Surfactant and allergic airway inflammation
DOI: https://doi.org/10.4414/smw.2013.13818
Carla
Winkler, Jens Michael
Hohlfeld
Abstract
Pulmonary surfactant is a complex mixture of unique proteins and lipids that covers the airway lumen. Surfactant prevents alveolar collapse and maintains airway patency by reducing surface tension at the air-liquid interface. Furthermore, it provides a defence against antigen uptake by binding foreign particles and enhancing cellular immune responses. Allergic asthma is associated with chronic airway inflammation and presents with episodes of airway narrowing. The pulmonary inflammation and bronchoconstriction can be triggered by exposure to allergens or pathogens present in the inhaled air. Pulmonary surfactant has the potential to interact with various immune cells which orchestrate allergen- or pathogen-driven episodes of airway inflammation. The complex nature of surfactant allows multiple sites of interaction, but also makes it susceptible to external alterations, which potentially impair its function. This duality of modulating airway physiology and immunology during inflammatory conditions, while at the same time being prone to alterations accompanied by restricted function, has stimulated numerous studies in recent decades, which are reviewed in this article.
Introduction
Pulmonary surfactant, which is composed of a complex mixture of phospholipids and proteins, lines the enormous alveolar surface at the air-liquid interface. It mainly reduces surface tension in the alveolus, thus stabilising the alveoli at end-expiration. Furthermore, it helps to keep the airways open and clean, and also regulates pulmonary host defence and immune responses. Surfactant is synthesised, assembled and finally released by type II pneumocytes present in the alveoli. Besides being present in the alveolar region, surfactant also lines the conducting airways, but with diminished surface tension-reducing properties [1].
Its primary function to reduce surface tension at the air-liquid interface was initially postulated in the 1920s by von Neergaard, whose findings suggested the presence of a “surface-active agent” for the first time [2]. It was not until the 1950s, however, that the unique properties and detailed composition of pulmonary surfactant were unravelled. At that time, a causal link between respiratory distress syndrome in newborn infants and pulmonary surfactant deficiency was established. This discovery emphasised the important contribution of pulmonary surfactant to the maintainence of lung integrity through stabilisation of the alveoli during lung ventilation. Thirty years later, Enhorning demonstrated in rabbit neonates that pulmonary surfactant was also essential for keeping the conducting airways open and preventing the alveoli from overexpanding [3]. Hitherto, several lung diseases have been described as associated with surfactant dysfunction, and therapies with surfactant preparations have been suggested for nonrespiratory-distress conditions [4, 5].
One focus of interest has been the biophysical properties of surfactant, which are mainly mediated by its phospholipid fraction. In addition, in recent decades the soluble surfactant proteins (SPs), SP-A and SP-D, have attracted more and more interest, because they participate in host defence and the immune responses of the lung [6].
In the context of allergic asthma, several studies have highlighted interactions of the soluble surfactant proteins with allergens and effector cells during allergic airway inflammation [7–10]. In addition, experimental asthma models in mice with either SP-A or SP-D deficiency revealed the importance of these proteins in dampening the allergic airway response. These mice have been shown to be more susceptible to induction of acute allergic airway inflammation, which results in a more severe inflammatory phenotype [11].
Vice versa, inflammatory mediators, proteases and reactive oxygen species, which are released during allergic inflammation, induce structural alterations in pulmonary surfactant components [12, 13]. These changes either lead to surfactant dysfunction or are associated with impaired immune function.
This review will discuss the effects of pulmonary surfactant components on the allergic airway inflammation and will also address implications of the allergic airway inflammatory response for constituents of the pulmonary surfactant system.
Pulmonary surfactant composition
Pulmonary surfactant is 80%–85% phospholipids and 5%–10% neutral lipids (mainly cholesterol). Besides lipids, a minor proportion is protein (5%–6%), which includes the four surfactant-associated proteins: SP-A, SP-B, SP-C and SP-D. Phospholipids have an amphipathic structure with a polar (hydrophilic) head and a nonpolar, hydrophobic, fatty acid chain, and self-organise into bilayers. At polarised interfaces like the air-liquid barrier in the alveoli, phospholipids form a monolayer in which the polar head groups are oriented towards the liquid phase and the hydrophobic fatty acid chains face the gaseous phase, that is, the airway lumen [14].
The phospholipid fraction of surfactant comprises different molecular species, with phosphatidylcholines being the most abundant. Disaturated dipalmitoyl-phosphotidylcholine accounts for more than 50% of total phosphotidylcholine and is significantly involved in achieving maximum reduction of surface tension at the air-liquid interface [15].
The term surfactant-associated protein is most applicable to the small hydrophobic proteins SP-B and SP-C (and to a minor extent also to SP-A), as they closely assemble with and within the surfactant film of phospholipids [16]. Mature SP-B is composed of 80 amino acid residues and exists as dimers permanently associated with the phospholipid monolayer, preferentially in highly disordered regions. It has been shown to be the most important surfactant protein for surfactant structure and function, as it is essential for packaging phospholipids into the lamellar bodies [17]. Furthermore, it regulates formation of tubular myelin after exocytosis and formation of the surfactant film [18]. Importantly, SP-B deficiency is lethal, in contrast to deficiency of the other surfactant proteins [19, 20]. The smallest surfactant protein, SP-C, which is hydrophobic and 35 amino acid residues in size, has a transmembrane orientation within the surfactant layer. SP-C interacts with the acylated phospholipid side chains and thereby stabilises the surfactant film [18, 21, 22].
Unlike SP-B and SP-C, SP-A and SP-D molecules are hydrophilic and occur as multimeric proteins consisting of monomers of 38 kDa and 43 kDa, respectively [23, 24]. In contrast to SP-D, SP-A is encoded by two genes, SP-A1and SP-A2. Both gene products have several polymorphic variants, and differences in structure and function at the protein level [25]. SP-A and SP-D belong to the family of collectins and comprise four analogue regions: the N-terminal cysteine-rich region is attached to a distinct collagen-like region and a short alpha-coiled neck region. Located at the C-terminus is a so-called carbohydrate recognition domain [26]. The carbohydrate recognition domain mediates binding to various ligands exhibiting glycosylation motifs (pathogens, lipids, cells, receptors) in a Ca2+-dependent manner [27]. SP-A and SP-D monomers undergo trimerisation by disulphide crosslinking within their N-terminal domain; further multimerisation is mediated by hydrogen bonding. Multimerised SP-D adopts a cruciform-like shape, whereas SP-A trimers assemble into a flower bouquet-like structure [6]. Carbohydrate recognition domain binding affinity depends on the degree of protein multimerisation, as less multimerised SP-D molecules possess weaker binding affinities to pathogens [28–31].
