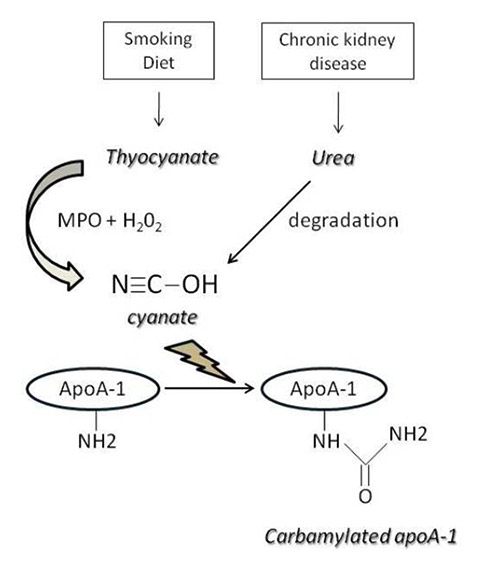
Figure 1
Mechanisms of apolipoprotein A-1 (apoA-1) carbamylation.
MPO = myeloperoxidase
DOI: https://doi.org/10.4414/smw.2013.13781
Apolipoprotein A-1 (apoA-1) constitutes the principal protein fraction of high-density lipoprotein (HDL) whose protective role in the cardiovascular system derives to a great extent from the inverse relationship between HDL-cholesterol and apoA-1 plasma concentrations, and a risk of myocardial infarction [1]. The atheroprotective role of HDL in the cardiovascular system has been attributed to its pleiotropic effects, including reverse cholesterol transport, vasodilatation, and antithrombotic, anticoagulant and anti-inflammatory effects [2]. Mirroring those versatile properties, mass spectrometry analyses revealed that HDL encompasses very heterogeneous macromolecular complexes of lipids and proteins. Only one-third of the up to 80 different proteins identified in HDL is dedicated to lipid transport. The remaining proteins are either acute-phase proteins, proteases, antioxidant or antithrombotic enzymes, or proteins involved in complement regulation [3].
As well as being the principal protein fraction of HDL and a limiting factor for HDL formation, apoA-1 per se has many of the HDL-related atheroprotective properties [4]. Apart from its pivotal role in reverse cholesterol transport (RCT), where it induces cholesterol efflux via an adenosine triphosphate (ATP) binding cassette transporter and activates lecithin:cholesterol acyltransferase (LCAT) [5], the second best-studied property of apoA-1 is its mitigating effect on inflammation through several pathways [2–4]. However, there is a growing body of evidence that both acute and chronic inflammatory conditions induce post-translational modifications of apoA-1. These, in turn, not only dampen the functions regulating lipid homoeostasis and the anti-inflammatory properties of HDL/ApoA-1, but even turn it into a proinflammatory molecule [4].
Thus, HDL and apoA-1 appear to contribute to host defence against many biological and chemical hazards. However, analogous to failing or erroneous host defences through leukocytes and their humoral mediators, the protective activities of HDL can be compromised and made to fail, or even perverted to harm. The literature regarding apoA-1 is complex and often blurred by the fact that biological activities are frequently attributed to apoA-1 alone, to apoA-1 in association with different lipids or even to HDLs. Therefore, the aim of the present paper is to review specifically the pathways by which apoA-1 per se or as part of HDL acts as an anti-inflammatory molecule, and to analyse the translational and post-translational modifications to apoA-1 that could lead to a proinflammatory phenotype. Several natural apoA-1 variants have been described and some are characterised by alterations in lipid binding, LCAT activation and cholesterol efflux stimulation in vitro, as well as defective HDL formation in vivo. However, these are out of the scope of this review, and will not be discussed here.
Human apoA-1 is a 28-kD protein with 243 amino acid residues encoded by the apolipoprotein multigene superfamily located on chromosome 11q23 [6]. The protein is synthesised as a preprosequence of apoA-1 (24 amino acid residues longer), primarily by hepatocytes in the liver and also by enterocytes. The largest part of its secondary structure consists of class A amphipathic α-helices separated by proline hinges. In these helices, apolar residues occupy one face whereas polar residues occupy the other face, and positively charged lysine and arginine residues separate the two faces of the helix. This amphipathic structure allows the interaction of apoA-1 with lipids through its hydrophobic faces and with the aqueous phase through its hydrophilic faces. The proline hinges confer a high steric flexibility on the apoA-1 molecule, allowing important conformational changes needed to occupy both discoidal and spherical HDL particles of different sizes [7].
Upon maturation, HDL undergoes conformational changes accompanied by important functional changes, enabling interaction with a wide range of different proteins including ATP binding cassette transporters A1 (ABCA1) and G1 (ABCG1), as well as LCAT and scavenger receptor B1 (SR-B1), which are expressed mostly by hepatocytes and macrophages [8–10].
Lipid-free apoA-1 represents up to 10% of circulating apoA-1 [9], which is present in plasma in normal conditions at a concentration of 90–250 mg/dl, slightly higher in women than in men. In pathological conditions apoA-1 behaves as a negative (“reverse”) acute-phase protein, that is, a protein whose level is lowered by more than 25% during the acute-phase response and thought to be displaced by serum amyloid A (SAA) which is a “positive” acute-phase protein [11]. However, it was demonstrated subsequently that during the acute-phase response, proinflammatory cytokines increase the expression of SAA, while simultaneously decreasing the expression of apoA-1 and paraoxonase 1 (PON-1), which suggests that the reduction in apoA-1 during the acute-phase response is not only due to physical displacement of apoA-1, but to inverse transcriptional regulation in the liver [12].
Site-directed mutagenesis studies resulted in the identification of structural domains which specifically modulate the functionalities of apoA-1. So far, most reports have dealt with the effects of lipid-free apoA-1 and apoA-1-related mimetic peptides on cholesterol efflux models mediated by ABCA1 as well as LCAT activation [9], for which the C-terminal (amino acid residues (aa): 187‒243) and central domains (aa: 143‒165), respectively, are important [13]. The N-terminal part of lipid-free apoA-1 (aa: 1‒98) has been shown to be essential for stabilising its lipid-free conformation, as well as for lipid interaction [14–16]. Furthermore, stabilising the α-helix of apoA-1 mimetic peptides translated into higher cholesterol efflux efficacy, further confirming that the conformational state of apoA-1 is crucial for its function in regard to cholesterol efflux [17].
HDLs have been considered to be anti-inflammatory molecules for 20 years [18]. However, whether these effects are mainly due to the lipid (for example, sphingosine-1-phosphate) or the protein constituents of HDL (apoA-1, but also quantitatively minor proteins such as paraoxonase (PON) or clusterin) is still a matter of debate. In the following paragraphs, we will review only the specific anti-inflammatory properties of HDL/apoA-1.
