Impact of diabetes on platelet activation in different manifestations of atherosclerosis
DOI: https://doi.org/10.4414/smw.2013.13800
Thomas
Gremmel, Christoph W
Kopp, Daniela
Seidinger, Renate
Koppensteiner, Sabine
Steiner, Simon
Panzer
Summary
BACKGROUND: Atherosclerosis affects many patients with type 2 diabetes. Both are associated with platelet activation, but it remains unclear how diabetes contributes to, or even enhances, platelet activation in patients with atherosclerosis. We therefore investigated the impact of diabetes on platelet activation and protease activated receptor-1 (PAR-1) mediated platelet response in patients with symptomatic coronary artery disease (CAD), as compared with other manifestations of atherosclerosis.
METHODS: Baseline P-selectin expression, thrombin receptor activating peptide-6 (TRAP-6) inducible P-selectin, and relative increase of platelet P-selectin after activation with TRAP-6 were measured using flow cytometry in platelets from 317 patients after angioplasty and stenting for symptomatic atherosclerotic disease, and from 50 healthy controls.
RESULTS: Patients with symptomatic atherosclerosis exhibited significantly higher levels of baseline P-selectin expression, TRAP-6-inducible P-selectin and relative increase of platelet P-selectin after stimulation with TRAP-6 than healthy controls. Patients with symptomatic peripheral artery disease or cerebrovascular disease (PAD/CVD) had higher levels of platelet activation and PAR-1-mediated platelet reactivity than patients with symptomatic CAD. Of interest, CAD patients with diabetes responded more strongly to TRAP-6 than those without diabetes, and their platelet activation and PAR-1-mediated platelet reactivity resembled those from PAD/CVD patients.
CONCLUSION: Compared with healthy controls, platelets from patients with symptomatic atherosclerotic disease are activated and susceptible to PAR-1-mediated activation. Diabetes affects platelet reactivity only in patients with symptomatic CAD, while other manifestations of atherosclerosis may have an overwhelming effect on platelet reactivity that is not further enhanced by diabetes.
Abbreviations
ACS acute coronary syndrome
CAD coronary artery disease
HbA1c glycated haemoglobin
MFI mean fluorescence intensity
PAD/CVD peripheral artery or cerebrovascular disease
PAR protease activated receptor
PCI percutaneous coronary intervention
TRAP-6 thrombin receptor activating peptide-6
Introduction
Patients with type 2 diabetes frequently suffer from atherosclerosis [1, 2], and it has been reported that diabetics have a two- to four-fold increased risk of coronary artery disease (CAD) and peripheral artery disease (PAD) [2–4], and a higher incidence of carotid plaques, than matched control populations [5]. In a population-based study, the 7-year incidence of first myocardial infarction or death was 20% in diabetics, but only 3.5% in nondiabetic patients [6]. Moreover, the relative risk of lower extremity amputation in diabetics was 12.7 compared with that of nondiabetic patients [7], and the risk of ischaemic stroke was increased by up to 400% in diabetics [8]. Platelets play a central role in the development of thrombotic events in these patients, as they are activated in association with impaired metabolic control [9, 10], which itself promotes thrombin generation and vulnerability of atherosclerotic plaques [11, 12]. Plaque rupture results in platelet activation and adhesion to the subendothelial structures with subsequent vessel occlusion.
P-selectin is a cell adhesion molecule which mediates binding to specific carbohydrate-containing ligands [13]. It allows tethering of leukocytes to activated endothelial cells and platelets, and mediates leukocyte rolling on the endothelial cell surface [14]. Platelet surface expression of P-selectin is one of the most sensitive indicators of platelet activation [15], and has been reported to be substantially enhanced in patients with acute and stable CAD [16, 17]. However, data on P-selectin expression in stable atherosclerosis are scarce, and limited to CAD.
Persistent thrombin generation is considered a major reason for adverse ischaemic events in patients on standard antiplatelet therapy. Thrombin activates platelets via protease-activated receptors (PARs) – PAR-1 and PAR-4 – and glycoproteins Ib-α and V [18, 19]. There is strong evidence that PAR-1-mediated platelet activation is clinically significant. For example, PAR-1 modulation has been shown in patients with ischaemic stroke [20].
Increased platelet activation in diabetic patients has been reported in the past [21, 22]. Further, it has been shown that improved metabolic control decreases platelet activation markers in patients with type 2 diabetes [23, 24]. These findings suggest, among others, a reduction in thrombin generation, which is particularly abundant in obese type 2 diabetic patients [11]. Since many patients with diabetes also have atherosclerosis, it is not clear how diabetes contributes to, or even enhances, platelet activation in patients with atherosclerosis. We therefore investigated the impact of diabetes on platelet activation and PAR-1-mediated platelet response in patients with symptomatic CAD, as compared with other manifestations of atherosclerosis.