Pulmonary surfactant synthesis
Type II pneumocytes present in the distal airways synthesise and secrete surfactant phospholipids and proteins. Synthesis takes place in the endoplasmic reticulum, with further processing by the Golgi apparatus [32]. Surfactant components are tightly packaged and stored in the so-called lamellar bodies, which are specific intracellular storage organelles. These lamellar bodies undergo exocytosis and release the surfactant components into the aqueous hypophase between the lung epithelium and the gaseous phase [33]. After exocytosis, phospholipids form tubular myelin, a network of phospholipid bilayers and proteins that replenish the air-liquid interface with surfactant components [15].
SP-B and SP-C are associated and exocytosed with lamellar bodies. In contrast, SP-A and SP-D are secreted from type II pneumocytes via a lamellar body-independent pathway [34]. SP-A and SP-D are also released by Clara cells in the more proximal airways and are additionally present at extrapulmonary sites [35–37].
Surfactant components removed from the surfactant film appear as small and inactive aggregates. After reuptake by type II pneumocytes, they are either catabolised or recycled by being reincorporated into the metabolic pathway of synthesis [38]. A minor fraction is phagocytosed and degraded by alveolar macrophages, and some surfactant is also cleared through the airways by mucocillilary clearance [39, 40].
How surfactant affects allergic inflammation
The course of the airway response upon allergen challenge in sensitised individuals can be divided into an early phase and a late phase. The early phase develops very rapidly after allergen exposure and is characterised by mast cell degranulation accompanied by histamine and leukotriene release, leading first and foremost to effects such as bronchoconstriction and mucus hypersecretion. The late phase develops hours after allergen contact, with severe inflammation caused by activation of allergen-specific effector cells and a massive influx of eosinophils into the airways. These effector cells in turn maintain an inflammatory environment in the lung, leading to airway remodelling and airway obstruction [41].
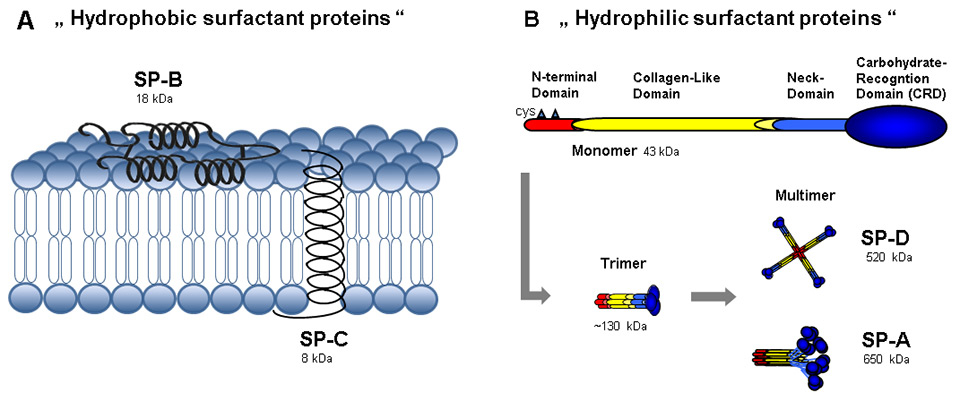
Figure 1
(A) The hydrophobic surfactant proteins B and C are small proteins with α-helices. SP-B is arranged in monolayers or bilayers on the surface of surfactant lipids. Together with SP-C, which is integrated into the phospholipid membrane, they facilitate the spreading and adsorption of lipids into the phospholipid membrane and enhance its stability. Thereby, they greatly contribute to the reduction of surface tension in the alveoli. (B) The hydrophilic surfactant proteins SP-A and SP-D are large multimeric proteins which are assembled from monomers with cysteinyl (cys) residues in the N-terminal domain. These monomers have a characteristic carbohydrate recognition domain in their C-terminal ending, with distinctive binding affinities to glycosylated structures. Trimers are established by covalent disulphide bridging of three monomers, which subsequently assemble into higher multimeric molecules.
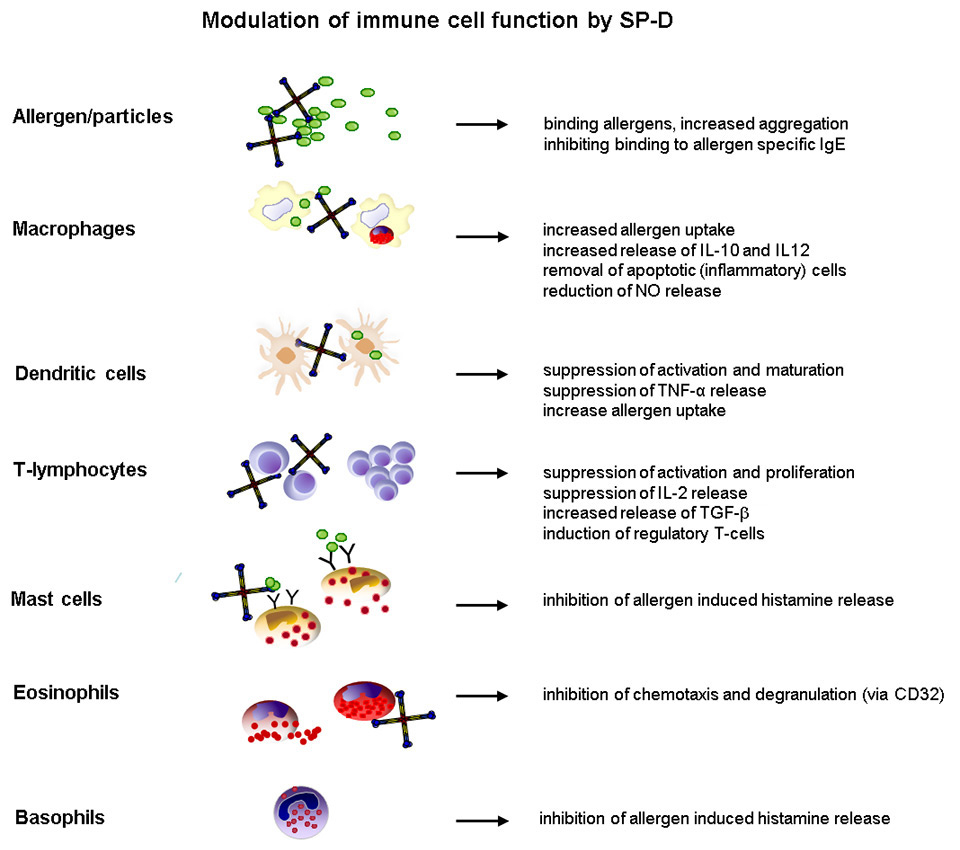
Figure 2
Overview of interactions between surfactant protein-D and immune effector cells, which orchestrate the pulmonary immune response during the onset of allergic airway inflammation.
Binding of allergens/allergen particles
A prerequisite for initiation of acute allergic airway inflammation is inhalation and deposition of airborne allergens / allergen particles in the lower respiratory tract. In this compartment, allergens come into contact with the surfactant-containing air-liquid interface that covers the epithelium of the airway mucosa underneath. Effective clearance of allergens is essential to avoid initiation of inflammation and/or to dampen it.