In the mid-nineties, apoA-1 was observed to display anti-inflammatory properties by inhibiting the cytokine-induced expression of cell adhesion molecules such as vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1) and E-selectin, which are key players in diapedesis of immunocompetent cells from the circulation into the artery wall [19]. Since the latter phenomenon is considered to be one of the earliest events in atherogenesis, those seminal findings raised the possibility that apoA-1 could represent an innovative therapeutic alternative for preventing atherosclerosis at an early stage. They also provided initial evidence for an atheroprotective role of apoA-1 independent of cellular cholesterol homeostasis regulation. Later on, the same group demonstrated that apoA-1 or phospholipids per se were not sufficient to exert the anti-inflammatory properties of lipid-poor apoA-1, so that for optimal function both must coexist on one particle. In line with this, the shape of apoA-1containing HDLs also influences the anti-inflammatory response, spherical apoA-1-containing HDLs being more anti-inflammatory than discoidal ones [19]. Those results were the first to indicate a significant role of the conformational state of apoA-1 and HDLs in ensuring the optimal mediation of their anti-inflammatory effects. Afterwards, it was shown that HDL/apoA-1 modulates the cytokine-induced integrin expression by activated endothelial cells, as extensively reviewed elsewhere [20].
ApoA-1 has been shown to inhibit both shear stress and phorbol-12 myristate 13-acetate (PMA) induced monocyte expression of CD11b, one of the integrins, complementary to VCAM-1 and ICAM-1, involved in the early stages of transendothelial migration. In this study, the dose-dependent inhibition of CD11b expression was accompanied by decreased transendothelial migration of monocytes toward chemotactic agents such as monocyte chemoattractant protein-1 (MCP-1). This activity was strongly dependent on ABCA1, but independent of SR-B1 signalling [21]. Those results confirmed and extended a previous observation that HDL inhibits LDL-induced MCP-1 production in monocytes as well as their subsequent transendothelial diapedesis [22]. Further insight into the molecular mechanisms underlying this phenomenon was gained from the observation that apoA-1 interaction with ABCA1 promoted signal transducer and activator of transcription (STAT-3) activation through janus kinase (JAK) 2, which ultimately abolished the lipopolysaccharide (LPS) induced proinflammatory response (such as interleukin (IL) 1β, IL-6, and tumour necrosis factor alpha (TNF-α) production by monocytes), without affecting cholesterol efflux [23]. Altogether, these results suggested that ABCA1 is not only involved in cholesterol metabolism, but also modulates the anti-inflammatory response of cells to apoA-1.
The charge and size of the hydrophobic face of synthetic apoA-1 peptides are major factors affecting their anti-inflammatory properties. More specifically, inhibition of VCAM-1 expression was maximised by increasing the hydrophobic face size and the number of positively charged residues. Inhibition of CD11b expression in monocytes increased with asymmetry and hydrophobicity, but diminished with increasing numbers of positively charged residues, further emphasising the importance of apoA-1 conformation to the deployment of its anti-inflammatory properties [19, 24].
In conclusion, in specific conformations, apoA-1 can inhibit transendothelial migration of immunocompetent cells, both by blocking the production of chemotactic cytokines by monocytes and by impeding the expression by endothelial and monocytic cells of integrins required for efficient diapedesis in response to chemotactic agents.
Once transendothelial migration of inflammatory cells has occurred, the interaction between monocyte-macrophages and T cells represents another important step in modulating the microenvironment of the chronic inflammatory response. Indeed, direct contact between activated T cells and monocytes/macrophages is a major driver for the production of several inflammatory cytokines, such as IL-1β, IL-6, IL-8, MCP-1 and TNF-α, which are well known for being involved in atherogenesis and tissue destruction [25, 26]. ApoA-1 has been shown to inhibit contact-mediated monocyte stimulation and the subsequent production of IL-1β and TNF-α. Although the exact mechanisms of action are not fully elucidated yet, the authors suggested that apoA-1 blocks T-cell/monocyte interactions at the T cell level [25]. This conclusion is in accordance with previous work suggesting the presence of a specific, but as yet unidentified, HDL binding site on human lymphocytes [27]. Additional studies in this model of cell-cell interaction demonstrated that HDL/apoA-1 inhibits T-cell contact-activated specific proinflammatory gene expression in monocytes [28]. In contrast, HDL/apoA-1 does not affect or inhibit - or only to a small extent - the expression of molecules displaying anti-inflammatory and regulatory functions such as IL-1 receptor antagonist (IL-1Ra). Microarray analysis shows that 437 out of 54,675 probe sets were enhanced in monocytes activated by contact with stimulated T cells of which 164 (i.e. 38%) were inhibited in the presence of HDL. These results were validated using real-time polymerase chain reaction (qPCR) [28].
These in-vitro observations were confirmed in vivo in various chronic and acute pathological conditions. For example, immunohistochemistry on synovial biopsies from rheumatoid arthritis patients revealed that apoA-1 was consistently present in inflamed synovial tissue that contained infiltrating T cells and macrophages, but was absent from noninflamed tissue samples obtained from treated patients and from normal subjects. ApoA-1 was not present in synovium from patients in apparent remission, which suggests that it has a specific role during phases of disease activity [29]. On the other hand, HDL/apoA-1 was markedly reduced in patients admitted to an intensive care unit for systemic inflammatory response syndrome. Patients with an exacerbated inflammatory state had significantly lower plasma levels of apoA-1 than those whose inflammation was not exacerbated, which suggests a protective role of apoA-1. Moreover, a correlation was established between the levels of apoA-1 in patients’ sera and their in-vitro inhibitory effect on the release of IL-1β by T-cell-stimulated monocytes [30]. There is currently no information available as to the conformational requirements for apoA-1 to modulate the cell-cell contact inhibition. However, a specific blocking antibody to apoA-1 proved to interfere with the inhibitory action of HDL [30].
Another important step in inflammation and atherogenesis is the uptake of lipid peroxidation products by macrophages, with oxidised low-density lipoproteins (oxLDL) being key players in early and late stages of atherogenesis [31–32]. The proatherogenic properties of oxLDLs consist mostly in fostering sterile inflammation through the activation of various innate immune receptors that trigger numerous signalling pathways, as extensively reviewed elsewhere [31–32]. In 1979 it was demonstrated that HDLs are able to inhibit LDL oxidation [33]. Subsequent studies demonstrated that this feature was related to the presence of apoA-1 on HDL molecules, and that it could be fully reproduced by apoA-1 mimetic peptides [2]. This antioxidant property of apoA-1 is believed to be mainly, but not exclusively, due to PON-1, an enzyme associated with apoA-1 and known to prevent different kinds of lipid peroxidation [34]. Preserving the proline bridge on bihelical apoA-1 synthethic peptides was found to be detrimental to the antioxidant properties of the peptides, whereas modifications of the charge, the hydrophobicity, or the size of hydrophobic face had only a limited impact on the antioxidant capacity of those peptides [24].
Considering that antioxidant agents failed to prevent cardiovascular disease in randomised controlled trials, the clinical benefit of these antioxidant properties remains to be determined [35].