Patients and methods
Study population
In this cross-sectional study, the patients were recruited consecutively at the Division of Angiology of the University Hospital Vienna between 2008 and 2010.
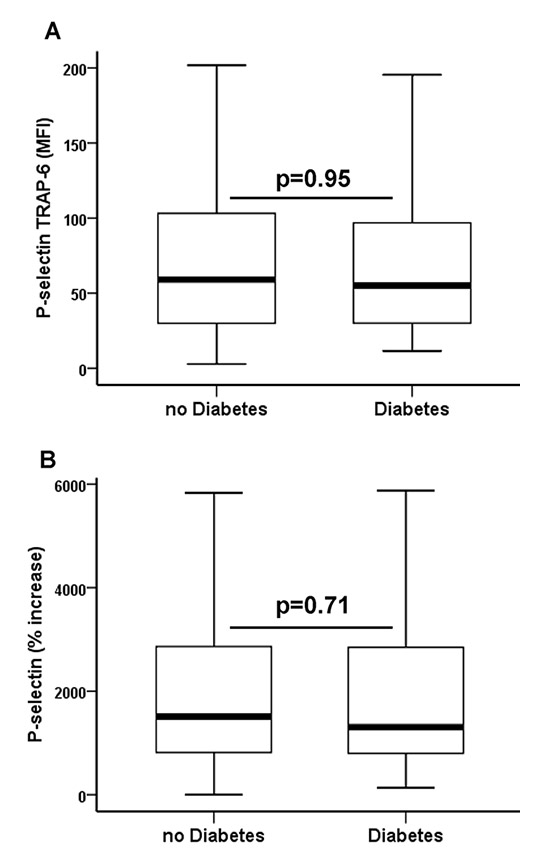
Figure 1
(A)Thrombin receptor activating peptide-6 (TRAP-6) inducible P-selectin (P-selectin TRAP-6) and (B) relative increase of platelet P-selectin (% increase) after activation with TRAP-6 in patients with peripheral artery or cerebrovascular disease without and with type 2 diabetes. The boundaries of the box show the lower and upper quartiles of data, the line inside the box represents the median. Whiskers are drawn from the edge of the box to the highest and lowest values that are outside the box but within 1.5 times the box length. MFI = mean fluorescence intensity.
Inclusion criteria were age >18 years, percutaneous angioplasty with stent implantation due to peripheral artery/cerebrovascular disease (PAD/CVD) or CAD, and written informed consent.
Exclusion criteria were: known aspirin or clopidogrel intolerance (allergic reactions, gastrointestinal bleeding); therapy with vitamin K antagonists (warfarin, phenprocoumon, acenocoumarol); treatment with ticlopidine, dipyridamol or nonsteroidal anti-inflammatory drugs; a family or personal history of bleeding disorders, malignant paraproteinaemias, myeloproliferative disorders or heparin-induced thrombocytopenia; severe hepatic failure; known qualitative defects in thrombocyte function; a major surgical procedure within 1 week before enrolment; a platelet count <100,000/µl or >450,000/µl; and a haematocrit <30%.
The healthy control population consisted of hospital staff members and their relatives, all from the urban area of Vienna.
The study protocol was approved by the Ethics Committee of the Medical University of Vienna in accordance with the Declaration of Helsinki, and written informed consent was obtained from all study participants.
Blood sampling
Blood was drawn by means of clean venipuncture from an antecubital vein with a 21-gauge butterfly needle (0.8 x 19 mm; Greiner Bio-One, Kremsmünster, Austria) 1 day after the percutaneous intervention. To avoid procedural deviations all blood samples were taken by the same physician. A light tourniquet was applied and immediately released, and the samples were mixed adequately by gently inverting the tubes. After the initial 3 ml of blood had been discarded to reduce procedurally induced platelet activation, blood was drawn into a 3.8% sodium citrate Vacuette tube (Greiner Bio-One; 9 parts of whole blood, 1 part of sodium citrate 0.129 M/L). P-selectin expression was determined 1 hour after blood sampling for all patients.
