DNA-PKcs to form the catalytically active DNA-PK holoenzyme which is a major component of the canonical NHEJ machinery during DSB repair. MRN on the other hand initiates HR (see also fig. 2). Once activated, the DNA damage signalling cascade extends through multiple phosphorylation events primarily via the cell-cycle checkpoint kinases CHK1 and CHK2. Their signals converge on downstream effectors such as the tumour suppressor protein p53 or the CDC25 protein phosphatase and WEE1 tyrosine kinase. As a result, CDK activity is inhibited, delaying cell cycle progression from G1 to S (the G1/S checkpoint) or from G2 to M phase (the G2/M checkpoint). The DNA damage response (DDR) thus orchestrates a variety of cellular outcomes: the transcriptional programme of the damaged cell is altered and the cell cycle is transiently arrested, thereby facilitating repair of the DNA lesions. In situations where DNA damage is too severe and cannot be repaired, the DDR triggers apoptosis or senescence.
ADP = adenosine diphosphate; ATM = ataxia telangiectasia mutated protein; ATR = ATM- and Rad3-related; ATRIP = ATR-interacting protein; CDK = cyclin-dependeant kinase; DNA = deoxyribonucleic acid; DNA-PK = DNA-dependeant protein kinase; DNA-PKcs = DNA-PK catalytic subunit; FANCM = Fanconi anaemia complementation group M; HR = homologous recombination; ICL = interstrand crosslink; MRN = MRE11-RAD50-NBS1 complex; NEHJ = nonhomologous end joining; PARP = poly(ADP-ribose) polymerase; RPA = replication protein A
DNA double-strand break repair
Thus far, four mechanistically distinct DSB repair mechanisms in mammalian cells have been described: nonhomologous end joining (NHEJ), alternative-NHEJ, single-strand annealing and HR [14]. NHEJ and HR represent the two major DSB repair pathways, with NHEJ operating throughout the cell cycle and HR being most active during S-phase (fig. 2) [15]. NHEJ starts with the binding of the Ku70/80 heterodimer to both ends of the break, followed by the recruitment of the catalytic subunit of DNA-dependent protein kinase (DNA-PKcs). Subsequent phosphorylation events mediated by the DNA-PK holoenzyme lead to appropriate DNA end processing by the Artemis nuclease. DSB repair by NHEJ is completed by rejoining of the ends catalysed by a complex consisting of X-ray repair cross-complementing protein 4 (XRCC4), XRCC4-like factor (XLF) and DNA ligase IV. HR takes over if NHEJ is unsuccessful in ligating the broken DNA ends or when the DSB is first recognised by the MRE11-RAD50-NBS1 (MRN) complex rather than by Ku70/80 [16]. Together with CtBP-interacting protein (CtIP; CtBP = C-terminal binding protein), MRN resects DSBs to generate short 3'-single-stranded-DNA (ssDNA) tails that get immediately coated with replication protein A (RPA) [17]. The BRCA2-PALB2 complex promotes RAD51 nucleation onto ssDNA, thereby replacing RPA. The RAD51 nucleoprotein filament then invades the homologous, intact DNA template forming a displacement loop. The second end of the broken chromosome is captured and anneals to the complementary strand of the donor DNA molecule, resulting in the formation of two Holliday junctions (HJs). After DNA synthesis and ligation of both strands, the double HJ is either dissolved or is dismantled by the catalytic action of resolvases in order to complete repair [18]. Thus, repair by HR is error-free since it copies the missing genetic information from the undamaged sister chromatid, whereas NHEJ is error-prone since DNA ends without sequence homology are religated with the risk of causing mutations [19]. Given that a single unrepaired DSB has the potential to kill a cell, inhibition of repair by compounds that target factors involved in NHEJ or HR will increase the sensitivity of cancer cells to DSB-inducing anticancer agents.
Harnessing DNA damage signalling and repair for cancer therapy
The fact that cells with a compromised DDR are hypersensitive to DNA damage-inducing agents is currently under vigorous investigation for use in targeted cancer therapy. More precisely, during their pathogenesis, many cancer cells acquire defects in a certain DNA repair pathway and become dependent on a compensatory mechanism in order to survive. Hence, pharmacological inhibition of the “backup” pathway in combination with DNA damage will selectively kill cancer cells but spare their normal counterparts. Furthermore, highly proliferative cancer cells are inherently hypersensitive to DNA damage because S-phase, in which DNA replication takes place, is the most vulnerable period of the cell cycle.
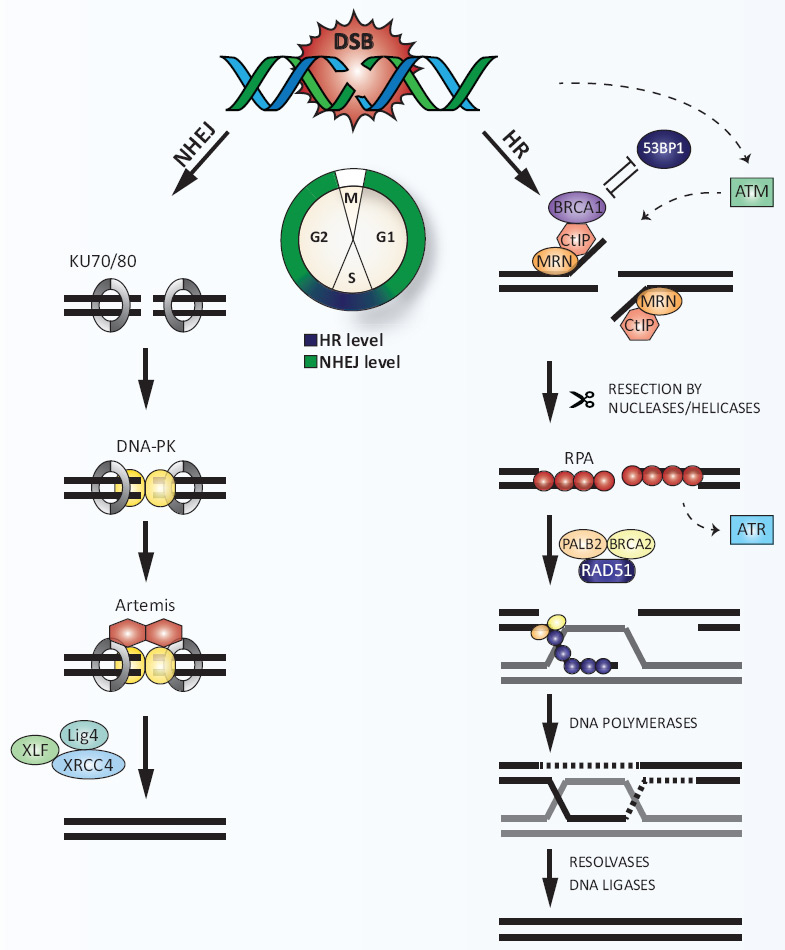
Figure 2
DNA double-strand break (DSB) repair.