The innate immune response represents the first line of defence against infectious agents and modified autoantigens (modified/oxidised lipids and apoptotic cells), allowing their rapid clearance through lysosomal degradation, and at the same time drives the development of a more specific and efficient adaptive response. The innate immune system consists of both cellular and humoral components. Major cellular components are neutrophils, monocyte/macrophages, natural killer (NK) and lymphokine-activated killer (LAK) cells, and eosinophils. The humoral counterpart is mostly represented by the pentraxin family, the complement system and various cytokines. Three main classes of innate immune receptors have been described: Toll-like receptors (TLRs), scavenger receptors (SRs) and Nod-like receptors (NLRs). The former two are considered important mediators of the immune-mediated inflammation that characterises atherogenesis [36–37]. Both HDL and apoA-1 have been shown to modulate either directly or indirectly the signalling of the innate immune system at different levels.
As early as 1979, the effect of HDL on the innate immune system proved to reduce LPS toxicity in vivo[38]. In vitro, LPS stimulates substantially more IL-1 mRNA and cell-associated IL-1 protein when monocytes are stimulated with LPS alone than with LPS-HDL [39]. This activity was later attributed to the fact that HDL sequesters LPS and thereby prevents the transduction of the proinflammatory cascade through TLR4/CD14 complex interaction [40]. By using several apoA-1 mutants, apoA-1 was found to be the key component of HDL-related LPS neutralisation. Indeed, the N-terminal region (aa: 52‒74) of apoA-1 was identified as being specific to this effect, without affecting cholesterol efflux from macrophages [41].
As well as preventing upstream TLR4 activation by impeding the interaction with its ligand, apoA-1 has also been shown to inhibit TLR4 signalling directly by impeding its transport into lipid rafts [42]. Although controversial, recent work identified apoA-1 not only as a circulating molecule, but also as a cellular molecule expressed in low amounts by macrophages. It has been localised in lipid rafts and in the vicinity of ABCA1 [43]. These authors reported that, by its spatially close association, apoA-1 stabilises the membrane expression of ABCA1. Moreover, suppressed endogenous apoA-1 was associated with increased expression of TLR4 at the mRNA level [43]. According to these findings, endogenous and circulating apoA-1 can modulate TLR4 signalling. To the best of our knowledge, such evidence is currently lacking for other TLRs.
SRs were the first innate immune receptors known to have apoA-1 as one of their numerous ligands. Among them, SR-B1 is known to be an important effector of the various anti-inflammatory functions of HDL. Indeed, apoA-1/SR-B1 interaction is crucial for cholesterol efflux [22], nitric oxide synthesis, glucocorticoid synthesis and TLR4 signalling [9–10, 44]. However, as discussed later in the proinflammatory section, apoA-1 interaction with SRs could have contradictory effects on inflammation control.
Another important mechanism for the functionality of innate immune receptors is their targeting on functional lipid rafts, which allows their interaction with other coreceptors for the optimal activation downstream of specific signalling pathways. Accordingly, the integrity of lipid rafts has been shown to be crucial for appropriate proinflammatory TLR signalling [45]. In this respect, apoA-1 has been shown to induce cholesterol depletion from lipid rafts, thereby decreasing TLR4 functionality and inhibiting LPS-induced inflammatory responses [46].
Because lipid rafts on antigen presenting cells (APCs) also concentrate major histocompatibility complex (MHC) class 2 molecules, which are known to play a crucial part in T-lymphocyte activation [47], it can be hypothesised that apoA-1 may have a significant impact on the adaptive aspect of the immune response. Nevertheless, to the best of our knowledge, this appealing hypothesis has never been formally validated.
Among other major players of the immune system, regulatory T cells (Tregs) are important components of the adaptive immune response, acting to maintain self-tolerance. Tregs suppress the activation of T cells by either a contact-dependent process involving APCs or secretion of anti-inflammatory cytokines such as IL-10 and transforming growth factor beta (TGF-β) [48]. The absence of, or defects in, Tregs can lead to inflammatory disease such as autoimmune disorders and atherosclerosis [48].
ApoA-1-/-x LDL-receptor-/- double knock-out mice exhibit an autoimmune phenotype. In this model, administration of exogenous apoA-1 was found to prevent the differentiation of T cells into a proinflammatory Th17 phenotype, and to increase the Treg population. These anti-inflammatory effects at the cellular level were accompanied macroscopically by the restoration of a close-to-normal phenotype of skin architecture, morphology and composition [49]. The mechanisms underlying the beneficial effect of apoA-1 administration on Treg function are still unknown.
As summarised in table 1, apoA-1 acts as a pleiotropic anti-inflammatory agent by modulating the inflammatory response at the prereceptor, receptor and postreceptor levels, mainly on innate APCs. It also appears to act on the adaptive compartment by decreasing Th17 polarisation and promoting Treg expansion. As a consequence, apoA-1 can inhibit the production of proinflammatory cytokines, and the cell contact-mediated inflammatory response. It also prevents the generation of self-modified antigens by inhibiting the formation or persistence of lipid peroxidation products. Together, these effects have the potential ultimately to translate into reduced chemotaxis, diapedesis and inflammatory activity of immunocompetent cells at the site of inflammation.
| Table 1: Summary of apolipoprotein A-1 (apoA-1) related anti-inflammatory properties in relation with apoA-1 specific regions and conformation. | |||
| ApoA-1 anti-inflammatory properties | ApoA-1 specific region | Conformation-dependent | Reference number |
| LPS binding | N-terminal (aa:52‒74) | Unknown | [38–40] |
| ABCA1 interaction | C-terminal | Yes, hydrophobic regions close to negatively charged cluster | [8, 9, 13] |
| SR-B1 interaction | C-terminal | Yes, positively charged residues | [10, 24] |
| TLR4 expression/signalling inhibition | No specified | Unknown | [41–43, 45, 46] |
| Inhibition of endothelial integrin expression (VCAM1, ICAM1, E-selectin) | Not specified | Yes, depends on the hydrophobic face size and the number of positively charged residues | [19–20, 24] |
| Inhibition of monocytes integrin expression (CD11b) | Not specified, but ABCA1 Dependent; C-terminal? | Yes, depends on the asymmetry and hydrophobic face size, and the negatively charged residues | [21, 22] |
| Lipid peroxidation | Not specified | Yes, depends on the presence of the proline bridge, determining the angle between helixes | [33, 34, 24] |
| T cell/Monocyte interaction | Not specified | Unknown | [25, 26, 28] |
| Increase Treg differentiation | Not specified | Unknown | [49] |
| Aa = amino acid residues; ABC = ATP binding cassette transporter; ATP = adenosine triphosphate; ICAM = intercellular adhesion molecule; LPS = lipopolysaccharide; SR = scavenger receptor; TLR = Toll-like receptor; Treg = regulatory T cell; VCAM = vascular cell adhesion molecule. | |||
In both acute and chronic inflammation, HDL can become proinflammatory [4], a phenomenon initially explained by the replacement of apoA-1 by serum amyloid A (SAA) protein, ceruloplasmin and haptoglobin [4], with SAA representing up to 87% of HDL proteins [50]. These SAA-containing HDLs may be retained in the arterial intima through facilitated interaction with vascular proteoglycans, and are thus more extensively exposed to oxidative modifications. Recent studies reviewed in the next paragraphs demonstrate that other translational and post-translational modifications can turn apoA-1 into a proinflammatory molecule.