In this process, SP-A and SP-D play a crucial role. SP-A and SP-D have been shown to bind effectively a common major allergen derived from house dust mite (Dermatophagoides pteronyssinus; Der p1) [42]. Nitric oxide (NO) release from Der p1-stimulated macrophages was subsequently inhibited in the presence of SP-D [43]. SP-D also bound and aggregated pollen starch granules from various grass species [44, 45]. Pollen starch granules are released from plant pollen and carry several major allergens on their surface. Importantly, cellular clearance of these particles by alveolar macrophages was augmented in vitro in the presence of SP-D [44]. In an in-vivo study using SP-D knock-out mice, treatment with recombinant SP-D accelerated the uptake and binding of pollen starch granules by alveolar macrophages. However, overall lung clearance of instilled pollen starch granules was not affected, indicating that SP-D mainly contributes to cellular clearance mechanisms [46]. In contrast to SP-D, the natural porcine-derived surfactant preparation Curosurf® (which contains phospholipids and hydrophobic SP-B and SP-C, but no SP-A or SP-D) has been shown to promote mucociliary clearance in dogs [47].
Binding and aggregation of free allergens also reduces mast cell activation induced by allergen binding to specific IgE on the surface of these cells. Their crosslinking through allergens finally initiates mast cell activation and degranulation. SP-D was shown to inhibit degranulation of mast cells and release of histamine, a potent mediator of bronchoconstriction, by binding and aggregating allergens [45]. Similarly, SP-A and SP-D inhibited binding of IgE to Aspergillus fumigatus and subsequent histamine release from basophils [48]. These observations indicate that surfactant proteins may regulate the severity of the early-phase reaction due to allergen binding.
Exacerbations of allergic airway inflammation can result from additional bacterial or viral infections. Among other environmental factors, bacterial or viral infections can trigger exacerbations of allergic asthma. Besides airborne allergens, SP-A and SP-D possess high binding-affinity to microbial pathogens and viruses (such as Streptococcus pneumoniae, Mycoplasma pneumonia and respiratory syncytial virus), and promote their clearance by alveolar macrophages [49–51]. This mechanism might also contribute to a reduction in the risk of pathogen-induced exacerbations of allergic airway inflammation.
Modulation of effector cell functions
Antigen-presenting cells
Pulmonary antigen-presenting cells represent the key effector cells for the uptake of inhaled antigens and their presentation to T-lymphocytes. Thus, they contribute to the induction of sensitisation or initiation of acute allergic airway inflammation. Antigen-presenting cells comprise different subsets of dendritic cells and macrophages, although only dendritic cells efficiently present antigens to naïve CD4+ T-lymphocytes [52]. SP-D and SP-A were shown to inhibit dendritic cell maturation, an important requirement for antigen presentation and pro-inflammatory tumour necrosis factor-alpha (TNF-α) release [53, 54]. Additionally, dendritic cells and macrophages from SP-D knock-out mice exhibited enhanced TNF-α release and also up-regulation of CD86 and MHCII molecules when compared with wild-type mice [54].
The role of surfactant proteins in modulating the antigen-presenting functions of dendritic cells is diverse. The presence of SP-A in a dendritic cell culture had an inhibitory effect on allostimulation of T-cells, whereas SP-D augmented presentation of bacterial antigen by bone marrow-derived dendritic cells [53, 55]. Furthermore, SP-D reduced the interaction of Der p1 with monocyte-derived dendritic cells [56]. When using antigen-presenting cells isolated from the lung, the opposite effect was observed for SP-D, indicating that the origin and subset of dendritic cells might matter [57].
Given that mucosal dendritic cells under noninflamed conditions sample the airway lumen mainly with their dendrites, it remains unclear to what extent surfactant proteins can modulate residential dendritic cell function, because of spatial compartmentalisation. During inflammation, however, when the epithelium is damaged, interaction between invading inflammatory dendritic cells and pulmonary SP-D might be increased. This, in turn, could then modulate and dampen dendritic cell activation and maturation.
Several interactions of surfactant proteins with alveolar macrophages have been described. SP-A and SP-D bind to multiple surface receptors such as Toll-like receptors (TLRs) 2 and 4, CD14, signal regulatory protein-alpha (SIRPα) and C1q [53, 58–61], and thereby modulate alveolar macrophage function. In allergic airway inflammation, SP-D has been observed to inhibit Der p1 allergen-induced NO release from macrophages triggered by activation of the TLR4/CD14 complex [43]. In further studies, interactions of SP-A and SP-D with these pattern recognition receptors reduced the release of proinflammatory cytokines in response to peptidoglycans and lipopolysaccharide [59, 60]. Alveolar macrophages isolated from mice after allergen challenge released increased amounts of interleukins (ILs) 10 and 12, and interferon-gamma (INF-γ) after restimulation with allergen in the presence of SP-D [10]. This emphasises the importance of SP-A and SP-D in regulating allergen-induced inflammatory responses via macrophage functions. Besides regulation of cytokines and NO release, the presence of surfactant proteins also contributes to macrophage phagocytosis of pathogens, allergens and apoptotic cells from the airway lumen [46, 62, 63]. In detail, C-terminal binding of lung collectins to the SIRPα receptor was shown to inhibit phagocytosis of apoptotic cells under noninflamed conditions. In contrast, N-terminal binding to CD91/calreticulin induced release of proinflammatory cytokines. Furthermore, phagocytosis was increased during inflammation [58].
Lymphocytes
T-lymphocyte activation by antigen-presenting cells mainly takes place in the draining lymph nodes and results in proliferation of antigen-specific CD4+ T-lymphocytes. During the onset of allergic inflammation, these cells release large amounts of IL-4, IL-5 and eotaxin, orchestrating the invasion of the lung by effector cells (predominantly eosinophils and mast cells). Furthermore, lymphocytes migrate into the lung tissue upon epithelial expression of thymus- and activation-regulated cytokine (TARC) [64]. Various studies have explored the role of surfactant proteins in T-lymphocyte proliferation. Surfactant proteins have a suppressive effect on T-lymphocyte function. For example, T-lymphocyte proliferation in response to nonspecific stimuli such as mitogens and to specific allergens was inhibited in the presence of SP-A and SP-D [65–67]. This was mirrored by reduced IL-2 release by T-lymphocytes in the presence of both proteins [65].
Moreover, SP-A and SP-D suppressed the allergen-specific proliferative response in a mixed lymphocyte preparation of blood from asthmatic children [68]. The mechanism by which surfactant proteins regulate T-lymphocyte proliferation remains unclear, since evidence for surfactant protein-specific receptors on T-lymphocytes is fragmentary. One study identified the high-affinity SP-A receptor SP-R210 as a receptor for N-terminal binding of SP-A, which inhibits T-cell proliferation [69]. A later investigation identified SP-R210 as unconventional myosin 18A, but the downstream signalling pathway of its activation by SP-A binding remains unknown [70].