DSBs are predominantly repaired by two distinct pathways: NHEJ or HR. NHEJ operates throughout the cell cycle, but mainly during the G1 and G2 phases, whereas HR peaks in S phase. Rapid association of the Ku70/80 heterodimer to DSBs promotes NHEJ by recruiting DNA-PKcs. DNA ends are processed by the nucleolytic activity of Artemis, followed by religation catalysed by a complex of XLF, Ligase IV (Lig4) and XRCC4. Alternatively, MRN, which is initially recruited to DSBs in competition with Ku70/80, initiates DSB resection together with CtIP thereby promoting HR. 53BP1 antagonises BRCA1 in DSB resection. Extensive DSB resection by other nucleases and formation of RPA-coated ssDNA stimulates the activation of ATR. Displacement of RPA by RAD51 is mediated by BRCA2 and PALB2, resulting in the formation of RAD51 nucleoprotein filaments. Subsequent strand invasion into the homologous DNA template and capturing of the second DNA end leads to the formation of a double Holliday junction, which is processed by resolvases. Finally, the DNA is sealed by ligases to accomplish error-free repair of the DSB.
53BP = p53 binding protein; ATM = ataxia telangiectasia mutated protein; CtBP = C-terminal binding protein; CtIP = CtBP-interacting protein; DNA = deoxyribonucleic acid; DNA-PK = DNA-dependent protein kinase; DNA-PKcs = DNA-PK catalytic subunit; HR = homologous recombination; MRN = MRE11-RAD50-NBS1 complex; NHEJ = nonhomologous end joining; PALB1/2 = partner and localiser of BRCA1/2; RPA = replication protein A; ssDNA = single-stranded DNA; XLF = XRCC4-like factor; XRCC4 = X-ray repair cross-complementing protein 4
Targeting DSB signalling pathways
As previously mentioned, cell-cycle checkpoint activation in response to DSBs gives a cell time for DNA repair before entry into S-phase or mitosis. Consequently, cell-cycle checkpoints reduce the efficacy of DNA-damaging agents used in cancer therapy. Therefore, selective abrogation of checkpoint signalling sensitises cancer cells to chemo- and radio-therapy, potentiating cancer treatment [20]. Importantly, more than 50% of human tumours are defective in p53 tumour suppressor function and cell-cycle checkpoint inhibitors have been demonstrated particularly to sensitise p53-deficient cancer cells to various anticancer agents in clinical use [21].
In the late 1960s, long before the discovery of cell-cycle checkpoints, the first attempts to sensitise cancer cells to standard cytotoxic therapy were made using ordinary compounds such as caffeine [22]. Later it was found that caffeine directly binds to and inhibits ATM and ATR in vitro and thus interferes with initiation of the DDR [23, 24]. However, since caffeine is a relatively nonselective agent, efforts have been made to develop more potent and selective inhibitors of the PIKK family members ATM, ATR and DNA-PKcs (table 1). In 2004, KuDOS Pharmaceuticals (now AstraZeneca) reported the identification of KU-55933, a specific SMI of ATM [25]. On the molecular level, KU-55933, like most kinase inhibitors, competes with the ATP-binding site of the enzyme, thereby inhibiting the catalytic activity of ATM [26]. Based on the promising preclinical results, KU-60019, a KU-55933 analogue with improved pharmacokinetics and bioavailability, was synthesised and shown to radiosensitise glioma cells approximately 10 times more efficiently than KU-55933 [27]. Compounds selectively targeting ATR have long been awaited, particularly when inhibitors of CHK1, a direct downstream target of ATR, had proven to be clinically effective [28]. Finally, in 2011, three ATR inhibitors, NU-6027, VE-821 and ETP-46464 were described. NU-6027 is a pyrimidine analogue originally discovered as an adenosine triphosphate (ATP) competitive inhibitor of CDKs, but recently reported also to inhibit ATR at low micromolar concentrations and to confer cisplatin cytotoxicity independently of CDK inhibition [29, 30]. The ATR inhibitor VE-821 (Vertex Pharmaceuticals) was identified using a high-throughput screen against full-length recombinant ATR [31]. Preclinical testing of VE-821 using pancreatic cancer cells demonstrated its chemo- and radio-sensitisation properties [32]. ETP-46464 was discovered by screening compounds with a previously reported activity against the related PI3Ks using a cell-based system assaying for ATR activity [33]. In the same study, NVP-BEZ235, a recognised dual PI3K/mTOR inhibitor (mTOR = mammalian target of rapamycin), was also reported to block efficiently ATM, ATR and DNA-PK activity. Furthermore, NVP-BEZ235 was found to act as a radio- and chemo-sensitiser in various cancer cell lines and is currently being tested as a single agent in various Phase I/II clinical trials [34, 35]. Importantly, most, if not all, of the aforementioned compounds are likely to inhibit additional protein kinases, especially when used at concentrations in the high micromolar range, thus potentially exhibiting “off-target” effects.
Downstream of ATM and ATR act the two transducer kinases CHK1 and CHK2, against which several inhibitors have emerged during recent years. One of the first SMIs, UCN-01, a derivative of staurosporine, was originally isolated from a Streptomycesstrain as a protein kinase antagonist with cytotoxic effects [36]. UCN-01 was later shown to be a potent inhibitor of CHK1 and to block its kinase activity by interacting with the ATP-binding pocket [37, 38]. Six Phase II clinical trials of UCN-01, either as a single agent or in combination with other drugs, in patients with different types of advanced cancer have already been completed. Recently, three novel CHK1 inhibitors, GDC-0425 (Genentech Inc.), SCH900776 (now renamed MK-8776, Merck) and LY-2606368 (Eli Lilly), have entered Phase I clinical trials either as single agents or in combination with gemcitabine, a nucleoside analogue [39].
Another promising drug that interferes with checkpoint activation is the WEE1 tyrosine kinase inhibitor MK-1775 (Merck), which was discovered by screening a chemical library [40]. MK-1775 is already under investigation in a Phase II trial combined with carboplatin in order to assess the benefit for patients with p53-mutated epithelial ovarian cancer. Last but not least, efforts to target CDC25 phosphatases, which also represent key molecules in checkpoint regulation, led to the discovery of several CDC25 inhibitors, amongst which the most potent are quinonoid-based derivatives such as the bis-quinone compound IRC-08386 [41, 42].
In summary, several SMIs that interfere with checkpoint activation show great promise of advancing in clinical studies and eventually being used as chemo- or radio-sensitisers as well as monotherapeutic agents in cancer treatment. Nevertheless, since many of the SMIs have only very recently been discovered, their safety, tolerability and efficacy when used alone or in combination has to be further investigated.