Figure 1
Mechanisms of apolipoprotein A-1 (apoA-1) carbamylation.
MPO = myeloperoxidase
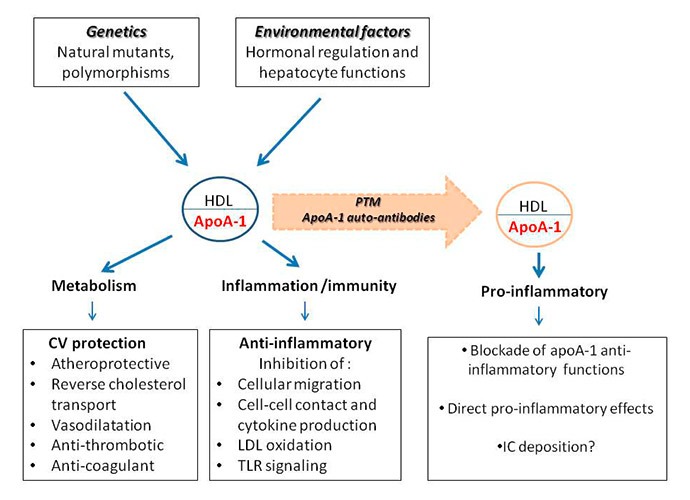
Figure 2
The pro- and anti-inflammatory role of apolipoprotein A-1 (apoA-1) / high-density lipoprotein (LDL).
CV = cardiovascular; IC = immune complexes; LDL = low-density lipoprotein; PTM = post-translational modifications; TLR = Toll-like receptor.
ApoA-1 is involved in two forms of amyloidosis, a disease affecting vital organs such as kidney, liver and heart; amyloidosis therefore constitutes a suitable human paradigm to investigate the pathophysiological consequences of apoA-1 modifications, both at the translational and post-translational levels. In both nonhereditary and hereditary amlyoidosis, modifications of apoA-1 (either through oxidation or proteolysis in the former, or through mutations in the latter, form of the disease) lead to the formation of amyloid fibrils/complexes found in various organs and in atherosclerotic plaques [51]. Although their involvement in inflammation and atherosclerosis is just beginning to be elucidated, it seems that newly formed amyloid complexes, fibrils or β-sheets are capable of inhibiting reverse cholesterol transport [52] and generate a strong proinflammatory response through the signalling receptor of advanced glycation end products (AGE) [53]. Nevertheless, those in vitro observations have not yet been validated by experimental evidence in vivo.
Functionally relevant post-translational modifications of apoA-1 include chlorination, nitration and oxidation of tyrosine residues, which are catalysed by myeloperoxidase (MPO). ApoA-1 and HDL derived from patients with cardiovascular disease had a significantly higher content of MPO-dependent oxidative modifications, which are functionally associated with a reduced capacity of apoA-1 to bind to ABCA1 and to induce ABCA1-mediated cholesterol efflux [54]. Physiologically relevant concentrations of MPO may generate dysfunctional HDLs characterised by a reduced cholesterol efflux capacity [55]. These findings were the first to demonstrate a causal link between specific apoA-1 modifications inducing dysfunctional HDL and a proinflammatory status through a specific MPO-driven pathway.
Another post-translational HDL modification driven by MPO reported recently is carbamylation [56], whose clinical relevance to cardiovascular disease, especially in renal failure patients, is well established [57]. Protein carbamylation is mediated by cyanate, which is in turn produced either by MPO or urea decomposition (fig. 1). Carbamylated apoA-1 was found to be significantly increased in advanced atherosclerotic lesions when compared with vessels with early lesions, but no such differences in plasma HDL levels were noted between those groups. The authors also demonstrated that in-vitro carbamylation of apoA-1 inhibits SR-B1 but not ABCA1-mediated cholesterol efflux, leading to a marked cholesterol accumulation in THP-1 macrophages [57]. Those results constitute further evidence of apoA-1 modifications affecting HDL atheroprotective properties.
Reactive carbonyls can also generate specific apoA-1 products derived from oxidation/glycation. Reactive carbonyls are generated either by carbohydrate oxidation, which gives rise to proinflammatory AGE molecules [58], or lipid peroxidation, which produces advanced lipoxidation end products (ALE) [58, 59]. Both ALE and AGE have been associated with diabetes and cardiovascular disease [58, 59], and were shown to modify native apoA-1 in different but specific ways. Nobecourt and co-workers demonstrated that, after exposure to glycation and subsequent incorporation into discoidal reconstituted HDLs, apoA-1 lost its capacity to activate LCAT. This loss was associated with modifications of arginine, lysine and tryptophan residues [60]. Recently, the same group showed that apoA-1 glycation induced by methylglyoxal in vitro or in vivo resulted in apoA-1 failing to inhibit both the infiltration of neutrophils into the intima of rabbit carotid arteries and the expression of ICAM-1 and VCAM-1 in endothelial cells. This effect was accompanied by increased nuclear NF-κB translocation when compared with native apoA-1, and a reduced ability to inhibit the formation of reactive oxygen species (ROS) [61]. When exposed to methylglyoxal, reconstituted HDLs displayed the same loss of function of their anti-inflammatory properties when compared with lipid-free apoA-1, suggesting that those apoA-1 modifications driven by carbonyl-reactive species can generate dysfunctional HDL in terms of cholesterol efflux, and anti-inflammatory and antioxidant properties. These appealing in-vitro findings still need to be validated in relevant human in vivo models.
In humans, low apoA-1 concentrations are associated not only with increased risk for cardiovascular disease, but also autoimmune diseases, such as rheumatoid arthritis and systemic lupus erythematosus (SLE) [29]. In accordance with these observations in humans, hypercholesterolaemic mice lacking plasma apoA-1 display an increased susceptibility to autoimmunity characterised by enlarged lymph nodes, cellular activation and proliferation, as well as lipid accumulation [62].
Autoantibodies to apoA-1 have been primarily shown to be increased in autoimmune conditions associated with increased cardiovascular risk, such as SLE [63, 64], primary antiphospholipid syndrome [65], and subsequently in patients with rheumatoid arthritis [66, 67], as well as in patients suffering from acute coronary syndromes (ACS) [68–70] and severe carotid stenosis [71]. In ACS and rheumatoid arthritis patients, high levels of these autoantibodies were found to predict independently poor cardiovascular prognosis [67, 72]. From a pathophysiological point of view, these autoantibodies elicit a direct proinflammatory effect through TLR2/CD14 complex signalling [73] and neutrophil chemotaxis in vitro, as well as promoting atherogenesis and atherosclerotic plaque vulnerability in vivo [71]. Furthermore, these autoantibodies have also been shown in vitro to act as a positive chronotropic agent in the presence of aldosterone [72], an effect recently attributed to a protein kinase A-dependent L-type calcium channel activation [74].