P-selectin expression
Flow cytometry was performed as previously described [25]. In brief, the expression of P-selectin was determined in whole blood without agonists and after in-vitro exposure to suboptimal concentrations of thrombin receptor activating peptide-6 (TRAP-6) (5.7 μM; Bachem, Bubendorf, Switzerland) for 10 minutes. The use of suboptimal concentrations of the agonist allows differentiation between platelets that are activated but can still respond further and platelets that are already devoid of the α-granule content of P-selectin owing to in-vivo exhaustion. The platelet population was identified by staining with anti-CD42b (clone HIP1, allophycocyanin labeled; Becton Dickinson, B.D., San José, California, USA), and P-selectin expression was determined by the binding of the monoclonal antibody anti-CD62p (clone CLB-Thromb6, phycoerythrin-labelled; Immunotech, Beckman Coulter, Fullerton, California, USA). The reaction was stopped by adding 500 µl PBS and samples were tested immediately in a FACS Calibur flow cytometer (B.D.) with excitation by an argon laser at 488 nm and a red diode laser at 635 nm at a rate of 200–600 events per second. Standard B.D. Calibrite beads were used for daily calibration of the cytometer.
Statistical analysis
A sample size calculation was based on the observed mean ± standard deviation (SD) (3.4 ± 1.5 MFI; mean fluorescence intensity) of P-selectin expression in a former population of 105 patients (84 male, 21 female; median age 60 years, interquartile range 53–68 years) under dual antiplatelet treatment 24 hours after percutaneous coronary intervention (PCI) with stent implantation [26]. We calculated that we needed to include 300 patients to be able to detect a 20% relative difference in P-selectin expression between patients with PAD/CVD and patients with CAD with a power of 96% (using a two-sided alpha level of 0.05). To compensate for potential technical problems we included 17 additional patients.
Statistical analysis was performed using the Statistical Package for Social Sciences (IBM SPSS version 19, Armonk, New York, USA). The relative increase in P-selectin expression was calculated as percentage of P-selectin expression before the addition of TRAP-6. Medians and interquartile ranges of continuous variables are shown. Categorical variables are given as number (%). We performed Mann Whitney U tests to detect differences in continuous variables. The chi-square test was used to detect differences in categorical variables. Two-sided p-values <0.05 were considered statistically significant.
Results
The study population comprised 317 patients undergoing percutaneous intervention with endovascular stent implantation for PAD/CVD (n = 202) and CAD (n = 115). All patients received dual antiplatelet therapy consisting of aspirin (100 mg/day) and an adenosine diphosphate P2Y12 receptor antagonist (clopidogrel 75 mg/day [n = 303] or prasugrel 10 mg/day [n = 14]). Clinical, laboratory and procedural characteristics of the overall patient population, of patients with PAD/CVD, and of patients with CAD are given in table 1. All patients were enrolled in a stable clinical condition 1 day after the angioplasty procedure. Patients with PAD suffered from intermittent claudication (n = 165), patients with CVD had signs of transient cerebral ischaemia within 4 weeks before study enrolment (n = 37), and patients with CAD suffered from stable angina (n = 47) or acute coronary syndromes (ACS; n = 68) prior to percutaneous intervention. Moreover, we included 50 healthy individuals (17 male, 33 female; aged 46 years [32–52 years]) without diabetes or clinical symptoms of atherosclerosis as controls for P-selectin expression. As expected, the healthy control population was significantly younger than the patient population, had lower levels of serum creatinine and C-reactive protein, and female gender was more common (all p <0.05).
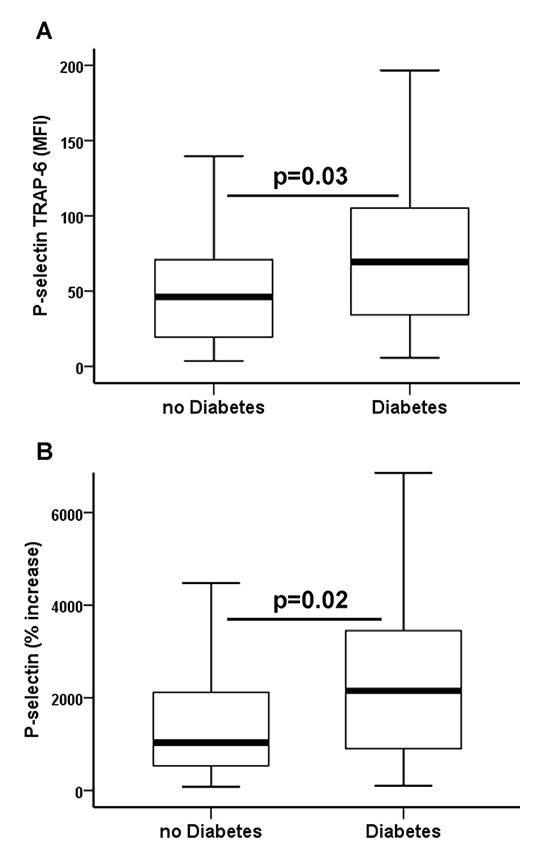
Figure 2
(A)Thrombin receptor activating peptide-6 (TRAP-6) inducible P-selectin (P-selectin TRAP-6) and (B) relative increase of platelet P-selectin (% increase) after activation with TRAP-6 in patients with coronary artery disease without and with type 2 diabetes. The boundaries of the box show the lower and upper quartiles of data, the line inside the box represents the median. Whiskers are drawn from the edge of the box to the highest and lowest values that are outside the box but within 1.5 times the box length. MFI = mean fluorescence intensity.