According to recent findings, the signal strength inducing T-lymphocyte proliferation determines whether SP-A promotes or inhibits proliferation, since SP-A effectively suppressed T-lymphocyte proliferation afer a strong stimulus [71]. Interestingly, SP-A enhanced the frequency of regulatory T-cells in extended lymphocyte cultures by inducing transforming growth factor-beta (TGF-β) [9].
Expression of the cytotoxic T-lymphocyte antigen CTLA4, a negative regulator of T-lymphocytes, was shown to be induced in the presence of SP-D [72]. This study suggests that SP-D might actively regulate T-lymphocyte proliferation at the gene level.
Eosinophils
Infiltration of the lung tissue with eosinophils is a hallmark of allergic airway inflammation. These key effector cells differentiate from the bone marrow in response to IL-5 and migrate to the lung. Upon activation and degranulation, eosinophils release high amounts of proinflammatory cytokines and cytotoxic proteins such as major basic protein (MBP), eosinophilic cationic protein (ECP) and eosinophilic peroxidase (EPO), and reactive oxygen species [73]. SP-A inhibited ionomycin-induced IL-8 release from eosinophils [74]. SP-D bound to eosinophils and inhibited eosinophilic chemotaxis towards eotaxin, as well as ECP degranulation of activated eosinophils [75]. Additionally, SP-D increased the uptake by a macrophage cell line of apoptotic eosinophils from asthmatic donors [76], which had been shown before for apoptotic neutrophils [63]. These studies illustrated the important modulatory role of pulmonary surfactant proteins during the resolution phase of pulmonary inflammation. However, local pulmonary application of the natural porcine-derived surfactant preparation, Curosurf®, in asthma patients after segmental allergen challenge was found to accelerate the eosinophilic inflammation. Eotaxin and IL-5 levels, as well as ECP release in bronchoalveolar lavage (BAL) were significantly increased in response to Curosurf® instillation [77]. Further in-vitro analyses revealed that Curosurf® indeed induced apoptosis and necrosis of eosinophils, accompanied by release of high levels of ECP and leukotriene C4 (LTC4) [78].
How allergic inflammation affects pulmonary surfactant
Alteration of surfactant protein expression
Besides modulating the immune response underlying allergic airway inflammation, pulmonary surfactant components have been found to be influenced and modified by the presence of activated effector cells and by the inflammatory cytokine milieu. SP-B, SP-C and SP-D levels were increased in BAL of allergic individuals after segmental allergen challenge, while SP-A levels were found to be reduced [79]. Similar observations have been made in animal models of allergic airway inflammation [80–82]. No alterations of surfactant proteins were observed in nonasthmatic patients, indicating that the regulation of surfactant proteins is a specific response that depends on the cytokine milieu during allergy-induced inflammation. Furthermore, serum levels of SP-D and SP-A in allergic subjects were elevated compared with those of healthy controls, and correlated with ECP concentrations in sputum after allergen challenge [83]. However, the source of increased serum SP-D levels (extrapulmonary secretion or leakage from the lung into the circulation) remains unknown.
Data from IL-4- and IL-13-overexpressing mice indicated that both of these type 2 T-cell helper (Th2) cytokines augmented SP-A, SP-B, SP-C and SP-D expression in type II pneumocytes [84]. Furthermore, chronic IL-13 exposure induced hypertrophy of type II pneumocytes and augmented phospholipid release [85]. In-vitro experiments, however, revealed conflicting results. While IL-13 and IL-4 stimulation of type II pneumocytes from neonatal rats induced an increase in SP-D expression [82, 86], a recent study could not confirm these results [87].
Alteration of surfactant components
Besides altered expression levels, inflammatory mediators such as proteases or reactive oxygen species can structurally modify components of pulmonary surfactant, leading to surfactant (protein) dysfunction.
In a model of segmental allergen provocation, we showed that airway inflammation induced surfactant dysfunction [88]. Vascular leakage of plasma proteins into the airways coincided with an elevated ratio of small (biophysically inactive) to large surfactant aggregates. Furthermore, the phosphatidylcholine composition of BAL surfactant was altered, resulting in lower surface tension at the air-liquid interface [89].
Besides plasma proteins, eosinophil-derived secretory phospholipase was shown to alter the phosphatidylglycerol content in large surfactant aggregates, and this correlated with the degree of surfactant dysfunction [90]. Additionally, ECP caused severe alterations of the ultrastructural composition of surfactant vesicles [91].
Several posttranslational modifications of SP-D as a result of reactive oxygen and nitrogen species released from activated macrophages, epithelial cells, and eosinophils have been identified [92]. Two cysteinyl residues (cys15/cys20) in the N-terminal domain of SP-D control oligomerisation by means of disulphide bridging. This was elegantly demonstrated by replacing these residues with serinyl residues, which hampered further oligomerisation [28]. Located in a hydrophobic region of the protein, these cysteinyl residues are particularly susceptible to oxidation and nitrosylation by reactive oxygen and nitrogen species. Nitrosylation induced a loss of the disulphide bonds and thereby promoted disintegration of the multimer, accompanied by impaired function of the protein [30].
Interestingly, S-nitrosylated SP-D molecules, in contrast to native SP-D, mediated proinflammatory cytokine release and chemotaxis of macrophages, and no longer prevented eotaxin-induced chemotaxis of eosinophils [13, 93]. Furthermore, we have shown that the severity of allergic inflammation correlated with disruption of the quaternary structure of SP-D. In addition, appearance of peroxynitrite induced covalently crosslinked SP-D isoforms of 100–200 kDa [13]. Others have demonstrated that oxidative damage to SP-D similarly resulted in less multimerisation, leading to a loss of effective bacteria agglutination capacity [94].
SP-A isolates from asthmatic patients were defective in abrogating the inflammatory response of stimulated epithelial cells in vitro. Further, these isolates exhibited a higher proportion of the lower multimeric SP-A1 gene variant product [95]. In addition to posttranslational modifications, surfactant proteins were also susceptible to the proteolytic activity of, for example, allergens. The proteases Der p1 and Der f1 induced degradation of SP-D, which resulted in its complete functional inactivation [96].
Therapeutic potential
Due to the fact that the hydrophilic surfactant proteins mediate and dampen several effector cell functions, their therapeutic use in allergic asthma has been explored in several animal studies.