Furthermore, in some circumstances anti-apoA-1 autoantibodies have been associated with a decrease in the beneficial anti-inflammatory effects of apoA-1. Indeed, clinical and animal studies indicate that those autoantibodies impeded some of the HDL-related antiatherogenic properties, leading to dysfunctional HDLs. In SLE and primary antiphospholipid syndrome patients, as well as in lupus-prone mice, the presence of these autoantibodies was associated with a decrease in the antioxidant properties of HDL due to decreased PON-1 activity [64, 75, 76], and increased proinflammatory ROS levels [65].
Whether the existence of these apoA-1 antibodies is the cause or the consequence of the aforementioned apoA-1 modifications is still unclear. In favour of the former hypothesis would be that MPO or apoA-1 modifications mediated by reactive carbonyls give rise to neo-epitopes, followed by the appearance of autoantibodies. On the other hand, antibodies may act as oxidative proteins generating a pro-oxidant microenvironment through their ability to generate ROS via water oxidation [77]. If this effect optimises the affinity of antibodies for their target [78], it is possible that such antibody-related oxidative properties could induce oxidative modifications in their target. In this scenario, autoantibodies to apoA-1/HDL may lead to the formation of proinflammatory apoA-1/HDLs. Further work is required to confirm or reject those hypotheses.
Finally, as hormones markedly influence autoimmune diseases, and the oestrogen/androgen balance determines the synthesis of apoA-1 in the liver [79], it is essential to find out whether the hormonal environment could modulate the occurrence of anti-apoA-1 antibodies. All these aspects are summarised in figure 2.
| Table 2:Summary of apolipoprotein A1 (apoA-1) post-translational modifications leading to dysfunctional apoA-1. | |||||
| ApoA-1 modificatios | ApoA-1 specific region | Effector | End products | Functional consequences | References Number |
| Non hereditary Amyloidosis-related oxidation/proteolysis | N and C-terminal regions | Unknown | Amyloid fibrils/complexes | AGE receptor activation | [51, 52] |
| Oxidation | Central region, methionine 148 | MPO | Oxidized methionine apoA-1 | LCAT inhibition | [52] |
| C-terminal, Tyrosine 192 | MPO | apoA-1 tyrosine chlorination | ABCA1-mediated cholesterol efflux inhibition | [54, 55] | |
| C-terminal, Lysine residues | Reactive carbonyls | ALE | ABCA1-mediated cholesterol efflux inhibition | [61] | |
| Nitration | Tyrosine 29, 166, 192, 236 | MPO | apoA-1 Nitration | ABCA1-mediated cholesterol efflux inhibition | [54] |
| Carbamylation | Lysine residues, unspecific | MPO | carbamallysine, homocitrulline | SR (A-1 and B1)-mediated cholesterol efflux inhibition | [56, 57] |
| Glycation | Arigine, lysine and tryptophane residues, unspecific | Reactive carbonyls | AGE | LCAT inhibition Enhanced neutrophil chemotaxis Enhanced ICAM-1 and VCAM-1 expression, NFκB activation, and AGE receptor activation | [58–60] |
| ABC = ATP binding cassette transporter; AGE = advanced glycation end products; ALE = advanced lipoxidation end products; ATP = adenosine triphosphate; ICAM = intercellular adhesion molecule; LCAT = lecithin:cholesterol acyltransferase; MPO = myeloperoxidase; SR = scavenger receptor; VCAM = vascular cell adhesion molecule. | |||||
ApoA-1 displays pleiotropic anti-inflammatory properties by modulating the innate inflammatory response at the prereceptor, receptor and postreceptors levels. It also appears to act on the adaptive compartment by decreasing Th17 polarisation and promoting Treg expansion. As a consequence, lipid-free apoA-1 inhibits the production of proinflammatory cytokines without affecting anti-inflammatory cytokines (i.e. IL-1Ra), cell contact-mediated inflammatory response, and the generation of self-modified antigen by inhibiting lipid peroxidation products. Together, these effects translate into impaired chemotaxis and diapedesis, and decreased pro-inflammatory activity of immunocompetent cells at the site of inflammation.
Nevertheless, different groups demonstrated in past years that specific post-translational modifications of apoA-1 (summarised in table 2) that occur in the context of systemic inflammation can transform this genuine anti-inflammatory molecule into a proinflammatory one. Oxidative damage mediated either by myeloperoxidase (MPO) or reactive carbonyls as well as glycation and MPO-driven carbamylation appear to be the most important pathways leading to the transformation of anti-inflammatory apoA-1 into a pro-inflammatory molecule. Furthermore, humoral autoimmunity to apoA-1 and HDL has been reported in populations at high cardiovascular risk and may constitute another mechanism potentially leading to proinflammatory apoA-1/HDL.
From a clinical perspective, the recent unfortunate failures of cholesteryl ester transfer protein (CETP) inhibitors (ILLUMINATE trial for torcetrapib and Dal-Outcomes trial for dalcetrapib) to prevent cardiovascular disease [80, 81] casts doubt on the “HDL-raising hypothesis”. The one-year cardiovascular mortality increase observed with torcetrapib (prompting study interruption) has been attributed to aldosterone-related off-target effects of this molecule and to deleterious effects on endothelial function [82, 83], but the reasons for the failure of dalcetrapib, devoid of such deleterious properties, to prevent cardiovascular disease despite raising HDL levels effectively are still unclear [81, 84]. The results of ongoing trials involving two other promising CETP inhibitors (anacetrapib and evacetrapib) are therefore eagerly awaited before any firm conclusion can be drawn about the efficacy of these therapeutic agents to prevent cardiovascular disease. In conclusion, the failures of CETP inhibitors to prevent cardiovascular disease despite inducing a sustained HDL increase stress the clinical importance of understanding the mechanisms and the circumstances driving apoA-1 and HDL towards pro- or anti-inflammatory molecules. These results also suggest that in parallel to in-vitro models of cholesterol efflux, models testing the proinflammatory response should be highly encouraged in order to determine the potential future therapeutic benefit of apoA-1 and apoA-1 mimetic peptides. Nevertheless, because these hypotheses are currently without any clinical evidence, and because no consensus exists on how HDL functionality should be determined, no rapid changes in the current management of dyslipidaemia and cardiovascular risk stratification [85] are expected to occur.
1 Emerging Risk Factors Collaboration, Di Angelantonio E, Sarwar N, Perry P, Kaptoge S, et al. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302(18):1993–2000.
2 Gordon SM, Davidson WS. Apolipoprotein A-I mimetics and high-density lipoprotein function. Curr Opin Endocrinol Diabetes Obes. 2012;19(2):109–14.