Patients with symptomatic atherosclerotic vascular disease exhibited significantly higher levels of baseline P-selectin expression, TRAP-6-inducible P-selectin and percentage increase of platelet P-selectin after activation with TRAP-6 than healthy controls (table 2).
Overall, patients with symptomatic PAD/CVD had higher levels of baseline P-selectin and TRAP-6-inducible P-selectin than patients with symptomatic CAD (table 2). Type 2 diabetes was present in 75 patients (37.1%) undergoing peripheral angioplasty and in 28 patients (24.3%) undergoing PCI (table 1; p = 0.03). All diabetics had been suffering from type 2 diabetes for at least 2 years at the time of study enrolment. Glycated haemoglobin (HbA1c) levels were 7% (6.4%–7.7%) and 5.7% (5.5%–6%) in diabetic and nondiabetic patients, respectively (p <0.001). Median HbA1c was similar in diabetics with PAD/CVD and diabetics with CAD (7.1% vs 7%; p = 0.4). Levels of platelet activation were similar in diabetic and nondiabetic patients within the group of PAD/CVD patients (table 2 and fig. 1). In contrast, diabetic CAD patients had a significantly increased susceptibility to PAR-1-mediated platelet activation and a more pronounced increase of P-selectin after activation with TRAP-6 when compared with nondiabetic CAD patients (table 2 and fig. 2). Thereby, their levels of TRAP-6-inducible P-selectin were similar to those in PAD/CVD patients without or with diabetes. Susceptibility to PAR-1-mediated platelet activation was significantly higher in nondiabetic patients with PAD/CVD than in nondiabetic patients with CAD (table 2). Levels of baseline and TRAP-6-inducible P-selectin were similar in diabetics with good metabolic control (HbA1c <7%; n = 50) and insufficient metabolic control (HbA1c ≥7%; n = 53) (both p ≥0.4).
|
Table 1:Clinical, laboratory and procedural characteristics of the overall study population, and of patients with peripheral artery / cerebrovascular disease (PAD/CVD) or coronary artery disease (CAD). |
|
Characteristic
|
All patients (n = 317)
|
PAD/CVD (n = 202)
|
CAD (n = 115)
|
p-value
|
| Age, years |
65 (57–74) |
66 (58–74) |
63 (54–73) |
0.1 |
| Male sex |
209 (65.9) |
128 (63.4) |
81 (70.4) |
0.2 |
| BMI, kg/m2
|
27 (24.4–29.8) |
26.8 (24.1–29.6) |
27.7 (24.7–30.3) |
0.2 |
|
Medical history
|
|
|
|
|
| Hypertension |
283 (89.3) |
184 (91.1) |
99 (86.1) |
0.2 |
| Hypercholesterolaemia |
297 (93.7) |
191 (94.6) |
106 (92.2) |
0.5 |
| Diabetes mellitus |
103 (27.8) |
75 (37.1) |
28 (24.3) |
0.03 |
| Active smoking |
133 (42) |
87 (43.1) |
46 (40) |
0.6 |
|
Laboratory data
|
|
|
|
|
| Platelet count, G/l |
209 (175–252) |
213 (177–261) |
203 (171–231) |
0.03 |
| Serum creatinine, µmol/l |
88.5 (79.7–106.2) |
88.5 (79.7–106.2) |
88.5 (79.7–97.4) |
0.1 |
| C-reactive protein, mg/l |
10 (4–19) |
10 (4–18) |
9 (4–26) |
0.5 |
|
Procedure
|
|
|
|
|
| Stent implantation |
317 (100) |
202 (100) |
115 (100) |
1 |
| Number of stents/patient |
1 (1–2) |
1 (1–2) |
1 (1–2) |
0.6 |
|
Medication preintervention
|
|
|
|
|
| Thienopyridine |
317 (100) |
202 (100) |
115 (100) |
1 |
| Aspirin |
317 (100) |
202 (100) |
115 (100) |
1 |
| Statins |
303 (95.6) |
191 (94.6) |
112 (97.4) |
0.3 |
| ACE inhibitors |
194 (61.2) |
111 (55) |
83 (72.2) |
0.003 |
| Angiotensin receptor blockers |
83 (26.2) |
59 (29.2) |
24 (20.9) |
0.1 |
| Beta blockers |
221 (69.7) |
113 (55.9) |
108 (93.9) |
<0.001 |
| Continuous data are shown as median (interquartile range). Dichotomous data are shown as n (%).