In a model ofAspergillus fumigatus-induced allergic bronchopulmonary aspergillosis, treatment of mice with SP-A or SP-D reduced blood eosinophila, pulmonary infiltration of effector cells and specific antibody titres in blood [8]. Our group has, furthermore, shown that treatment with a human SP-D fragment before allergen challenge with Aspergillus fumigatus inhibits the early airway response and airway hyperresponsivness [7]. Studying SP-A- and SP-D-deficient mice revealed increased BAL eosinophils, and IL-5 and IL-13 levels compared with wild-type animals, indicating a Th2 shift of the immune response [8]. In a model of allergic asthma, SP-D-deficient mice revealed increased numbers of eosinophils and IL-13 levels in the lung, whereas INF-γ was reduced compared with wild-type animals [97]. Moreover, eosinophilia was markedly reduced after treatment with either a truncated trimeric recombinant fragment of human SP-D or recombinant rat SP-D in a mouse model of asthma [98; 99]. Another study using an animal asthma model showed that SP-D treatment normalised airway function, reduced goblet cell hyperplasia and induced production of regulatory cytokines such as IL-10, IL-12 and INF-γ by macrophages [10]. These findings demonstrate that SP-D beneficially influences allergic inflammation in the airways.
Although increasing evidence from animal models indicates that SP-A and SP-D treatment dampens allergic airway inflammation, these proteins are not available for clinical trials in humans at present. This is in contrast to exogenous surfactant preparations, which are well established in, for example, the treatment of respiratory distress syndrome in infants. Evaluation of these preparations in models of airway obstruction resulted in an inhibition of acetylcholine-induced airway obstruction. This was shown in rats and guinea pigs by prior application of the aerosolised surfactant preparations Alveofact® and Surfactant TA, respectively [100, 101]. A recent study indicated that administration of a natural exogenous surfactant (Infasurf®) in an ovalbumin rat model reduced airway narrowing after allergen challenge. Release of mast cell-derived mediators such as cysteinyl-leukotrienes and amphiregulin was also reduced [102].
The use of surfactant preparations in asthmatic patients revealed conflicting results. Beneficial effects on respiratory functions have been reported after use of aerosolised dry-powder synthetic phospholipids in humans during an asthma attack [103]. Inhalation of two doses of synthetic phospholipid dry-powder surfactant (Pumacant®) prior to allergen challenge also prevented the early asthmatic response [104]. In contrast, instillation of Curosurf® prior to segmental allergen provocation in asthmatic patients elevated eosinophil numbers in BAL. In addition, eosinophilia was accompanied by increased levels of Th2-promoting mediators such as eotaxin and IL-5 [77].
Conclusion
In summary, the pulmonary surfactant system is essential for reducing surface tension at the air-liquid interface and for regulating pulmonary immune responses. The soluble surfactant proteins in particular bind to and regulate a variety of immune/effector cells present in the course of allergic airway inflammation. Both SP-A and SP-D dampen the allergic inflammation, making them interesting molecules for treatment of allergic asthma. Administration of SP-A and SP-D would also replenish the pool of surfactant proteins that become inactivated owing to structural modifications induced by inflammatory mediators.
Finally, in patients with asthma administration of synthetic surfactant preparations (lacking SP-A and SP-D for human use) has been shown to improve lung function, but at the same time to worsen the underlying allergic immune response. Theoretically, development of synthetic surfactant preparations including SP-A and SP-D proteins might be a promising way to establish a surfactant-based therapy in allergic asthma. It has to be evaluated, however, whether or not pharmaceutical production of such complex surfactant preparations is economically feasible.
References
1 Bernhard W, Haagsman HP, Tschernig T, Poets CF, Postle AD, van Eijk ME, et al. Conductive airway surfactant: surface-tension function, biochemical composition, and possible alveolar origin. Am J Respir Cell Mol Biol. 1997;17(1):41–50.
2 von Neergaard K. Neue Auffassungen über einen Grundbegriff der Atemmechanik: die Retraktionskraft der Lunge, abhängig von der Oberflächenspannung der Alveolen. Z Ges Exp Med. 1929;66:373–94.
3 Enhorning G. Photography of peripheral pulmonary airway expansion as affected by surfactant. J Appl Physiol. 1977;42(6):976–9.
4 Whitsett JA, Wert SE, Weaver TE. Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease. Annu Rev Med. 2010;61:105–19.
5 Erpenbeck VJ, Krug N, Hohlfeld JM. Therapeutic use of surfactant components in allergic asthma. Naunyn Schmiedebergs Arch Pharmacol. 2009;379(3):217–24.
6 Wright JR. Immunoregulatory functions of surfactant proteins. Nat Rev Immunol. 2005;5(1):58–68.
7 Erpenbeck VJ, Ziegert M, Cavalet-Blanco D, Martin C, Baelder R, Glaab T, et al. Surfactant protein D inhibits early airway response in Aspergillus fumigatus-sensitized mice. Clin Exp Allergy. 2006;36(7):930–40.
8 Madan T, Kishore U, Singh M, Strong P, Clark H, Hussain EM, et al. Surfactant proteins A and D protect mice against pulmonary hypersensitivity induced by Aspergillus fumigatus antigens and allergens. J Clin Invest. 2001;107(4):467–75.
9 Mukherjee S, Giamberardino C, Thomas JM, Gowdy K, Pastva AM, Wright JR. Surfactant Protein A Modulates Induction of Regulatory T Cells via TGF-beta. J Immunol. 2012;188(9):4376–84.
10 Takeda K, Miyahara N, Rha YH, Taube C, Yang ES, Joetham A, et al. Surfactant protein D regulates airway function and allergic inflammation through modulation of macrophage function. Am J Respir Crit Care Med. 2003;168(7):783–9.
11 Madan T, Reid KB, Singh M, Sarma PU, Kishore U. Susceptibility of mice genetically deficient in the surfactant protein (SP)-A or SP-D gene to pulmonary hypersensitivity induced by antigens and allergens of Aspergillus fumigatus. J Immunol. 2005;174(11):6943–54.
12 Atochina-Vasserman EN, Beers MF, Gow AJ. Review: Chemical and structural modifications of pulmonary collectins and their functional consequences. Innate Immun. 2010;16(3):175–82.
13 Atochina-Vasserman EN, Winkler C, Abramova H, Schaumann F, Krug N, Gow AJ, et al. Segmental allergen challenge alters multimeric structure and function of surfactant protein D in humans. Am J Respir Crit Care Med. 2011;183(7):856–64.
14 Serrano AG, Perez-Gil J. Protein-lipid interactions and surface activity in the pulmonary surfactant system. Chem Phys. Lipids. 2006;141(1–2):105–18.
15 Perez-Gil J. Structure of pulmonary surfactant membranes and films: the role of proteins and lipid-protein interactions. Biochim. Biophys Acta. 2008;1778(7-8):1676–95.
16 Whitsett JA, Weaver TE. Hydrophobic surfactant proteins in lung function and disease. N Engl J Med. 2002;347(26):2141–8.
17 Foster CD, Zhang PX, Gonzales LW, Guttentag SH. In vitro surfactant protein B deficiency inhibits lamellar body formation. Am J Respir Cell Mol Biol. 2003;29(2):259–66.
18 Stahlman MT, Gray MP, Falconieri MW, Whitsett JA, Weaver TE. Lamellar body formation in normal and surfactant protein B-deficient fetal mice. Lab Invest. 2000;80(3):395–403.