3 Heinecke JW. The HDL proteome: a marker – and perhaps mediator – of coronary artery disease. J Lipid Res. 2009;50(Suppl.):S167–71.
4 Navab M, Reddy ST, Van Lenten BJ, Fogelman AM. HDL and cardiovascular disease: atherogenic and atheroprotective mechanisms. Nat Rev Cardiol. 2011;8:222–32.
5 Khera AV, Rader DJ. Future therapeutic directions in reverse cholesterol transport. Curr Atheroscler Rep. 2010;12:73–81.
6 Li WH, Tanimura M, Luo CC, Datta S, Chan L. The apolipoprotein multigene family: biosynthesis, structure function relationships, and evolution. J Lipid Res. 1988;29(3):245–71.
7 Segrest JP, Garber DW, Brouillette CG, Harvey SC, Anantharamaiah GM. The amphipathic a helix: A multifunctional structural motif in plasma apolipoproteins. Adv Protein Chem. 1994;45:303–69.
8 Huang R, Silva RA, Jerome WG, Kontush A, Chapman MJ, Curtiss LK, et al. Apolipoprotein A-I structural organization in high-density lipoproteins isolated from human plasma. Nat Struct Mol Biol. 2011;18(4):416−22.
9 Frank PG, Marcel YL. Apolipoprotein A-I: structure–function relationships. J Lipid Res. 2000;41:853–72.
10 Yuhanna IS, Zhu Y, Cox BE, Hahner LD, Osborne-Lawrence S, Lu P, et al. High-density lipoprotein binding to scavenger receptor-BI activates endothelial nitric oxide synthase. Nat Med. 2001;7:853–7.
11 Van Lenten BJ, Wagner AC, Nayak DP, Hama S, Navab M, Fogelman AM. High-density lipoprotein loses its anti-inflammatory properties during acute influenza a infection. Circulation. 2001;8;103(18):2283–8.
12 Han CY, Chiba T, Campbell JS, Fausto N, Chaisson M, Orasanu G, et al. Reciprocal and coordinate regulation of serum amyloid A versus apolipoprotein A-I and paraoxonase-1 by inflammation in murine hepatocytes. Arterioscler Thromb Vasc Biol. 2006;26(8):1806–13.
13 Cho KH, Durbin DM, Jonas A. Role of individual amino acids of apolipoprotein A-I in the activation of lecithin:cholesterol acyltransferase and in HDL rearrangements. J Lipid Res. 2001;42(3):379–89.
14 Ajees AA, Anantharamaiah GM, Mishra VK, Hussain MM, Murthy HM. Crystal structure of human apolipoprotein A-I: insights into its protective effect against cardiovascular diseases. Proc Natl Acad Sci USA. 2006;103(7):2126–31.
15 Rogers DP, Brouillette CG, Engler JA, Tendian SW, Roberts L, Mishra VK, et al. Truncation of the amino terminus of human apolipoprotein A-I substantially alters only the lipid-free conformation. Biochemistry. 1997;36(2):288–300.
16 Thomas MJ, Bhat S, Sorci-Thomas MG. Three-dimensional models of HDL apoA-I: implications for its assembly and function. J Lipid Res. 2008;49(9):1875–83.
17 Sviridov DO, Ikpot IZ, Stonik J, Drake SK, Amar M, Osei-Hwedieh DO, et al. Helix stabilization of amphipathic peptides by hydrocarbon stapling increases cholesterol efflux by the ABCA1 transporter. Biochem Biophys Res Commun. 2011;410(3):446–51.
18 Cockerill GW, Rye KA, Gamble JR, Vadas MA, Barter PJ. High-density lipoproteins inhibit cytokine-induced expression of endothelial cell adhesion molecules. Arterioscler Thromb Vasc Biol. 1995;15(11):1987–94.
19 Baker PW, Rye KA, Gamble JR, Vadas MA, Barter PJ. Ability of reconstituted high density lipoproteins to inhibit cytokine-induced expression of vascular cell adhesion molecule-1 in human umbilical vein endothelial cells. J Lipid Res. 1999;40(2):345–53.
20 Barter PJ, Baker PW, Rye KA. Effect of high-density lipoproteins on the expression of adhesion molecules in endothelial cells. Curr Opin Lipidol. 2002;13(3):285–8.
21 Murphy AJ, Woollard KJ, Hoang A, Mukhamedova N, Stirzaker RA, McCormick SP, et al. High-density lipoprotein reduces the human monocyte inflammatory response. Arterioscler. Thromb Vasc Biol. 2008;28(11):2071–7.
22 Navab M, Imes SS, Hama SY, Hough GP, Ross LA, Bork RW, et al. Monocyte transmigration induced by modification of low density lipoprotein in cocultures of human aortic wall cells is due to induction of monocyte chemotactic protein 1 synthesis and is abolished by high density lipoprotein. J Clin Invest. 1991;88(6):2039–46.
23 Tang C, Liu Y, Kessler PS, Vaughan AM, Oram JF. The macrophage cholesterol exporter ABCA1 functions as an anti-inflammatory receptor. J Biol Chem. 2009;284:32336–43.
24 D’Souza W, Stonik JA, Murphy A, Demosky SJ, Sethi AA, Moore XL, et al. Structure/function relationships of apolipoprotein a-I mimetic peptides: implications for antiatherogenic activities of high-density lipoprotein. Circ Res. 2010;107(2):217–27.
25 Hyka N, Dayer JM, Modoux C, Kohno T, Edwards CK 3rd, Roux-Lombard P, et al. Apolipoprotein A-I inhibits the production of interleukin-1beta and tumor necrosis factor-alpha by blocking contact-mediated activation of monocytes by T lymphocytes. Blood. 2001;97(8):2381–9.
26 Burger D, Dayer JM. The role of human T-lymphocyte-monocyte contact in inflammation and tissue destruction. Arthritis Res. 2002;4(Suppl 3):S169–76.
27 Jurgens G, Xu QB, Huber LA, Böck G, Howanietz H, Wick G, et al. Promotion of lymphocyte growth by high-density lipoproteins (HDL). Physiological significance of the HDL binding site. J Biol Chem. 1989;264(15):8549–56.
28 Gruaz L, Delucinge-Vivier C, Descombes P, Dayer JM, Burger D. Blockade of T cell contact-activation of human monocytes by high-density lipoproteins reveals a new pattern of cytokine and inflammatory genes. PLoS One. 2010;5(2):e9418.
29 Bresnihan B, Gogarty M, Fitzgerald O, Dayer JM, Burger D. Apolipoprotein A-I infiltration in rheumatoid arthritis synovial tissue: a control mechanism of cytokine production? Arthritis Res Ther. 2004;6:R563–R566.
30 Chenaud C, Merlani PG, Roux-Lombard P, Burger D, Harbarth S, Luyasu S, et al. Low apolipoprotein A-I level at intensive care unit admission and systemic inflammatory response syndrome exacerbation. Crit Care Med. 2004;32(3):632–7.