ACE inhibitors = angiotensin converting enzyme inhibitors; BMI = body mass index. |
|
Table 2:Median and interquartile range of baseline P-selectin, thrombin receptor activating peptide-6 (TRAP-6) inducible P-selectin (P-selectin TRAP-6), and relative increase of platelet P-selectin after stimulation with TRAP-6 (% increase P-selectin) in the overall patient population, in healthy controls, in patients with peripheral artery disease / cerebrovascular disease (PAD/CVD) and coronary artery disease (CAD), in diabetics and nondiabetics with PAD/CVD, and in diabetics and nondiabetics with CAD. |
| |
All patients (n = 317)
|
Healthy controls (n = 50)
|
p-value
|
|
Baseline P-selectin (MFI)
|
3.2 (2.7–4) |
3 (2.6–3.4) |
0.02 |
|
P-selectin TRAP-6 (MFI)
|
54.9 (27.8–93.3) |
25.5 (15.6–40.7) |
<0.001 |
|
% increase P-selectin
|
1407 (691–2767) |
766 (403–1392) |
<0.001 |
|
|
Patients with PAD/CVD (n = 202)
|
Patients with CAD (n = 115)
|
p-value
|
|
Baseline P-selectin (MFI)
|
3.3 (2.9–4) |
3 (2.6–3.9) |
0.01 |
|
P-selectin TRAP-6 (MFI)
|
57.1 (29.7–102) |
50.2 (21.6–81.2) |
0.04 |
|
% increase P-selectin
|
1490 (805–2930) |
1155 (549–2535) |
0.1 |
|
|
Diabetics with PAD/CVD (n = 75)
|
Nondiabetics with PAD/CVD (n = 127)
|
p-value
|
|
Baseline P-selectin (MFI)
|
3.4 (2.9–4.2) |
3.3 (2.8–3.9) |
0.3 |
|
P-selectin TRAP-6 (MFI)
|
54.9 (29.7–99.5) |
59.1 (29.7–103.9) |
0.9 |
|
% increase P-selectin
|
1305 (793–2930) |
1522 (817–3169) |
0.7 |
|
|
Diabetics with CAD (n = 28)
|
Nondiabetics with CAD (n = 87)
|
p-value
|
|
Baseline P-selectin (MFI)
|
3 (2.6–3.7) |
3 (2.6–3.9) |
0.9 |
|
P-selectin TRAP-6 (MFI)
|
69.5 (34.2–102.2) |
46.3 (18.9–71.5) |
0.03 |
|
% increase P-selectin
|
2148 (904–3450) |
1033 (52 –2169) |
0.02 |
|
|
Diabetics with PAD/CVD (n = 75)
|
Diabetics with CAD (n = 28)
|
p-value
|
|
Baseline P-selectin (MFI)
|
3.4 (2.9–4.2) |
3 (2.6–3.7) |
0.1 |
|
P-selectin TRAP-6 (MFI)
|
54.9 (29.7–99.5) |
69.5 (34.2–102.2) |
0.5 |
|
% increase P-selectin
|
1305 (793–2930) |
2148 (904–3450) |
0.3 |
|
|
Nondiabetics with PAD/CVD (n = 127)
|
Nondiabetics with CAD (n = 87)
|
p-value
|
|
Baseline P-selectin (MFI)
|
3.3 (2.8–3.9) |
3 (2.6–3.9) |
0.08 |
|
P-selectin TRAP-6 (MFI)
|
59.1 (29.7–103.9) |
46.3 (18.9–71.5) |
0.009 |
|
% increase P-selectin
|
1522 (817–3169) |
1033 (524–2169) |
0.02 |
| MFI = mean fluorescence intensity. |
Discussion
Increased platelet activation has been reported in the past in diabetic patients [21, 22], and also in patients with atherosclerosis. Many patients with symptomatic atherosclerosis have diabetes, and it is therefore not clear if diabetes further affects platelet reactivity in these patients. We confirm that atherosclerosis is associated with increased platelet activation. Further, our data show that patients with PAD/CVD have higher levels of platelet activation, and respond more strongly to platelet activation by PAR-1, than CAD patients. However, in the latter group, patients with type 2 diabetes exhibit more pronounced platelet activation by PAR-1 than nondiabetic patients. Thereby, their increased levels of platelet activation resemble those from patients with PAD/CVD.