|
Table 1: Small molecule inhibitors of DNA damage response factors in preclinical or clinical development for cancer therapy. |
|
Target
|
Inhibitor
|
Mono- or combination therapy / clinical study stage
|
Clinical trial identifier/reference
|
| ATM |
KU-55933 |
IR, etoposide, doxorubicin, camptothecin, in preclinical testing |
[25, 95] |
| KU-60019 |
IR in preclinical testing using glioma cells |
[27] |
| ATR |
NU-6027 |
Hydroxyurea, cisplatin, temozolomide, rucaparib in preclinical testing |
[29] |
| VE-821 |
Cisplatin in breast and ovarian cell lines
IR, gemcitabine in pancreatic cancer cells in preclinical testing |
[31, 96]
[32] |
| ETP-46464 |
Single agent in p53-deficient cancer cells in preclinical testing |
[33] |
| DNA-PKcs |
NU-7441 |
IR, etoposide in preclinical testing of cancer cell lines and tumour xenografts |
[44, 97] |
| NU-7026 |
IR and combined with AG14361 (PARPi) in preclinical testing
Anthracyclines, mitoxantrone, etoposide in preclinical testing using leukaemia cells |
[43]
[98] |
| DNA-PKcs/PI3K |
KU-60648 |
Etoposide, doxorubicin in preclinical testing |
[45] |
| DNA-PKcs/mTOR |
CC-115 |
Single agent in Phase I safety and tolerability study (recruiting) |
NCT01353625 |
| PI3K/mTOR/PIKK |
NVP-BEZ235 |
Single agent in several clinical trials
IR, cisplatin in preclinical testing |
http://www.clinicaltrials.gov
[34, 35] |
| CHK1/(CHK2) |
UCN-01 |
Single agent in Phase II for relapsed T-cell lymphoma (completed)
Single agent in Phase II for metastatic melanoma (completed)
Five-fluorouracil in Phase II for metastatic pancreatic cancer (completed)
Topotecan in Phase II for various forms of ovarian cancer (completed)
Topotecan in Phase II for small cell lung cancer (completed)
Olaparib in pre-clinical testing for multiple mammary tumour types |
NCT00082017
NCT00072189
NCT00045747
NCT00072267
NCT00098956
[99] |
| GDC-0425 |
Single agent or with gemcitabine in Phase I dose-escalation study (recruiting) |
NCT01359696 |
| MK-8776 |
Single agent or with gemcitabine in Phase I dose-escalation study (completed) |
NCT00779584 |
| LY-2606368 |
Single agent in Phase I study in patients with advanced cancer (recruiting) |
NCT01115790 |
| WEE1 |
MK-1775 |
Carboplatin in Phase II for epithelial ovarian cancer |
NCT01164995 |
| CDC25 |
IRC-083864 |
Single agent in preclinical testing using pancreatic and prostate cancer cells |
[42] |
| MRE11 |
mirin |
Single agent or with olaparib (PARPi) in preclinical testing using BRCA2-deficient cells |
[47] |
| RPA |
MCl13E |
Single agent or with cisplatin in preclinical testing |
[48] |
| RAD51 |
B02 |
IR, mitomycin C, cisplatin in preclinical testing |
[100] |
| RI-1 |
Mitomyin C in preclinical testing |
[50] |
| ADP = adenosine diphosphate; ATM = ataxia telangiectasia mutated protein; ATR = ATM- and Rad3-related; CHK = checkpoint kinase; DNA = deoxyribonucleic acid; DNA-PK = DNA-dependent protein kinase; DNA-PKcs = DNA-PK catalytic subunit; IR = ionising radiation; mTOR = mammalian target of rapamycin; PAR = poly(ADP-ribose); PARP = PAR polymerase; PARPi = PARP inhibitor; PI3K = phosphatidylinositol-3-kinase; PIKK = PI3K-related kinase; RPA = replication protein A |
Targeting DSB repair
Impairing the repair of DSBs using drugs that either inhibit the enzymatic activity or interfere with protein-protein interactions of repair factors provides another approach to sensitising cancer cells for chemo- and radio-therapy. A key player in DSB repair by NHEJ is DNA-PK (see fig. 1), which, like ATM and ATR, belongs to the PIKK family of protein kinases. In 2003, two DNA-PKcs-specific inhibitors, NU-7026 and NU-7441, were reported, both of which are practically inactive against ATM and ATR [43, 44]. Unfortunately, neither of them has progressed into clinical development. However, Celgene Corporation is currently recruiting patients with advanced tumours unresponsive to standard therapies in order to test the pharmacokinetics and preliminary efficacy of the dual DNA-PK/mTOR inhibitor, CC-115, in a Phase I trial. Moreover, a very recent preclinical study reported that KU-60648 (AstraZeneca), a dual inhibitor of DNA-PK and PI3K, acts as a chemosensitiser in cell-based assays and in mice xenografts [45]. More recent attempts to find novel DSB repair inhibitors led to the identification of mirin, the first inhibitor of the MRN complex that acts by blocking the nuclease activity of MRE11 [46]. Interestingly, mirin was shown to kill BRCA2-deficient cells, an effect that was even more pronounced when combined with a PARP inhibitor [47]. However, since mirin has to be applied at high micromolar concentrations to inhibit MRN, such treatment is prone to increase the risks of undesired “off-target” effects and the generation of more selective derivatives is eagerly anticipated.
During the course of DSB repair by HR, ssDNA is generated and immediately coated by RPA, which later on is replaced by the RAD51 recombinase (see fig. 2). Inhibiting the DNA-binding activity of RPA by the SMI MCI13E yielded encouraging preclinical results in combination with cisplatin [48]. Moreover, several means to prevent RAD51 action have been reported, including SMIs (B02, RI-1) as well as inhibitory peptides that interfere with the binding of BRCA2 to RAD51 [49–52]. Although peptides blocking protein-protein interactions represent an interesting concept for inhibiting DSB repair, their potential application in the clinics has yet to be established.
In summary, safety, tolerability, pharmacokinetics and efficacy of most of the aforementioned SMIs have still to be carefully validated before they may enter clinical trials to examine their benefit for cancer therapy.
Synthetic lethality approaches to target DSB repair-deficient cancers
Mutations in DSB repair genes render cancer cells dependent on alternative DNA repair pathways. Thus, compromised abilities to repair DSBs confer a weakness that can be therapeutically exploited on the basis of the concept of synthetic lethality, whereby inhibition of the “back-up” pathway induces greater toxicity in DSB repair-deficient cancer cells as compared with normal cells (fig. 3).
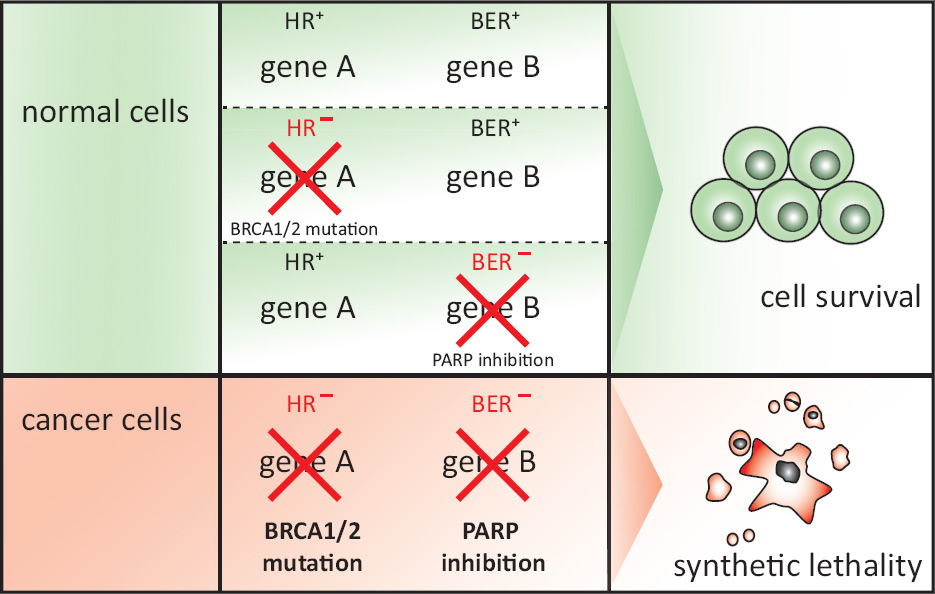
Figure 3
Synthetic lethality.