31 Navab M, Hama SY, Anantharamaiah GM, Hassan K, Hough GP, Watson AD, et al. Normal high density lipoprotein inhibits three steps in the formation of mildly oxidized low density lipoprotein: steps 2 and 3. J Lipid Res. 2000;41(9):1495–508.
32 Navab M, Hama SY, Cooke CG, Anantharamaiah GM, Chaddha M, Jin L, et al. Normal high density lipoprotein inhibits three steps in the formation of mildly oxidized low density lipoprotein: step 1. J Lipid Res. 2000;41(9):1481–94.
33 Hessler JR, Robertson LA, Chisolm GA. LDL-induced cytotoxicity and its inhibition by HDL in human vascular smooth muscle and endothelial cells in culture. Atherosclerosis. 1979; 32(3): 213–29.
34 Tward A, Xia YR, Wang XP, Shi YS, Park C, Castellani LW, et al. Decreased atherosclerotic lesion formation in human serum paraoxonase transgenic mice. Circulation. 2002;106(4):484–90.
35 US Preventive Services Task Force. Routine vitamin supplementation to prevent cancer and cardiovascular disease: recommendations and rationale. Ann Intern Med. 2003;139(1):51–5.
36 Frantz S, Ertl G, Bauersachs J. Mechanisms of disease: Toll-like receptors in cardiovascular disease. Nat Clin Pract Cardiovasc Med. 2007;4(8):444–54.
37 Kzhyshkowska J, Neyen C, Gordon S. Role of macrophage scavenger receptors in atherosclerosis. Immunobiology. 2012;217(5):492–502.
38 Mathison JC, Ulevitch RJ. The clearance, tissue distribution, and cellular localization of intravenously injected lipopolysaccharide in rabbits. J Immunol. 1979;123(5):2133–43.
39 Baumberger C, Ulevitch RJ, Dayer JM. Modulation of endotoxic activity of lipopolysaccharide by high-density lipoprotein. Pathobiology. 1991;59(6):378–83.
40 Levine DM, Parker TS, Donnelly TM, Walsh A, Rubin AL. In vivo protection against endotoxin by plasma high density lipoprotein. Proc Natl Acad Sci USA. 1993;90(24):12040–4.
41 Wang Y, Zhu X, Wu G, Shen L, Chen B. Effect of lipid-bound apoA-I cysteine mutants on lipopolysaccharide-induced endotoxemia in mice. J Lipid Res. 2008;49(8):1640–5.
42 Cheng AM, Handa P, Tateya S, Schwartz J, Tang C, Mitra P, et al. Apolipoprotein A-I attenuates palmitate-mediated NF-κB activation by reducing Toll-like receptor-4 recruitment into lipid rafts. PLoS One. 2012;7(3):e33917.
43 Mogilenko DA, Orlov SV, Trulioff AS, Ivanov AV, Nagumanov VK, Kudriavtsev IV, et al. Endogenous apolipoprotein A-I stabilizes ATP-binding cassette transporter A1 and modulates Toll-like receptor 4 signaling in human macrophages. FASEB J. 2012;26(5):2019–30.
44 Guo L, Song Z, Li M, Wu Q, Wang D, Feng H, et al. Scavenger receptor BI protects against septic death through its role in modulating inflammatory response. J Biol Chem. 2009;284(30):19826–34.
45 Triantafilou M, Miyake K, Golenbock DT, Triantafilou K. Mediators of innate immune recognition of bacteria concentrate in lipid rafts and facilitate lipopolysaccharide-induced cell activation. J Cell Sci. 2002;115:2603–11.
46 Smythies LE, White CR, Maheshwari A, Palgunachari MN, Anantharamaiah GM, Chaddha M, et al. Apolipoprotein A-I mimetic 4F alters the function of human monocyte-derived macrophages. Am J Physiol Cell Physiol. 2010;298(6):C1538–C1548.
47 Zilber MT, Setterblad N, Vasselon T, Doliger C, Charron D, Mooney N, et al. MHC class II/CD38/CD9: a lipid-raft-dependent signaling complex in human monocytes. Blood. 2005;106(9):3074–81.
48 Mallat Z, Taleb S, Ait-Oufella H, Tedgui A. The role of adaptive T cell immunity in atherosclerosis. J Lipid Res. 2009;50(Suppl):S364–9.
49 Wilhelm AJ, Zabalawi M, Owen JS, Shah D, Grayson JM, Major AS, et al. Apolipoprotein A-I modulates regulatory T cells in autoimmune LDLr-/-, ApoA-I-/- mice. J Biol Chem. 2010;285(46)36158–69.
50 Van Lenten BJ, Hama SY, de Beer FC, Stafforini DM, McIntyre TM, Prescott SM, et al. Anti-inflammatory HDL becomes pro-inflammatory during the acute phase response. Loss of protective effect of HDL against LDL oxidation in aortic wall cell cocultures. J Clin Invest. 1995;96(6):2758–67.
51 Obici L, Franceschini G, Calabresi L, Giorgetti S, Stoppini M, Merlini G, et al. Structure, function and amyloidogenic propensity of apolipoprotein A-I. Amyloid. 2006;13(4):191–205.
52 Shao B, Cavigiolio G, Brot N, Oda MN, Heinecke JW. Methionine oxidation impairs reverse cholesterol transport by apolipoprotein A-I. Proc Natl Acad Sci U S A. 2008;105(34):12224–9.
53 Bierhaus A, Stern DM, Nawroth PP. RAGE in inflammation: a new therapeutic target? Curr Opin Investig Drugs. 2006;7(11):985–91.
54 Zheng L, Settle M, Brubaker G, Schmitt D, Hazen SL, Smith JD, et al. Localization of nitration and chlorination sites on apolipoprotein A-I catalyzed by myeloperoxidase in human atheroma and associated oxidative impairment in ABCA1-dependent cholesterol efflux from macrophages. J Biol Chem. 2005;280(1):38–47.
55 Undurti A, Huang Y, Lupica JA, Smith JD, DiDonato JA, Hazen SL. Modification of high density lipoprotein by myeloperoxidase generates a pro-inflammatory particle. J Biol Chem. 2009;284(45):30825–35.
56 Holzer M, Gauster M, Pfeifer T, Wadsack C, Fauler G, Stiegler P, et al. Protein carbamylation renders high-density lipoprotein dysfunctional. Antioxid Redox Signal. 2011;14(12):2337–46.
57 Wang Z, Nicholls SJ, Rodriguez ER, Kummu O, Hörkkö S, Barnard J, et al. Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat Med. 2007;13(10):1176–84.
58 Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414(6865):813–20.
59 Brownlee M, Cerami A, Vlassara H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N Engl J Med. 1988;318(20):1315–21.
60 Nobecourt E, Davies MJ, Brown BE, Curtiss LK, Bonnet DJ, Charlton F, et al. The impact of glycation on apolipoprotein A-I structure and its ability to activate lecithin:cholesterol acyltransferase. Diabetologia. 2007;50(3):643–53.