We selected platelet surface expression of P-selectin without addition of an agonist and in response to TRAP-6 for our study because these parameters are only affected to a small extent by antiplatelet treatment with aspirin and thienopyridines [27]. This approach of adding an agonist in vitro thus resembles a real life scenario when thrombin is generated.
The response of already activated platelets to additional exogenous stimuli is stronger than that of platelets that are not preactivated [28, 29]. Our finding that diabetic patients with CAD have increased levels of PAR-1-mediated platelet activation when compared with nondiabetic CAD patients may therefore explain their increased risk for repeated cardiovascular events. On the other hand, in patients with PAD/CVD, the already enhanced levels of platelet activation due to the greater extent of atherosclerosis may not be significantly further increased by diabetes. The worse long-term prognosis of patients with PAD/CVD, as compared with CAD patients, may at least in part be attributable to their enhanced platelet activation [30]. Our observation that baseline platelet activation in patients with CAD resembles that in healthy controls is in line with a report by Michelson et al., who found similar levels of platelet P-selectin expression in patients with myocardial infarction and healthy controls [31].
Various mechanisms have been proposed to explain platelet dysfunction and increased platelet reactivity in diabetics. Hyperglycaemia may impair membrane fluidity through glycation of platelet surface proteins and thereby enhance platelet adhesion [9]. Further, it activates protein kinase C, which is a mediator of platelet activation [10]. Insulin deficiency leads to an increased intracellular calcium concentration, resulting in more pronounced platelet degranulation [32]. Moreover, insulin deficiency is linked to an impaired response to nitric oxide and prostacyclin leading to higher platelet reactivity [33]. An upregulation of the P2Y12 pathway [34], increased platelet turnover [35], endothelial dysfunction [36] and oxidative stress may further contribute to the enhanced platelet activation in diabetics [37]. Since we exclusively enrolled type 2 diabetics, who are in many cases not insulin-deficient but have high insulin levels as a consequence of insulin resistance, increased platelet activation in the diabetics in our study population may mainly be caused by the other mechanisms. Finally, diabetic patients often suffer from additional metabolic disorders, in particular obesity, hyperlipidaemia and systemic inflammation. These conditions are independently associated with increased platelet adhesion and aggregation [38, 39].
In a recent study, Stellos et al. found no significant difference in platelet-bound P-selectin between diabetics and nondiabetics presenting with an ACS [16]. In accord with our results, these findings indicate that in the clinical setting of already increased platelet reactivity, diabetes may not have the same effect on platelet activation as in patients with a stable clinical condition. The already high platelet reactivity in patients with PAD/CVD may also be the reason why type 2 diabetes had no additional influence on platelet activation in these patients in our study population.
A limitation of our study is its cross-sectional design. Systemic atherosclerosis affects, more or less, arteries of all vascular beds. Therefore, we cannot exclude the possibility that patients with CAD also had other manifestations of atherosclerosis. Likewise, we have no proof that patients with PAD/CVD did not have CAD. The latter may even be too limited in their physical activity to experience symptoms of CAD in daily life. Thus, only clinical presentation guided the differentiation between CAD and PAD/CVD. Moreover, we have no data on plaque burden, the exact duration of diabetes and end-organ damage due to diabetes in the different subgroups. Finally, the healthy controls did not undergo arterial puncture, which may affect platelet activation.
Conclusion
In diabetic patients with atherosclerosis it is difficult to attribute increased platelet activation to diabetes alone. Rather, atherosclerosis and diabetes together, and mutually dependent, may contribute to increased platelet activation. Our data indicate that diabetes significantly enhances platelet activation in patients with CAD, who have less disseminated disease, whereas no such effect of diabetes is discernible if atherosclerosis affects larger parts of the vascular system. Future studies specifically addressing platelet activation and plaque burden in diabetic and nondiabetic patients are warranted to elucidate further the impact of diabetes on platelet activation in CAD as compared with different manifestations of atherosclerotic vascular disease.
Acknowledgement:The authors would like to thank Beate Eichelberger for expert technical support.
References
1 Creager MA, Luscher TF, Cosentino F, Beckman JA. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: Part I. Circulation. 2003;108:1527–32.