Synthetic lethality is defined as a combination of mutations or perturbations in two or more genes that leads to cell death, whereas inactivation of any one of the genes alone does not. Perturbation of genes can occur through genetic mutation or silencing, depletion by RNAi or inhibition by SMIs and is depicted by a red cross. If genes that are essential for a certain DNA repair pathway (e.g. gene A) are inactivated in normal cells, alternative pathways with functional genes (e.g. gene B) are utilised to respond to DNA damage. Conversely, cancer cells mutated or silenced for a component of a DDR pathway are compromised in their ability to process DSBs. These cells then rely on alternative DNA repair pathways to repair the breaks. Therefore, inhibition of the alternative pathway will cause cell death due to persisting DSBs. Please refer to main text for details about the example given for synthetic lethality between BRCA1/2 mutation and PARP inhibition.
ADP = adenosine diphosphate; BER = base excision repair; DDR = DNA damage response; DNA = deoxyribonucleic acid; DSB = double-strand break; HR = homologous recombination; PARP = poly(ADP-ribose) polymerase; RNA = ribonucleic acid; RNAi = RNA interference; SMI = small molecule inhibitor
PARP inhibitors
The first “proof-of-principle” study verifying synthetic lethality as a suitable approach for targeted cancer therapy was published in 2005, after it had been demonstrated that HR-defective BRCA1- or BRCA2-deficient cell lines display dramatically increased sensitivity to inhibition of the SSB repair enzyme PARP [53, 54]. Subsequently, clinical development of potent small molecule PARP inhibitors (PARPis) rapidly advanced, and the first Phase II results in 2009 showed that monotherapy with the PARPi olaparib (AZD-2281; AstraZeneca) achieved encouraging response rates of 41% and 33% in patients with BRCA1- or BRCA2-mutated advanced breast and ovarian cancers, respectively [55, 56]. Furthermore, preclinical studies suggested the potential use of PARPi also in sporadic cancers that share phenotypical features with cancers arising from hereditary BRCA mutations, a phenomenon that is referred to as “BRCAness” [57]. Reasons for “BRCAness” can be the inactivation of BRCA1 or BRCA2 function caused by aberrant epigenetic or posttranslational modifications, or a wider range of mutations in other genes resulting in defective DSB signalling and HR. For example, it was reported that depletion of factors such as ATR, ATM, CHK1, CHK2, NBS1, CtIP and RAD51 in cultured cells synergistically increases PARPi cytotoxicity to an extent similar to mutations in BRCA1/2. This indicates that BRCA-deficient cells are, at least in part, sensitive to PARP inhibition because of a defect in HR [58, 59]. The current understanding suggests that inhibition of PARP leads to the accumulation of SSBs which are converted into DSBs upon encountering DNA replication forks during S-phase when HR is most active [60]. Consequently, in the absence of functional HR, such as in cancer cells lacking BRCA1 or BRCA2, PARP inhibition results in the accumulation of DSBs and, ultimately, in apoptosis or mitotic catastrophe. Importantly, normal cells survive the treatment owing to functional HR, providing the kind of selectivity that is considered the ultimate goal of cancer therapy. Nowadays, most PARPi in preclinical and clinical trials belong to the third generation of SMIs designed to compete with the substrate nicotine adenine dinucleotide (NAD+) resulting in reversible inhibition of PARP. Recent reports indicate that in addition to catalytic inhibition, some PARPi induce cytotoxic PARP–DNA complexes, trapping PARP proteins on damaged DNA. Currently, PARPi are divided into two classes: catalytic inhibitors and dual inhibitors that not only block the enzymatic activity but also act as so-called PARP “poisons” [61, 62].
Today, 7 years after PARPi were first established for cancer therapy and despite some quite promising clinical studies, none of them has gained official approval for the treatment of cancer patients. In 2011, encouraging results from a Phase II trial with iniparib (BSI-201, Sanofi-Aventis) in patients with triple negative breast cancer, which shares many features with BRCA-associated breast cancer, failed to translate into overall patient survival in a Phase III trial [63]. Later that year, AstraZeneca announced that olaparib would not progress into Phase III for hereditary BRCA mutation-associated breast cancer. This decision was possibly driven by economic concerns rather than by clinical issues [64]. Notwithstanding all setbacks, clinical development and research on the mechanism of action of PARPi is still ongoing (table 2). Despite controversies about its effectiveness as a PARPi, Sanofi's iniparib is under clinical investigation as a single agent and in combination with chemotherapeutic regimens in patients with recurrent solid tumours (NCT01455532), nonsmall-cell lung cancer (NCT01082549) and ovarian cancer (NCT01033292) [65, 66]. Likewise, AstraZeneca is continuing Phase II trials with olaparib to treat serous ovarian cancer, since it shares many features with BRCA1/2-mutated cancers. Indeed, activity of olaparib as a monotherapy was evident in women with pretreated high-grade serous ovarian cancer without germline BRCA1/2 mutations [67]. This finding clearly demonstrates positive responses of a subpopulation of sporadic cancers to PARPi therapy and also underlines the importance of classifying patients according to biomarkers in order to predict the efficacy of PARPi. Such potential biomarkers also include deficiency of the phosphatase and tensin homologue (PTEN) tumour suppressor. Interestingly, due to its role in the regulation of RAD51 transcription, loss of PTEN is associated with defective HR [68, 69]. In general, detection of compromised HR provides a rationale to stratify patients for PARPi treatment. Several ways of identifying HR defects are under investigation, including gene expression profiling and gene copy number analysis of DNA repair factors [70, 71]. Further approaches assess the DSB repair capacity of tumours by measuring expression of the MRN complex, monitoring RAD51 foci formation and poly(ADP-ribosyl)ation as surrogate markers for DSB repair proficiency [72]. As for most cancer therapies, a major challenge of using PARPis is the acquired resistance of initially PARPi-sensitive cancer cells due, for example, to the loss of 53BP1 (a p53 binding protein) or to overexpression of multidrug-resistance efflux transporters [72, 73]. In addition, secondary BRCA2 mutations have been identified, which restore the full-length protein thereby re-establishing BRCA2 functions and conferring PARPi resistance [74].
Thus, despite considerable efforts to develop PARPi for clinical use, conventional DNA-damaging chemo- and radio-therapy largely remains the mainstay of cancer treatment. However, several onging preclinical and clinical studies employ PARPi both as monotherapy and as chemo- or radio-sensitisers, because an improvement of current anti-cancer regimes is long-awaited.