19 Clark JC, Wert SE, Bachurski CJ, Stahlman MT, Stripp BR, Weaver TE, et al. Targeted disruption of the surfactant protein B gene disrupts surfactant homeostasis, causing respiratory failure in newborn mice. Proc Natl Acad Sci. U S A. 1995;92(17):7794–8.
20 Melton KR, Nesslein LL, Ikegami M, Tichelaar JW, Clark JC, Whitsett JA, et al. SP-B deficiency causes respiratory failure in adult mice. Am J Physiol Lung Cell Mol Physiol. 2003;285(3):L543–9.
21 Weaver TE, Conkright JJ. Function of surfactant proteins B and C. Annu Rev Physiol. 2001;63:555–78.
22 Beers MF, Mulugeta S. Surfactant protein C biosynthesis and its emerging role in conformational lung disease. Annu Rev Physiol. 2005;67:663–96.
23 Crouch E, Persson A, Chang D, Heuser J. Molecular structure of pulmonary surfactant protein D (SP-D). J Biol Chem. 1994;269(25):17311–9.
24 McCormack FX. Structure, processing and properties of surfactant protein A. Biochim Biophys Acta. 1998;1408(2-3):109–31.
25 Wang G, Myers C, Mikerov A, Floros J. Effect of cysteine 85 on biochemical properties and biological function of human surfactant protein A variants. Biochemistry. 2007;46(28):8425–35.
26 Kishore U, Greenhough TJ, Waters P, Shrive AK, Ghai R, Kamran MF, et al. Surfactant proteins SP-A and SP-D: structure, function and receptors. Mol Immunol. 2006;43(9):1293–315.
27 Veldhuizen EJ, van EM, Haagsman HP. The carbohydrate recognition domain of collectins. FEBS J. 2011;278(20):3930–41.
28 Brown-Augsburger P, Hartshorn K, Chang D, Rust K, Fliszar C, Welgus HG, et al. Site-directed mutagenesis of Cys-15 and Cys-20 of pulmonary surfactant protein D. Expression of a trimeric protein with altered anti-viral properties. J Biol Chem. 1996;271(23):13724–30.
29 Matalon S, Shrestha K, Kirk M, Waldheuser S, McDonald B, Smith K, et al. Modification of surfactant protein D by reactive oxygen-nitrogen intermediates is accompanied by loss of aggregating activity, in vitro and in vivo. FASEB J. 2009;23(5):1415–30.
30 White M, Kingma P, Tecle T, Kacak N, Linders B, Heuser J, et al. Multimerization of surfactant protein D, but not its collagen domain, is required for antiviral and opsonic activities related to influenza virus. J Immunol. 2008;181(11):7936–43.
31 Zhang L, Ikegami M, Crouch EC, Korfhagen TR, Whitsett JA. Activity of pulmonary surfactant protein-D (SP-D) in vivo is dependent on oligomeric structure. J Biol Chem. 2001;276(22):19214–19.
32 Mason RJ. Surfactant synthesis, secretion, and function in alveoli and small airways. Review of the physiologic basis for pharmacologic intervention. Respiration. 1987;51 Suppl 13–9.
33 Weaver TE, Na CL, Stahlman M. Biogenesis of lamellar bodies, lysosome-related organelles involved in storage and secretion of pulmonary surfactant. Semin Cell Dev Biol. 2002;13(4):263–70.
34 Rooney SA. Regulation of surfactant secretion. Comp Biochem Physiol A Mol Integr Physiol. 2001;129(1):233–43.
35 Madsen J, Kliem A, Tornoe I, Skjodt K, Koch C, Holmskov U. Localization of lung surfactant protein D on mucosal surfaces in human tissues. J Immunol. 2000;164(11):5866–70.
36 Miyamura K, Malhotra R, Hoppe HJ, Reid KB, Phizackerley PJ, Macpherson P, et al. Surfactant proteins A (SP-A) and D (SP-D): levels in human amniotic fluid and localization in the fetal membranes. Biochim Biophys Acta. 1994;1210(3):303–7.
37 Mori K, Kurihara N, Hayashida S, Tanaka M, Ikeda K. The intrauterine expression of surfactant protein D in the terminal airways of human fetuses compared with surfactant protein A. Eur J Pediatr. 2002;161(8):431–4.
38 Veldhuizen R, Possmayer F. Phospholipid metabolism in lung surfactant. Subcell Biochem. 2004;37:359–88.
39 Grabner R, Meerbach W. Phagocytosis of surfactant by alveolar macrophages in vitro. Am J Physiol. 1991;261(6 Pt 1):L472–7.
40 Wright JR. Clearance and recycling of pulmonary surfactant. Am J Physiol. 1990;259(2 Pt 1):L1–12.
41 Galli SJ, Tsai M. IgE and mast cells in allergic disease. Nat Med. 2012;18(5):693–704.
42 Wang JY, Kishore U, Lim BL, Strong P, Reid KB. Interaction of human lung surfactant proteins A and D with mite (Dermatophagoides pteronyssinus) allergens. Clin Exp Immunol. 1996;106(2):367–73.
43 Liu CF, Chen YL, Chang WT, Shieh CC, Yu CK, Reid KB, et al. Mite allergen induces nitric oxide production in alveolar macrophage cell lines via CD14/toll-like receptor 4, and is inhibited by surfactant protein D. Clin Exp Allergy. 2005;35(12):1615–24.
44 Erpenbeck VJ, Malherbe DC, Sommer S, Schmiedl A, Steinhilber W, Ghio AJ, et al. Surfactant protein D increases phagocytosis and aggregation of pollen-allergen starch granules. Am J Physiol Lung Cell Mol Physiol. 2005;288(4):L692–8.
45 Malherbe DC, Erpenbeck VJ, Abraham SN, Crouch EC, Hohlfeld JM, Wright JR. Surfactant protein D decreases pollen-induced IgE-dependent mast cell degranulation. Am J Physiol Lung Cell Mol Physiol. 2005;289(5):L856–66.
46 Winkler C, Huper K, Wedekind AC, Rochlitzer S, Hartwig C, Muller M, et al. Surfactant protein D modulates pulmonary clearance of pollen starch granules. Exp Lung Res. 2010;36(9):522–30.
47 De Sanctis GT, Tomkiewicz RP, Rubin BK, Schurch S, King M. Exogenous surfactant enhances mucociliary clearance in the anaesthetized dog. Eur Respir J. 1994;7(9):1616–21.
48 Madan T, Kishore U, Shah A, Eggleton P, Strong P, Wang JY, et al. Lung surfactant proteins A and D can inhibit specific IgE binding to the allergens of Aspergillus fumigatus and block allergen-induced histamine release from human basophils. Clin Exp Immunol. 1997;110(2):241–9.