2 Beckman JA, Creager MA, Libby P. Diabetes and atherosclerosis: epidemiology, pathophysiology and management. JAMA. 2002;287:2570–81.
3 Feskens EJ, Kromhout D. Glucose tolerance and the risk of cardiovascular disease: the Zutphen Study. J Clin Epidemiol. 1992;45:1327–34.
4 Newman AB, Siscovick DS, Manolio TA, Polak J, Fried LP, Borhani NO, et al. Ankle-arm index as a marker of atherosclerosis in the Cardiovascular Health Study. Circulation. 1993;88:837–45.
5 Fabris F, Zanocchi M, Bo M, Fonte G, Poli L, Bergoglio I, et al. Carotid plaque, aging, and risk factors. A study of 457 subjects. Stroke. 1994;25:1133–40.
6 Haffner SM, Lehto S, Rönnemaa T, Pyörälä K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med. 1998;339:229–34.
7 Diabetes-related amputations of lower extremities in the Medicare population- Minnesota, 1993–1995. MMWR Morb Mortal Wkly Rep. 1998;47:649–52.
8 Folsom AR, Rasmussen ML, Chambless LE, Howard G, Cooper LS, Schmidt MI, et al. Prospective associations of fasting insulin, body fat distribution, and diabetes with risk of ischemic stroke. Diabetes Care. 1999;22:1077–83.
9 Winocour PD, Watala C, Perry DW, Kinlough-Rathbone RL. Decreased platelet membrane fluidity due to glycation or acetylation of membrane proteins. Thromb Haemost. 1992;68:577–82.
10 Assert R, Scherk G, Bumbure A, Pirags V, Schatz H, Pfeiffer AF. Regulation of protein kinase C by short term hyperglycaemia in human platelets in vivo and in vitro. Diabetologia. 2001;44:188–95.
11 Ay L, Kopp HP, Brix JM, Ay C, Quehenberger P, Schernthaner GH, et al. Thrombin generation in morbid obesity: significant reduction after weight loss. J Thromb Haemost. 2010;8:759–65.
12 Hong YJ, Jeong MH, Choi YH, Song JA, Kim DH, Lee KH, et al. Impact of diabetes mellitus on plaque vulnerability and clinical outcome in patients with acute myocardial infarction with plaque rupture. Int J Cardiol. 2012;154:197–8.
13 McEver RP. Selectins. Current opinion in immunology. 1994;6:75–84.
14 Dunlop LC, Skinner MP, Bendall LJ, Favaloro EJ, Castaldi PA, Gorman JJ, et al. Characterization of GMP-140 (P-selectin) as a circulating plasma protein. J Exp Med. 1992;175:1147–50.
15 Michelson AD and Furman MI. Laboratory markers of platelet activation and their clinical significance. Curr Opin Hematol. 1999;6:342–8.
16 Stellos K, Bigalke B, Stakos D, Henkelmann N, Gawaz M. Platelet-bound P-selectin expression in patients with coronary artery disease: impact on clinical presentation and myocardial necrosis, and effect of diabetes mellitus and anti-platelet medication. J Thromb Haemost. 2010;8:205–7.
17 Furman MI, Benoit SE, Barnard MR, Valeri CR, Borbone ML, Becker RC, et al. Increased platelet reactivity and circulating monocyte-platelet aggregates in patients with stable coronary artery disease. J Am Coll Cardiol. 1998;31:352–8.
18 Leger AJ, Covic L and Kuliopulos A. Protease-activated receptors in cardiovascular diseases. Circulation. 2006;114:1070–7.
19 Dormann D, Clemetson KJ and Kehrel BE. The GPIb thrombin-binding site is essential for thrombin-induced platelet procoagulant activity. Blood. 2000;96:2469–78.
20 Jurk K, Jahn UR, Van Aken H, Schriek C, Droste DW, Ritter MA, et al. Platelets in patients with acute ischemic stroke are exhausted and refractory to thrombin, due to cleavage of the seven-transmembrane thrombin receptor (PAR-1). Thromb Haemost. 2004;91:334–44.
21 Ferroni P, Basili S, Falco A, Davi G. Platelet activation in type 2 diabetes mellitus. J Thromb Haemost. 2004;2:1282–91.
22 Liani R, Halvorsen B, Sestili S, Handberg A, Santilli F, Vazzana N, et al. Plasma levels of soluble CD36, platelet activation, inflammation, and oxidative stress are increased in type 2 diabetic patients. Free radical biology & medicine. 2012;52:1318–24.