|
Table 2: PARP inhibitors in preclinical or clinical development for cancer therapy*. |
|
Inhibitor
|
Mono- or combination therapy
|
Preclinical and clinical study stage
|
Clinical trial identifier/reference
|
| Olaparib
(AZD-2281, KU-59436)
AstraZeneca |
Single agent |
Phase II trials showing with promising response rates in patients with BRCA1/ or BRCA2 mutated advanced breast and ovarian cancers |
NCT01078662, NCT00494234
[55, 56] |
| Single agent |
Phase II trial demonstrating efficacy for advanced high-grade serous ovarian cancer without germline BRCA1/2 mutations, but not with TNBC |
NCT00679783
[67] |
| Cediranib |
Phase I/II study for treatment of recurrent serous ovarian cancer and TNBC |
NCT01116648 |
| Single agent and combinations with other drugs |
Several ongoing Phase I/II trials for various cancers, dose-limiting adverse effects for combination of olaparib and topotecan |
NCT00516438, NCT00819221,
NCT01296763, NCT00912743, [101] |
| Cisplatin, radiation |
Phase I trial to test olaparib as a radio- and/or chemo-sensitiser in combination with high-dose radiotherapy with or without a daily cisplatin dose in locally advanced NSCLC |
NCT01562210 |
| Iniparib**
(BSI-201)
Sanofi-Aventis |
Single agent and combinations with other drugs |
Ongoing Phase I/II trials in solid tumours such as sarcomas as well as breast, uterine and ovarian cancers |
NCT01455532, NCT01033292,
NCT00687687 |
| Gemcitabine/carboplatin |
Promising results from a Phase II trial failed to translate into survival benefit for TNBC patients with unselected BRCA1/2 status in Phase III |
NCT00938652, NCT01130259,
[63] |
| Radiotherapy |
Ongoing Phase I trial of iniparib as radiosensitiser in nonoperable brain metastases |
NCT01551680 |
| Temozolomide and radiotherapy |
Ongoing Phase I/II trials for newly diagnosed malignant glioblastoma |
NCT00687765 |
| Gemcitabine/carboplatin |
Ongoing Phase III trial in advanced squamous NSCLC |
NCT01082549 |
| Veliparib
(ABT-888)
Abbott Laboratories |
Temozolomide, carboplatin/paclitaxel |
International randomised Phase II trial of veliparib combined with chemotherapy in BRCA1/2-mutated, metastatic breast cancer |
NCT01506609 |
| Single agent |
Phase I trial for refractory BRCA 1/2-mutated solid cancers; platinum-refractory ovarian, fallopian tube, or primary peritoneal cancer or basal-like breast cancer
Additional evaluation of BRCA1/2 expression and changes in PAR and γ-H2AX in peripheral blood mononuclear cells as diagnostic biomarkers |
NCT00892736 |
| Single agent and combinations with chemo- and radio-therapy |
Phase I and II trials to identify efficient combinatorial regimens in various solid and lymphoid tumours, promising results in combination with topotecan and cyclophosphamide |
NCT01154426, NCT01282333, NCT01386385 and more, [102-104] |
| Rucaparib
(AG-014699,
PF-01367338, CO-338)
Cancer Research UK
Clovis Oncology
Pfizer |
Single agent |
Phase I/II trials for BRCA1/2-mutated breast or ovarian cancer |
NCT01482715 |
| Temozolomide |
Initial Phase I trial as enhancer for chemotherapy in unselected solid tumours; severe myelosuppression of the combination in Phase II study for previously untreated metastatic melanoma |
[105] |
| Single agent and with carboplatin |
Phase I/II testing in advanced solid tumors with and without BRCA mutations
Several biomarkers for therapeutic response are being evaluated concurrently. |
NCT01009190, NCT00664781 |
| Niraparib
(MK-4827)
Merck
Tesaro Inc. |
Single agent |
Phase I trial in advanced solid tumours showing that MK-4827 is well tolerated, blocks PARP and has promising antitumour activity in both BRCA-deficient and sporadic cancer |
[106] |
| Single agent and with temozolomide |
Phase I dose-escalation study for solid tumours and haematological malignancies |
NCT00749502, NCT01294735 |
| * For a complete overview of PARPi currently used in clinical trials, we direct the reader to [61, 107].
** Since the primary mechanism of action for iniparib is likely not via inhibition of PARP activity, it is no longer considered to be a bona fide PARPi [65].
ADP = adenosine diphosphate; γ-H2AX = phosphorylated histone variant H2AX; NSCLC = nonsmall cell lung cancer; PARP = poly(ADP-ribose) polymerase; PARPi = PARP inhibitor; TNBC = triple negative breast cancer |
Synthetic lethal strategies emerging from preclinical research
As intensive basic research is leading towards a better understanding of cellular functions and their underlying genetic networks, more and more genetic interactions become apparent as potential targets for synthetic lethality in cancer therapy. Beyond BRCA1 and BRCA2, their joint interaction partner PALB2 is emerging as a breast cancer susceptibility gene, thus providing another opportunity for PARPi-based therapies [75].
PARP inhibition is not the only approach that takes advantage of synthetic lethal interactions between two DNA repair pathways, as inhibition of apurinic/apyrimidinic (AP) endonuclease 1, an essential component of BER, was recently shown to eliminate cancer cells with HR defects [76]. Moreover, synthetic lethality with components of the cell-cycle checkpoint machinery could be exploited in cancers harbouring activated oncogenes, since oncogene-induced replication stress activates the ATR-CHK1 signalling pathway. For example, exacerbated toxicity was reported upon inhibition of CHK1 in lymphoma cells with upregulated c-Myc expression [77]. This finding underscores the concept that cancers with elevated levels of replication stress rely on intact checkpoint signalling for cell survival. Replicative stress induces pan-nuclear distribution of phosphorylated histone variant H2AX (γ-H2AX), which is a useful biomarker for classification of tumour biopsies in order to stratify patients [78].
Finally, disruption of the FA repair pathway was shown to be synthetically lethal with abrogated checkpoint signalling. More precisely, inactivation of ATM or CHK1 resulted in reduced viability of FA-deficient cells, illustrating the concept that checkpoint signalling and FA are mutually compensatory pathways in the maintenance of genome integrity [79, 80]. These observations highlight the usefulness of SMIs, as currently tested for CHK1, to treat tumours bearing a specific genetic background. Although many of the strategies that are based on the concept of synthetic lethality have so far only been investigated in preclinical settings, some hold great promise of entering clinical trials soon.
Haploinsufficiency of DDR factors
There is increasing evidence that haploinsufficiency of DDR components promotes genome instability and drives tumourigenesis. Dosage insufficiencies of DNA repair genes might, however, only be unmasked once a cell is challenged with an increased load of DNA damage such as oncogene-induced replicative stress [81, 82]. Synthetic lethal approaches might therefore be applicable not only in cancer cells with deficiencies, but also in those bearing haploinsufficiencies for DDR factors. Evidence from gene targeting studies in mice revealed that, for example, the loss of one allele of ATR or CtIP is sufficient to cause increased chromosomal aberrations, genomic instability and tumour susceptibility [83, 84]. This indicates that heterozygous carriers of DDR defects are more prone to develop tumours once the threshold of endogenous DNA damage is increased as, for example, in precancerous lesions [85]. However, scientists are just beginning to unravel how haploinsufficiency of DDR genes contributes to carcinogenesis and how these may be exploited for novel synthetic lethal approaches in cancer therapy.
Conclusions and future perspectives
To date, DSB-inducing agents have been the core components of conventional cancer therapy, confirming the rationale of inflicting excessive DNA damage in order to kill cancer cells. However, most chemotherapeutic regimens cause severe side effects that limit their therapeutic potential. As summarised in this review, SMIs and synthetic lethal approaches targeting the individual genetic profile of the tumours are under clinical development, with the aim to improve the patients' benefit by increasing the efficacy while lowering the toxicity of cancer treatments. A prerequisite for personalised therapy is the molecular characterisation of tumours with reliable biomarkers to assign patients the appropriate treatment. In order to stratify cancer patients according to their DNA repair status, tumour biopsies can be analysed with immunohistochemistry, fluorescence in-situ hybridisation (FISH), gene sequencing, expression profiling and other methods [86]. Relevant biomarker assays should ideally predict the functionality of DNA repair pathways, rather than just providing information about mutations or expression levels of proteins involved in the DDR. Certainly, such a detailed molecular profiling of cancer versus normal tissue from a given patient is critical to maximise the potential of personalised cancer drugs in terms of both therapeutic success and cost-effectiveness.