49 Kuronuma K, Sano H, Kato K, Kudo K, Hyakushima N, Yokota S, et al. Pulmonary surfactant protein A augments the phagocytosis of Streptococcus pneumoniae by alveolar macrophages through a casein kinase 2-dependent increase of cell surface localization of scavenger receptor A. J Biol Chem. 2004;279(20):21421–30.
50 Ledford JG, Goto H, Potts EN, Degan S, Chu HW, Voelker DR, et al. SP-A preserves airway homeostasis during Mycoplasma pneumoniae infection in mice. J Immunol. 2009;182(12):7818–27.
51 Levine AM, Gwozdz J, Stark J, Bruno M, Whitsett J, Korfhagen T. Surfactant protein-A enhances respiratory syncytial virus clearance in vivo. J Clin Invest. 1999;103(7):1015–21.
52 Lambrecht BN, Hammad H. Taking our breath away: dendritic cells in the pathogenesis of asthma. Nat Rev Immunol. 2003;3(12):994–1003.
53 Brinker KG, Garner H, Wright JR. Surfactant protein A modulates the differentiation of murine bone marrow-derived dendritic cells. Am J Physiol Lung Cell Mol Physiol. 2003;284(1):L232–41.
54 Hortobagyi L, Kierstein S, Krytska K, Zhu X, Das AM, Poulain F, et al. Surfactant protein D inhibits TNF-alpha production by macrophages and dendritic cells in mice. J Allergy Clin Immunol. 2008;122(3):521–8.
55 Brinker KG, Martin E, Borron P, Mostaghel E, Doyle C, Harding CV et al. Surfactant protein D enhances bacterial antigen presentation by bone marrow-derived dendritic cells. Am J Physiol Lung Cell Mol Physiol. 2001;281(6):L1453–63.
56 Liu CF, Rivere M, Huang HJ, Puzo G, Wang JY. Surfactant protein D inhibits mite-induced alveolar macrophage and dendritic cell activations through TLR signalling and DC-SIGN expression. Clin Exp Allergy. 2010;40(1):111–22.
57 Hansen S, Lo B, Evans K, Neophytou P, Holmskov U, Wright JR. Surfactant protein D augments bacterial association but attenuates major histocompatibility complex class II presentation of bacterial antigens. Am J Respir Cell Mol Biol. 2007;36(1):94–102.
58 Gardai SJ, Xiao YQ, Dickinson M, Nick JA, Voelker DR, Greene KE, et al. By binding SIRPalpha or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cell. 2003;115(1):13–23.
59 Sano H, Chiba H, Iwaki D, Sohma H, Voelker DR, Kuroki Y. Surfactant proteins A and D bind CD14 by different mechanisms. J Biol Chem. 2000;275(29):22442–51.
60 Sato M, Sano H, Iwaki D, Kudo K, Konishi M, Takahashi H, et al. Direct binding of Toll-like receptor 2 to zymosan, and zymosan-induced NF-kappa B activation and TNF-alpha secretion are down-regulated by lung collectin surfactant protein A. J Immunol. 2003;171(1):417–25.
61 Vandivier RW, Ogden CA, Fadok VA, Hoffmann PR, Brown KK, Botto M, et al. Role of surfactant proteins A, D, and C1q in the clearance of apoptotic cells in vivo and in vitro: calreticulin and CD91 as a common collectin receptor complex. J Immunol. 2002;169(7):3978–86.
62 Pandit H, Madhukaran SP, Nayak A, Madan T. SP-A and SP-D in host defense against fungal infections and allergies. Front Biosci (Elite.Ed). 2012;4651–61.
63 Schagat TL, Wofford JA, Wright JR. Surfactant protein A enhances alveolar macrophage phagocytosis of apoptotic neutrophils. J Immunol. 2001;166(4):2727–33.
64 Lloyd CM, Hessel EM. Functions of T cells in asthma: more than just T(H)2 cells. Nat Rev Immunol. 2010;10(12):838–48.
65 Borron P, Veldhuizen RA, Lewis JF, Possmayer F, Caveney A, Inchley K, et al. Surfactant associated protein-A inhibits human lymphocyte proliferation and IL-2 production. Am J Respir Cell Mol Biol. 1996;15(1):115–21.
66 Borron PJ, Crouch EC, Lewis JF, Wright JR, Possmayer F, Fraher LJ. Recombinant rat surfactant-associated protein D inhibits human T lymphocyte proliferation and IL-2 production. J Immunol. 1998;161(9):4599–603.
67 Scanlon ST, Milovanova T, Kierstein S, Cao Y, Atochina EN, Tomer Y, et al. Surfactant protein-A inhibits Aspergillus fumigatus-induced allergic T-cell responses. Respir Res. 2005;6:97.
68 Wang JY, Shieh CC, You PF, Lei HY, Reid KB. Inhibitory effect of pulmonary surfactant proteins A and D on allergen-induced lymphocyte proliferation and histamine release in children with asthma. Am J Respir Crit Care Med. 1998;158(2):510–8.
69 Borron P, McCormack FX, Elhalwagi BM, Chroneos ZC, Lewis JF, Zhu S, et al. Surfactant protein A inhibits T cell proliferation via its collagen-like tail and a 210-kDa receptor. Am J Physiol. 1998;275(4 Pt 1):L679–86.
70 Yang CH, Szeliga J, Jordan J, Faske S, Sever-Chroneos Z, Dorsett B, et al. Identification of the surfactant protein A receptor 210 as the unconventional myosin 18A. J Biol Chem. 2005;280(41):34447–57.
71 Mukherjee S, Giamberardino C, Thomas J, Evans K, Goto H, Ledford JG, et al. Surfactant protein a integrates activation signal strength to differentially modulate T cell proliferation. J Immunol. 2012;188(3):957–67.
72 Lin KW, Jen KY, Suarez CJ, Crouch EC, Perkins DL, Finn PW. Surfactant protein D-mediated decrease of allergen-induced inflammation is dependent upon CTLA4. J Immunol. 2010;184(11):6343–9.
73 Wegmann M. Targeting eosinophil biology in asthma therapy. Am J Respir Cell Mol Biol. 2011;45(4):667–74.
74 Cheng G, Ueda T, Nakajima H, Nakajima A, Kinjyo S, Motojima S, et al. Suppressive effects of SP-A on ionomycin-induced IL-8 production and release by eosinophils. Int Arch Allergy Immunol. 1998;117:Suppl 159–62.
75 von Bredow C, Hartl D, Schmid K, Schabaz F, Brack E, Reinhardt D, et al. Surfactant protein D regulates chemotaxis and degranulation of human eosinophils. Clin Exp Allergy. 2006;36(12):1566–74.
76 Mahajan L, Madan T, Kamal N, Singh VK, Sim RB, Telang SD, et al. Recombinant surfactant protein-D selectively increases apoptosis in eosinophils of allergic asthmatics and enhances uptake of apoptotic eosinophils by macrophages. Int Immunol. 2008;20(8):993–1007.