61 Shao B, Pennathur S, Pagani I, Oda MN, Witztum JL, Oram JF, et al. Modifying apolipoprotein A-I by malondialdehyde, but not by an array of other reactive carbonyls, blocks cholesterol efflux by the ABCA1 pathway. J Biol Chem. 2010;285(24):18473–84.
62 Zabalawi M, Bharadwaj M, Horton H, Cline M, Willingham M, Thomas MJ, et al. Inflammation and skin cholesterol in LDLr-/-, apoA-I-/- mice: link between cholesterol homeostasis and self-tolerance? J Lipid Res. 2007;48(1):52–65.
63 Dinu AR, Merill JT, Shen C, Antonov IV, Myones BL, Lahita RG. Frequency of antibodies to the cholesterol transport protein apolipoprotein A1 in patients with SLE Lupus. 1998;7:355–60.
64 Batuca JR, Ames PR, Isenberg DA, Alves JD. Antibodies toward high-density lipoprotein components inhibit paraoxonase activity in patients with systemic lupus erythematosus. Ann N Y Acad Sci. 2007;1108:137–46.
65 Ames PR, Matsuura E, Batuca JR, Ciampa A, Lopez LL, Ferrara F, et al. High-density lipoprotein inversely relates to its specific autoantibody favoring oxidation in thrombotic primary antiphospholipid syndrome. Lupus. 2010;19(6):711–6.
66 Vuilleumier N, Bratt J, Alizadeh R, Jogestrand T, Hafström I, Frostegård J. Anti-apoA-1 IgG and oxidized LDL are raised in rheumatoid arthritis (RA): potential associations with cardiovascular disease and RA disease activity. Scand J Rheumatol. 2010;39(6):447–53.
67 Vuilleumier N, Bas S, Pagano S, Montecucco F, Guerne PA, Finckh A, et al. Anti-apolipoprotein A-1 IgG predicts major cardiovascular events in patients with rheumatoid arthritis. Arthritis Rheum. 2010;62(9):2640–50.
68 Vuilleumier N, Reber G, James R, Burger D, de Moerloose P, Dayer JM, Roux-Lombard P. Presence of autoantibodies to apolipoprotein A-1 in patients with acute coronary syndrome further links autoimmunity to cardiovascular disease. J Autoimmun. 2004;23(4):353–60.
69 Vuilleumier N, Charbonney E, Fontao L, Alvarez M, Turck N, Sanchez JC, et al. Anti-(apolipoprotein A-1) IgGs are associated with high levels of oxidized low-density lipoprotein in acute coronary syndrome. Clin Sci (Lond). 2008;115(1):25–33.
70 Keller PF, Pagano S, Roux-Lombard P, Sigaud P, Rutschmann OT, Mach F, et al. Autoantibodies against apolipoprotein A-1 and phosphorylcholine for diagnosis of non-ST-segment elevation myocardial infarction. J Intern Med. 2012;271(5):451–62.
71 Montecucco F, Vuilleumier N, Pagano S, Lenglet S, Bertolotto M, Braunersreuther V, et al. Anti-Apolipoprotein A-1 auto-antibodies are active mediators of atherosclerotic plaque vulnerability. Eur Heart J. 2011;32(4):412–21.
72 Vuilleumier N, Rossier MF, Pagano S, Python M, Charbonney E, Nkoulou R, et al. Anti-apolipoprotein A-1 IgG as an independent cardiovascular prognostic marker affecting basal heart rate in myocardial infarction. Eur Heart J. 2010;31(7):815–23.
73 Pagano S, Satta N, Werling D, Offord V, de Moerloose P, Charbonney E, et al. Anti-apolipoprotein A-1 IgG in patients with myocardial infarction promotes inflammation through TLR2/CD14 complex. J Intern Med. 2012 Feb 13. doi: 10.1111/j.1365-2796.2012.02530.x
74 Rossier MF, Pagano S, Python M, Maturana AD, James RW, Mach F, et al. Antiapolipoprotein A-1 IgG chronotropic effects require nongenomic action of aldosterone on L-type calcium channels. Endocrinology. 2012;153(3):1269–78.
75 Srivastava R, Yu S, Parks BW, Black LL, Kabarowski JH. Autoimmune-mediated reduction of high-density lipoprotein-cholesterol and paraoxonase 1 activity in systemic lupus erythematosus-prone gld mice. Arthritis Rheum. 2011;63(1):201–11.
76 Kaplan M, Aviram M. Oxidized low density lipoprotein: atherogenic and proinflammatory characteristics during macrophage foam cell formation. An inhibitory role for nutritional antioxidants and serum paraoxonase. Clin Chem Lab Med. 1999;37(8):777–87.
77 Wentworth P Jr, McDunn JE, Wentworth AD, Takeuchi C, Nieva J, Jones T, et al. Evidence for antibody-catalyzed ozone formation in bacterial killing and inflammation. Science. 2002;298(5601):2195–99.
78 Omersel J, Jurgec I, Cucnik S, Kveder T, Rozman B, Sodin-Semrl S, et al. Autoimmune and proinflammatory activity of oxidized immunoglobulins. Autoimmun Rev. 2008;7(7):523–9.
79 Burger D, Dayer JM. Cytokines, acute-phase proteins, and hormones: IL-1 and TNF-alpha production in contact-mediated activation of monocytes by T lymphocytes. Ann N Y Acad Sci. 2002;966:464–73.
80 Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJ, Komajda M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357(21):2109–22.
81 Davidson MH.HDL and CETP Inhibition: Will This DEFINE the Future? Curr Treat Options Cardiovasc Med. 2012;14(4):384–90.
82 Vergeer M, Bots ML, van Leuven SI, Basart DC, Sijbrands EJ, Evans GW, et al. Cholesteryl ester transfer protein inhibitor torcetrapib and off-target toxicity: a pooled analysis of the rating atherosclerotic disease change by imaging with a new CETP inhibitor (RADIANCE) trials. Circulation. 2008;118(24):2515–22.
83 Funder JW. Aldosterone, sodium, and hypertension: lessons from torcetrapib? Hypertension. 2010;55(2):221–3.
84 Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, et al. dal-OUTCOMES Investigators.Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367(22):2089–99.
85 Durrington P, Erdine S, Halcox J, Hobbs R, Kjekshus J, Filardi PP, et al. ESC Committee for Practice Guidelines (CPG) 2008–2010 and 2010–2012 Committees.ESC/EAS Guidelines for the management of dyslipidaemias: the Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). European Association for Cardiovascular Prevention & Rehabilitation, Eur Heart J. 2011;32(14):1769–818.
Funding / potential competing interests: This work was supported by grants from the Swiss National Foundation (SNF No 310030-140736 to NV and SNF 3100A0-130836/1 to AvE), the Swiss Heart Foundation (to NV) and Ernst and Lucie Schmidheiny foundation (to PRL), and the Zurich Center of Integrative Physiology.