Recent in-vitro and in-vivo research has deepened our knowledge about synthetic genetic interactions and put forward alternative ways to treat cancer. Furthermore, by utilising ribonucleic acid (RNA) interference technologies, screens for synthetic lethal interactions of cancer-specific defects in DNA repair pathways have augmented the discovery of targets for cancer therapy. For example, studies using MMR-deficient cells lacking human muts homologue 2 (MSH2) revealed synthetic sickness with POLB, a DNA polymerase acting in BER [87]. Since MSH2 is mutated in 40% of patients with hereditary nonpolyposis colorectal cancer, targeted inhibition of POLB potentially opens new therapeutic applications. Moreover, gaining further insights into the structure and mechanism of action of DNA repair factors such as CtIP will aid the design of new and more efficient SMIs of the DDR. Recently discovered inhibitors of RPA and RAD51 are promising candidates, which are in preclinical testing in order to be approved for the use in clinical trials soon [48, 50].
Interestingly, the latest scientific progress in the field of microRNAs (miRNAs) has demonstrated an intensive interplay of these small regulatory RNAs with the DDR, including DSB repair. Recent studies revealed that DNA damage globally induces miRNA biogenesis and, vice versa, that numerous miRNAs modulate the expression of DDR factors [88–90]. Notably, BRCA1 expression was shown to be downregulated by miR-182, conferring hypersensitivity to PARPi [91]. Conversely, BRCA1 was demonstrated to suppress expression of miR-155, an oncogenic miRNA that is overexpressed in many cancers [92, 93]. These observations highlight the therapeutic potential of miRNA mimics or inhibitors in future approaches for cancer therapy [94].
In summary, as the concept of personalised medicine emerges, tumour-specific defects of DSB repair pathways represent a promising therapeutic target to be exploited for the selective elimination of cancer cells. Thus, there is an air of optimism for targeted cancer therapy through exploiting the DDR of tumour cells in the clinics.
Acknowledgement:We wish to thank D. Nadal (University Children’s Hospital of Zurich) and J. Peña-Diaz for critical reading of the manuscript and helpful discussions. We also would like to apologize to all authors whose significant contributions could not be cited due to space limitations.
References
1 Bundesamt für Statistik – Todesfälle: Anzahl, Entwicklung und Ursachen. http://www.bfs.admin.ch .
2 Krebsliga Schweiz – Krebs in der Schweiz: wichtige Zahlen (Stand: Juni 2012). http://www.krebsliga.ch .
3 Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
4 Barnes DE, Lindahl T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet. 2004;38:445–76.
5 Hoeijmakers JHJ. DNA damage, aging, and cancer. N Engl J Med. 2009;361(15):1475–85.
6 Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071–8.
7 Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat Rev Cancer. 2012;12(9):587–98.
8 Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481(7381):287–94.
9 Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer. 2012;12(12):801–17.
10 Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434(7033):605–11.
11 Derheimer FA, Kastan MB. Multiple roles of ATM in monitoring and maintaining DNA integrity. FEBS Lett. 2010;584(17):3675–81.
12 Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol. 2006;7(5):335–46.
13 Schärer OD. DNA interstrand crosslinks: natural and drug-induced DNA adducts that induce unique cellular responses. Chembiochem. 2005;6(1):27–32.
14 Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40(2):179–204.
15 Chapman JR, Taylor MRG, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012;47(4):497–510.
16 Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet. 2011;45:247–71.
17 Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, et al. Human CtIP promotes DNA end resection. Nature. 2007;450(7169):509–14.
18 Moynahan ME, Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol. 2010;11(3):196–207.
19 Jackson SP. Sensing and repairing DNA double-strand breaks. Carcinogenesis. 2002;23(5):687–96.
20 Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–60.
21 Ma CX, Cai S, Li S, Ryan CE, Guo Z, Schaiff WT, et al. Targeting Chk1 in p53-deficient triple-negative breast cancer is therapeutically beneficial in human-in-mouse tumor models. J Clin Invest. 2012;122(4):1541–52.
22 Gaudin D, Yielding KL. Response of a “resistant” plasmacytoma to alkylating agents and x-ray in combination with the ‘excision’ repair inhibitors caffeine and chloroquine. Proc Soc Exp Biol Med. 1969;131(4):1413–6.
23 Sarkaria JN, Busby EC, Tibbetts RS, Roos P, Taya Y, Karnitz LM, et al. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999;59(17):4375–82.
24 Blasina A, Price BD, Turenne GA, McGowan CH. Caffeine inhibits the checkpoint kinase ATM. Curr Biol. 1999;9(19):1135–8.
25 Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NMB, Orr AI, et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64(24):9152–9.
26 Knight ZA. Small molecule inhibitors of the PI3-kinase family. Curr Top Microbiol Immunol. 2010;347:263–78.
27 Golding SE, Rosenberg E, Valerie N, Hussaini I, Frigerio M, Cockcroft XF, et al. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol Cancer Ther. 2009;8(10):2894–902.
28 Wagner JM, Kaufmann SH. Prospects for the Use of ATR Inhibitors to Treat Cancer. Pharmaceuticals. 2010;3(5):1311–34.
29 Peasland A, Wang L-Z, Rowling E, Kyle S, Chen T, Hopkins A, et al. Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br J Cancer. 2011;105(3):372–81.
30 Arris CE, Boyle FT, Calvert AH, Curtin NJ, Endicott JA, Garman EF, et al. Identification of novel purine and pyrimidine cyclin-dependent kinase inhibitors with distinct molecular interactions and tumor cell growth inhibition profiles. J Med Chem. 2000;43(15):2797–804.
31 Reaper PM, Griffiths MR, Long JM, Charrier J-D, MacCormick S, Charlton PA, et al. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol. 2011;7(7):428–30.
32 Prevo R, Fokas E, Reaper PM, Charlton PA, Pollard JR, McKenna WG, et al. The novel ATR inhibitor VE-821 increases sensitivity of pancreatic cancer cells to radiation and chemotherapy. Cancer Biol Ther. 2012;13(11):1072–81.
33 Toledo LI, Murga M, Zur R, Soria R, Rodriguez A, Martinez S, et al. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat Struct Mol Biol. 2011;18(6):721–7.
34 Yang F, Qian X-J, Qin W, Deng R, Wu X-Q, Qin J, et al. Dual Phosphoinositide 3-Kinase/Mammalian Target of Rapamycin Inhibitor NVP-BEZ235 Has a Therapeutic Potential and Sensitizes Cisplatin in Nasopharyngeal Carcinoma. PLoS ONE. 2013;8(3):e59879.
35 Mukherjee B, Tomimatsu N, Amancherla K, Camacho CV, Pichamoorthy N, Burma S. The dual PI3K/mTOR inhibitor NVP-BEZ235 is a potent inhibitor of ATM- and DNA-PKCs-mediated DNA damage responses. Neoplasia. 2012;14(1):34–43.
36 Takahashi I, Saitoh Y, Yoshida M, Sano H, Nakano H, Morimoto M, et al. UCN-01 and UCN-02, new selective inhibitors of protein kinase C. II. Purification, physico-chemical properties, structural determination and biological activities. J Antibiot. 1989;42(4):571–6.
37 Graves PR, Yu L, Schwarz JK, Gales J, Sausville EA, O'Connor PM, et al. The Chk1 protein kinase and the Cdc25C regulatory pathways are targets of the anticancer agent UCN-01. J Biol Chem. 2000;275(8):5600–5.
38 Zhao B, Bower MJ, McDevitt PJ, Zhao H, Davis ST, Johanson KO, et al. Structural basis for Chk1 inhibition by UCN-01. J Biol Chem. 2002;277(48):46609–15.