77 Erpenbeck VJ, Hagenberg A, Dulkys Y, Elsner J, Balder R, Krentel H, et al. Natural porcine surfactant augments airway inflammation after allergen challenge in patients with asthma. Am J Respir Crit Care Med. 2004;169(5):578–86.
78 Erpenbeck VJ, Fischer I, Wiese K, Schaumann F, Schmiedl A, Nassenstein C, et al. Therapeutic surfactants modulate the viability of eosinophils and induce inflammatory mediator release. Int Arch Allergy Immunol. 2009;149(4):333–42.
79 Erpenbeck VJ, Schmidt R, Gunther A, Krug N, Hohlfeld JM. Surfactant protein levels in bronchoalveolar lavage after segmental allergen challenge in patients with asthma. Allergy. 2006;61(5):598–604.
80 Kasper M, Sims G, Koslowski R, Kuss H, Thuemmler M, Fehrenbach H et al. Increased surfactant protein D in rat airway goblet and Clara cells during ovalbumin-induced allergic airway inflammation. Clin Exp Allergy. 2002;32(8):1251–8.
81 Atochina EN, Beers MF, Tomer Y, Scanlon ST, Russo SJ, Panettieri RA, Jr. et al. Attenuated allergic airway hyperresponsiveness in C57BL/6 mice is associated with enhanced surfactant protein (SP)-D production following allergic sensitization. Respir Res. 2003;415.
82 Haczku A, Cao Y, Vass G, Kierstein S, Nath P, Atochina-Vasserman EN, et al. IL-4 and IL-13 form a negative feedback circuit with surfactant protein-D in the allergic airway response. J Immunol. 2006;176(6):3557–65.
83 Koopmans JG, van der Zee JS, Krop EJ, Lopuhaa CE, Jansen HM, Batenburg JJ. Serum surfactant protein D is elevated in allergic patients. Clin Exp Allergy. 2004;34(12):1827–33.
84 Jain-Vora S, Wert SE, Temann UA, Rankin JA, Whitsett JA. Interleukin-4 alters epithelial cell differentiation and surfactant homeostasis in the postnatal mouse lung. Am J Respir Cell Mol Biol. 1997;17(5):541–51.
85 Homer RJ, Zheng T, Chupp G, He S, Zhu Z, Chen Q et al. Pulmonary type II cell hypertrophy and pulmonary lipoproteinosis are features of chronic IL-13 exposure. Am J Physiol Lung Cell Mol Physiol. 2002;283(1):L52–9.
86 Cao Y, Tao JQ, Bates SR, Beers MF, Haczku A. IL-4 induces production of the lung collectin surfactant protein-D. J Allergy Clin Immunol. 2004;113(3):439–44.
87 Ito Y, Mason RJ. The effect of interleukin-13 (IL-13) and interferon-gamma (IFN-gamma) on expression of surfactant proteins in adult human alveolar type II cells in vitro. Respir Res. 2010;11:157.
88 Hohlfeld JM, Ahlf K, Enhorning G, Balke K, Erpenbeck VJ, Petschallies J, et al. Dysfunction of pulmonary surfactant in asthmatics after segmental allergen challenge. Am J Respir Crit Care Med. 1999;159(6):1803–9.
89 Heeley EL, Hohlfeld JM, Krug N, Postle AD. Phospholipid molecular species of bronchoalveolar lavage fluid after local allergen challenge in asthma. Am J Physiol Lung Cell Mol Physiol. 2000;278(2):L305–11.
90 Hite RD, Seeds MC, Bowton DL, Grier BL, Safta AM, Balkrishnan R, et al. Surfactant phospholipid changes after antigen challenge: a role for phosphatidylglycerol in dysfunction. Am J Physiol Lung Cell Mol Physiol. 2005;288(4):L610–7.
91 Hohlfeld JM, Schmiedl A, Erpenbeck VJ, Venge P, Krug N. Eosinophil cationic protein alters pulmonary surfactant structure and function in asthma. J Allergy Clin Immunol. 2004;113(3):496–502.
92 Henricks PA, Nijkamp FP. Reactive oxygen species as mediators in asthma. Pulm Pharmacol Ther. 2001;14(6):409–20.
93 Guo CJ, Atochina-Vasserman EN, Abramova E, Foley JP, Zaman A, Crouch E, et al. S-nitrosylation of surfactant protein-D controls inflammatory function. PLoS Biol. 2008;6(11):e266.
94 Starosta V, Griese M. Oxidative damage to surfactant protein D in pulmonary diseases. Free Radic Res. 2006;40(4):419–25.
95 Wang Y, Voelker DR, Lugogo NL, Wang G, Floros J, Ingram JL, et al. Surfactant protein A is defective in abrogating inflammation in asthma. Am J Physiol Lung Cell Mol Physiol. 2011;301(4):L598–L606.
96 Deb R, Shakib F, Reid K, Clark H. Major house dust mite allergens Dermatophagoides pteronyssinus 1 and Dermatophagoides farinae 1 degrade and inactivate lung surfactant proteins A and D. J Biol Chem. 2007;282(51):36808–19.
97 Schaub B, Westlake RM, He H, Arestides R, Haley KJ, Campo M, et al. Surfactant protein D deficiency influences allergic immune responses. Clin Exp Allergy. 2004;34(12):1819–26.
98 Singh M, Madan T, Waters P, Parida SK, Sarma PU, Kishore U. Protective effects of a recombinant fragment of human surfactant protein D in a murine model of pulmonary hypersensitivity induced by dust mite allergens. Immunol Lett. 2003;86(3):299–307.
99 Liu CF, Chen YL, Shieh CC, Yu CK, Reid KB, Wang JY. Therapeutic effect of surfactant protein D in allergic inflammation of mite-sensitized mice. Clin Exp Allergy. 2005;35(4):515–21.
100 Hohlfeld J, Hoymann HG, Molthan J, Fabel H, Heinrich U. Aerosolized surfactant inhibits acetylcholine-induced airway obstruction in rats. Eur Respir J. 1997;10(10):2198–2203.
101 Kurashima K, Fujimura M, Tsujiura M, Matsuda T. Effect of surfactant inhalation on allergic bronchoconstriction in guinea pigs. Clin Exp Allergy. 1997;27(3):337–42.
102 Siddiqui S, Jo T, Tamaoka M, Shalaby KH, Ghezzo H, Bernabeu M, et al. Sites of allergic airway smooth muscle remodeling and hyperresponsiveness are not associated in the rat. J Appl Physiol. 2010;109(4):1170–8.
103 Kurashima K, Ogawa H, Ohka T, Fujimura M, Matsuda T, Kobayashi T. A pilot study of surfactant inhalation in the treatment of asthmatic attack. Arerugi. 1991;40(2):160–3.
104 Babu KS, Woodcock DA, Smith SE, Staniforth JN, Holgate ST, Conway JH. Inhaled synthetic surfactant abolishes the early allergen-induced response in asthma. Eur Respir J. 2003;21(6):1046–9.