23 Eibl N, Krugluger W, Streit G, Schrattbauer K, Hopmeier P, Schernthaner G. Improved metabolic control decreases platelet activation markers in patients with type-2 diabetes. Eur J Clin Invest. 2004;34:205–9.
24 Yngen M, Norhammar A, Hjemdahl P, Wallen NH. Effects of improved metabolic control on platelet reactivity in patients with type 2 diabetes mellitus following coronary angioplasty. Diabetes & vascular disease research: official journal of the International Society of Diabetes and Vascular Disease. 2006;3:52–6.
25 Eslam RB, Reiter N, Kaider A, Eichinger S, Lang IM, Panzer S. Regulation of PAR-1 in patients undergoing percutaneous coronary intervention: effects of unfractionated heparin and bivalirudin. Eur Heart J. 2009;30:1831–6.
26 Gremmel T, Eslam RB, Koppensteiner R, Lang IM, Panzer S. Prasugrel reduces agonists’ inducible platelet activation and leukocyte-platelet interaction more efficiently than clopidogrel. Cardiovasc Ther. 2013; Epub ahead of print.
27 Eikelboom JW, Weitz JI, Budaj A, Zhao F, Copland I, Maciejewski P, et al. Clopidogrel does not suppress blood markers of coagulation activation in aspirin-treated patients with non-ST-elevation acute coronary syndromes. Eur Heart J. 2002;23:1771–9.
28 Frelinger AL, Li Y, Linden MD, Tarnow I, Barnard MR, Fox ML, et al. Aspirin “resistance”: role of pre-existent platelet reactivity and correlation between tests. J Thromb Haemost. 2008;6:2035–44.
29 Frelinger AL 3rd, Michelson AD, Wiviott SD, Trenk D, Neumann FJ, Miller DL, et al. Intrinsic platelet reactivity before P2Y12 blockade contributes to residual platelet reactivity despite high-level P2Y12 blockade by prasugrel or high-dose clopidogrel. Results from PRINCIPLE-TIMI 44. Thromb Haemost. 2011;106:219–26.
30 Welten GM, Schouten O, Hoeks SE, Chonchol M, Vidakovic R, van Domburg RT, et al. Long-term prognosis of patients with peripheral arterial disease: a comparison in patients with coronary artery disease. J Am Coll Cardiol. 2008;51:1588–96.
31 Michelson AD, Barnard MR, Krueger LA, Valeri CR, Furman MI. Circulating monocyte-platelet aggregates are a more sensitive marker of in vivo platelet activation than platelet surface P-selectin: studies in baboons, human coronary intervention, and human acute myocardial infarction. Circulation. 2001;104:1533–7.
32 Ishida M, Ishida T, Ono N, Matsuura H, Watanabe M, Kajiyama G, et al. Effects of insulin on calcium metabolism and platelet aggregation. Hypertension. 1996;28:209–12.
33 Anfossi G, Mularoni EM, Burzacca S, Ponziani MC, Massucco P, Mattiello L, et al. Platelet resistance to nitrates in obesity and obese NIDDM, and normal platelet sensitivity to both insulin and nitrates in lean NIDDM. Diabetes Care. 1998;21:121–6.
34 Matsuno H, Tokuda H, Ishisaki A, Zhou Y, Kitajima Y, Kozawa O. P2Y12 receptors play a significant role in the development of platelet microaggregation in patients with diabetes. The Journal of clinical endocrinology and metabolism. 2005;90:920–7.
35 Guthikonda S, Alviar CL, Vaduganathan M, Arikan M, Tellez A, DeLao T, et al. Role of reticulated platelets and platelet size heterogeneity on platelet activity after dual antiplatelet therapy with aspirin and clopidogrel in patients with stable coronary artery disease. J Am Coll Cardiol. 2008;52:743–9.
36 Schafer A and Bauersachs J. Endothelial dysfunction, impaired endogenous platelet inhibition and platelet activation in diabetes and atherosclerosis. Current vascular pharmacology. 2008;6:52–60.
37 Jardin I, Redondo PC, Salido GM, Pariente JA, Rosado JA. Endogenously generated reactive oxygen species reduce PMCA activity in platelets from patients with non-insulin-dependent diabetes mellitus. Platelets. 2006;17:283–8.
38 Anfossi G, Russo I and Trovati M. Platelet dysfunction in central obesity. Nutr Metab Cardiovasc Dis. 2009;19:440–9.
39 Lim HS, Blann AD and Lip GY. Soluble CD40 ligand, soluble P-selectin, interleukin-6, and tissue factor in diabetes mellitus: relationships to cardiovascular disease and risk factor intervention. Circulation. 2004;109:2524–8.