39 Guzi TJ, Paruch K, Dwyer MP, Labroli M, Shanahan F, Davis N, et al. Targeting the replication checkpoint using SCH 900776, a potent and functionally selective CHK1 inhibitor identified via high content screening. Mol Cancer Ther. 2011;10(4):591–602.
40 De Witt Hamer PC, Mir SE, Noske D, Van Noorden CJF, Würdinger T. WEE1 kinase targeting combined with DNA-damaging cancer therapy catalyzes mitotic catastrophe. Clin Cancer Res. 2011;17(13):4200–7.
41 Lavecchia A, Di Giovanni C, Novellino E. CDC25 phosphatase inhibitors: an update. Mini Rev Med Chem. 2012;12(1):62–73.
42 Brezak M-C, Valette A, Quaranta M, Contour-Galcera M-O, Jullien D, Lavergne O, et al. IRC-083864, a novel bis quinone inhibitor of CDC25 phosphatases active against human cancer cells. Int J Cancer. 2009;124(6):1449–56.
43 Veuger SJ, Curtin NJ, Richardson CJ, Smith GCM, Durkacz BW. Radiosensitization and DNA repair inhibition by the combined use of novel inhibitors of DNA-dependent protein kinase and poly(ADP-ribose) polymerase-1. Cancer Res. 2003;63(18):6008–15.
44 Leahy JJJ, Golding BT, Griffin RJ, Hardcastle IR, Richardson C, Rigoreau L, et al. Identification of a highly potent and selective DNA-dependent protein kinase (DNA-PK) inhibitor (NU7441) by screening of chromenone libraries. Bioorg Med Chem Lett. 2004;14(24):6083–7.
45 Munck JM, Batey MA, Zhao Y, Jenkins H, Richardson CJ, Cano C, et al. Chemosensitization of cancer cells by KU-0060648, a dual inhibitor of DNA-PK and PI-3K. Mol Cancer Ther. 2012;11(8):1789–98.
46 Dupré A, Boyer-Chatenet L, Sattler RM, Modi AP, Lee J-H, Nicolette ML, et al. A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat Chem Biol. 2008;4(2):119–25.
47 Ying S, Hamdy FC, Helleday T. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res. 2012;72(11):2814–21.
48 Neher TM, Bodenmiller D, Fitch RW, Jalal SI, Turchi JJ. Novel irreversible small molecule inhibitors of replication protein A display single-agent activity and synergize with cisplatin. Mol Cancer Ther. 2011;10(10):1796–806.
49 Huang F, Motlekar NA, Burgwin CM, Napper AD, Diamond SL, Mazin AV. Identification of specific inhibitors of human RAD51 recombinase using high-throughput screening. ACS Chem Biol. 2011;6(6):628–35.
50 Budke B, Logan HL, Kalin JH, Zelivianskaia AS, Cameron McGuire W, Miller LL, et al. RI-1: a chemical inhibitor of RAD51 that disrupts homologous recombination in human cells. Nucleic Acids Res. 2012;40(15):7347–57.
51 Nomme J, Renodon-Cornière A, Asanomi Y, Sakaguchi K, Stasiak AZ, Stasiak A, et al. Design of potent inhibitors of human RAD51 recombinase based on BRC motifs of BRCA2 protein: modeling and experimental validation of a chimera peptide. J Med Chem. 2010;53(15):5782–91.
52 Pessetto ZY, Yan Y, Bessho T, Natarajan A. Inhibition of BRCT(BRCA1)-phosphoprotein interaction enhances the cytotoxic effect of olaparib in breast cancer cells: a proof of concept study for synthetic lethal therapeutic option. Breast Cancer Res Treat. 2012;134(2):511–7.
53 Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–21.
54 Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–7.
55 Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376(9737):235–44.
56 Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376(9737):245–51.
57 Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4(10):814–9.
58 McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66(16):8109–15.
59 Huehls AM, Wagner JM, Huntoon CJ, Karnitz LM. Identification of DNA repair pathways that affect the survival of ovarian cancer cells treated with a poly(ADP-ribose) polymerase inhibitor in a novel drug combination. Mol Pharmacol. 2012;82(4):767–76.
60 Karanam K, Kafri R, Loewer A, Lahav G. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol Cell. 2012;47(2):320–9.
61 Park SR, Chen A. Poly(Adenosine diphosphate-ribose) polymerase inhibitors in cancer treatment. Hematol Oncol Clin North Am. 2012;26(3):649–70–ix.
62 Murai J, Huang S-YN, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012;72(21):5588–99.
63 O’Shaughnessy J, Osborne C, Pippen JE, Yoffe M, Patt D, Rocha C, et al. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N Engl J Med. 2011;364(3):205–14.
64 Guha M. PARP inhibitors stumble in breast cancer. Nat Biotechnol. 2011;29(5):373–4.
65 Liu X, Shi Y, Maag DX, Palma JP, Patterson MJ, Ellis PA, et al. Iniparib nonselectively modifies cysteine-containing proteins in tumor cells and is not a bona fide PARP inhibitor. Clin Cancer Res. 2012;18(2):510–23.
66 Patel AG, De Lorenzo SB, Flatten KS, Poirier GG, Kaufmann SH. Failure of iniparib to inhibit poly(ADP-Ribose) polymerase in vitro. Clin Cancer Res. 2012;18(6):1655–62.
67 Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12(9):852–61.
68 Mendes-Pereira AM, Martin SA, Brough R, McCarthy A, Taylor JR, Kim J-S, et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med. 2009;1(6-7):315–22.
69 Dedes KJ, Wetterskog D, Mendes-Pereira AM, Natrajan R, Lambros MB, Geyer FC, et al. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci Transl Med. 2010;2(53):53ra75.
70 Konstantinopoulos PA, Spentzos D, Karlan BY, Taniguchi T, Fountzilas E, Francoeur N, et al. Gene expression profile of BRCAness that correlates with responsiveness to chemotherapy and with outcome in patients with epithelial ovarian cancer. J Clin Oncol. 2010;28(22):3555–61.
71 Tung N, Wang Y, Collins LC, Kaplan J, Li H, Gelman R, et al. Estrogen receptor positive breast cancers in BRCA1 mutation carriers: clinical risk factors and pathologic features. Breast Cancer Res. 2010;12(1):R12.
72 Oplustilova L, Wolanin K, Mistrik M, Korinkova G, Simkova D, Bouchal J, et al. Evaluation of candidate biomarkers to predict cancer cell sensitivity or resistance to PARP-1 inhibitor treatment. Cell Cycle. 2012;11(20):3837–50.
73 Jaspers JE, Kersbergen A, Boon U, Sol W, van Deemter L, Zander SA, et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 2013;3(1):68–81.
74 Barber LJ, Sandhu S, Chen L, Campbell J, Kozarewa I, Fenwick K, et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J Pathol. 2013;229(3):422–9.
75 Poumpouridou N, Kroupis C. Hereditary breast cancer: beyond BRCA genetic analysis; PALB2 emerges. Clin Chem Lab Med. 2012;50(3):423–34.
76 Sultana R, McNeill DR, Abbotts R, Mohammed MZ, Zdzienicka MZ, Qutob H, et al. Synthetic lethal targeting of DNA double-strand break repair deficient cells by human apurinic/apyrimidinic endonuclease inhibitors. Int J Cancer. 2012;131(10):2433–44.
77 Ferrao PT, Bukczynska EP, Johnstone RW, McArthur GA. Efficacy of CHK inhibitors as single agents in MYC-driven lymphoma cells. Oncogene. 2012;31(13):1661–72.
78 Ivashkevich A, Redon CE, Nakamura AJ, Martin RF, Martin OA. Use of the γ-H2AX assay to monitor DNA damage and repair in translational cancer research. Cancer Lett. 2012;327(1-2):123–33.
79 Kennedy RD, Chen CC, Stuckert P, Archila EM, la Vega De MA, Moreau LA, et al. Fanconi anemia pathway-deficient tumor cells are hypersensitive to inhibition of ataxia telangiectasia mutated. J Clin Invest. 2007;117(5):1440–9.
80 Chen CC, Kennedy RD, Sidi S, Look AT, D’Andrea A. CHK1 inhibition as a strategy for targeting Fanconi Anemia (FA) DNA repair pathway deficient tumors. Mol Cancer. 2009;8:24.
81 Bartek J, Lukas J, Bartkova J. DNA damage response as an anti-cancer barrier: damage threshold and the concept of 'conditional haploinsufficiency'. Cell Cycle. 2007;6(19):2344–7.
82 Kerzendorfer C, O'Driscoll M. Human DNA damage response and repair deficiency syndromes: linking genomic instability and cell cycle checkpoint proficiency. DNA Repair (Amst.). 2009;8(9):1139–52.
83 Chen P-L, Liu F, Cai S, Lin X, Li A, Chen Y, et al. Inactivation of CtIP leads to early embryonic lethality mediated by G1 restraint and to tumorigenesis by haploid insufficiency. Mol Cell Biol. 2005;25(9):3535–42.
84 Brown EJ, Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000;14(4):397–402.
85 Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319(5868):1352–5.
86 Vollebergh MA, Jonkers J, Linn SC. Genomic instability in breast and ovarian cancers: translation into clinical predictive biomarkers. Cell Mol Life Sci. 2012;69(2):223–45.
87 Martin SA, McCabe N, Mullarkey M, Cummins R, Burgess DJ, Nakabeppu Y, et al. DNA polymerases as potential therapeutic targets for cancers deficient in the DNA mismatch repair proteins MSH2 or MLH1. Cancer Cell. 2010;17(3):235–48.
88 Zhang X, Wan G, Berger FG, He X, Lu X. The ATM kinase induces microRNA biogenesis in the DNA damage response. Mol Cell. 2011;41(4):371–83.
89 Wang Y, Huang J-W, Li M, Cavenee WK, Mitchell PS, Zhou X, et al. MicroRNA-138 modulates DNA damage response by repressing histone H2AX expression. Mol Cancer Res. 2011;9(8):1100–11.
90 Landau D-A, Slack FJ. MicroRNAs in mutagenesis, genomic instability, and DNA repair. Semin Oncol. 2011;38(6):743–51.
91 Moskwa P, Buffa FM, Pan Y, Panchakshari R, Gottipati P, Muschel RJ, et al. miR-182-mediated downregulation of BRCA1 impacts DNA repair and sensitivity to PARP inhibitors. Mol Cell. 2011;41(2):210–20.
92 Chang S, Wang R-H, Akagi K, Kim K-A, Martin BK, Cavallone L, et al. Tumor suppressor BRCA1 epigenetically controls oncogenic microRNA-155. Nat Med. 2011;17(10):1275–82.
93 Jiang S, Zhang H-W, Lu M-H, He X-H, Li Y, Gu H, et al. MicroRNA-155 functions as an OncomiR in breast cancer by targeting the suppressor of cytokine signaling 1 gene. Cancer Res. 2010;70(8):3119–27.
94 Broderick JA, Zamore PD. MicroRNA therapeutics. Gene Ther. 2011;18(12):1104–10.
95 Li Y, Yang D-Q. The ATM inhibitor KU-55933 suppresses cell proliferation and induces apoptosis by blocking Akt in cancer cells with overactivated Akt. Mol Cancer Ther. 2010;9(1):113–25.
96 Charrier J-D, Durrant SJ, Golec JMC, Kay DP, Knegtel RMA, MacCormick S, et al. Discovery of potent and selective inhibitors of ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase as potential anticancer agents. J Med Chem. 2011;54(7):2320–30.
97 Zhao Y, Thomas HD, Batey MA, Cowell IG, Richardson CJ, Griffin RJ, et al. Preclinical evaluation of a potent novel DNA-dependent protein kinase inhibitor NU7441. Cancer Res. 2006;66(10):5354–62.
98 Willmore E, de Caux S, Sunter NJ, Tilby MJ, Jackson GH, Austin CA, et al. A novel DNA-dependent protein kinase inhibitor, NU7026, potentiates the cytotoxicity of topoisomerase II poisons used in the treatment of leukemia. Blood. 2004;103(12):4659–65.
99 Tang Y, Hamed HA, Poklepovic A, Dai Y, Grant S, Dent P. Poly(ADP-ribose) polymerase 1 modulates the lethality of CHK1 inhibitors in mammary tumors. Mol Pharmacol. 2012;82(2):322–32.
100 Huang F, Mazina OM, Zentner IJ, Cocklin S, Mazin AV. Inhibition of homologous recombination in human cells by targeting RAD51 recombinase. J Med Chem. 2012;55(7):3011–20.
101 Samol J, Ranson M, Scott E, Macpherson E, Carmichael J, Thomas A, et al. Safety and tolerability of the poly(ADP-ribose) polymerase (PARP) inhibitor, olaparib (AZD2281) in combination with topotecan for the treatment of patients with advanced solid tumors: a phase I study. Invest New Drugs. 2012;30(4):1493–500.
102 Kummar S, Ji J, Morgan R, Lenz H-J, Puhalla SL, Belani CP, et al. A phase I study of veliparib in combination with metronomic cyclophosphamide in adults with refractory solid tumors and lymphomas. Clin Cancer Res. 2012;18(6):1726–34.
103 Kummar S, Chen A, Ji J, Zhang Y, Reid JM, Ames M, et al. Phase I study of PARP inhibitor ABT-888 in combination with topotecan in adults with refractory solid tumors and lymphomas. Cancer Res. 2011;71(17):5626–34.
104 Patel AG, Flatten KS, Schneider PA, Dai NT, McDonald JS, Poirier GG, et al. Enhanced killing of cancer cells by poly(ADP-ribose) polymerase inhibitors and topoisomerase I inhibitors reflects poisoning of both enzymes. J Biol Chem. 2012;287(6):4198–210.
105 Plummer R, Lorigan P, Steven N, Scott L, Middleton MR, Wilson RH, et al. A phase II study of the potent PARP inhibitor, Rucaparib (PF-01367338, AG014699), with temozolomide in patients with metastatic melanoma demonstrating evidence of chemopotentiation. Cancer Chemother Pharmacol. 2013;71(5):1191–9.
106 Sandhu SK, Wenham RM, Wilding G, McFadden M, Sun L, Toniatti C, et al. First-in-human trial of a poly (ADP-ribose) polymerase (PARP) inhibitor MK-4827 in advanced cancer patients (pts) with antitumor activity in BRCA-deficient and sporadic ovarian cancers. J Clin Oncol. (Meeting Abstracts). 2010;28(15_suppl):3001.
107 Javle M, Curtin NJ. The role of PARP in DNA repair and its therapeutic exploitation. Br J Cancer. 2011;105(8):1114–22.