DNA methylation analysis
DOI: https://doi.org/10.4414/smw.2013.13799
Thomas
von Känel, Andreas R
Huber
Summary
DNA methylation, the addition of a methyl group to cytosines and adenosines, regulates gene expression on a level that is usually referred to as epigenetic, that is, stably maintained during cell divisions. In humans, aberrant DNA methylation is associated with several malignancies, including cancer and so-called imprinting disorders, making it an attractive target for diagnostic purposes. Here we give a brief introduction to the biology of DNA methylation and present the use of methylation biomarkers in laboratory medicine. DNA methylation assays have become the standard procedure in the diagnosis of imprinting disorders, and they are about to shift cancer diagnostics and prognostics to the next level of molecular medicine. However, there is evidence of problems associated with the introduction of such cancer assays in routine diagnostics. We review several assays that have been proposed for DNA methylation analysis. The assays presented analyse the methylation status of single loci and are based either on a bisulphite-treatment or on methylation-sensitive restriction of the DNA under investigation.
Abbreviations
AS Angelman syndrome
BWS Beckwith-Wiedemann syndrome
CI confidence interval
CRC colorectal cancer
DNA deoxyribonucleic acid
DNMT DNA methyltransferase
HNPCC hereditary nonpolyposis colorectal cancer
IC imprinting centre
MDS myelodysplastic syndrome
MGMT O6-methylguanine-DNA methyltransferase
MLPA multiplex ligation-dependent probe amplification
MMR mismatch repair
MS methylation sensitive
MSP methylation-specific PCR
NGS next generation sequencing
PCR polymerase chain reaction
PWS Prader-Willi syndrome
qPCR quantitative PCR
RNA ribonucleic acid
SRS Silver-Russell syndrome
UPD uniparental disomy
Introduction
Deoxyribonucleic acid (DNA) methylation is characterised by the addition of a methyl group to the C5 position of the cytosine pyrimidine ring or the N6 position of the adenine purine ring. Even though DNA methylation was discovered before the structure of DNA was resolved [1], work to reveal its multiple functions in living organisms is still ongoing. The latter can be attributed to the complexity of the processes associated with DNA methylation, and to the fact that DNA methylation is challenging to analyse.
In bacteria, methylation of adenines and cytosines is used as a defence mechanism that allows differentiation between (unmethylated) DNA of invading bacteriophages and (methylated) “self” DNA. As newly synthesised DNA strands are immediately methylated, this signature of “self” is stably maintained during cell division [2]. In addition, bacteria use DNA methylation patterns to pass information regarding gene expression from the mother cell to the daughter cells, allowing maintainance of acquired environmental adaptations in successive generations [3]. Accordingly, DNA methylation can be denoted as an “epigenetic” (Greek for “above genetics”) modification, because it transmits relevant information, which is not contained within the DNA sequence itself, to the next generation.
In humans, methylation is restricted to cytosine residues and mainly encountered in cytosine-guanine (CG) dinucleotides (usually abbreviated as “CpG”). This dinucleotide is underrepresented throughout the genome, yet encountered at increased rates in stretches of 0.3–3 kb, the so-called CpG islands. About 40% of the human genes contain a CpG island in their promoter region [4], and their methylation is usually associated with silenced expression. DNA methylation is established and maintained by DNA methyltransferases (DNMTs), with DNMT3A and DNMT3B mediating de-novo methylation and DNMT1 maintaining established DNA methylation [5].
DNA methylation fulfills the following functions in humans [6]:
(1.) Regulation of specific genes:
In order to maintain proper functioning of cellular processes, gene expression must be tightly controlled. Short-term changes in expression patterns are usually induced by transcription factors, whereas long-term gene regulation is achieved by a complex interplay of chromatin remodelling, modifications of DNA-bound histones, nuclear positioning of chromosomal regions, regulatory ribonucleic acids (RNAs) and, last but not least, DNA methylation [7–10].
(2.) Genomic imprinting:
Genomic imprinting refers to genomic regions from which only the paternal or the maternal allele is expressed. This parent-of-origin-specific expression pattern is mediated by epigenetic modifications, including DNA methylation, of either the paternal or maternal allele [11].
(3.) X-chromosomal inactivation (lyonisation):
DNA methylation is involved in inactivation of one of the two X-chromosome copies present in females, which ensures similar amounts of X-chromosomal gene products in both males and females [12].
(4.) Genome defence:
Parasitic genomic elements like retrotransposons are inactivated by methylation of their DNA sequence, making DNA methylation a key player in the maintenance of genome integrity [13].
Considering its pivotal role in the regulation of genes and other genetic elements, it is not astonishing that aberrant DNA methylation is associated with several disease conditions (table 1). For example, nonrandom X-inactivation might lead to manifestation of X-linked recessive disorders in female carriers, because of a preferential inactivation of the healthy allele [14]. Alternatively, constitutional mutations in the gene expression regulation machinery might lead to aberrant DNA methylation and altered gene expression patterns [15, 16], while genetic changes like noncoding trinucleotide expansions can cause disease by silencing the corresponding gene [17, 18]. Finally, the role of DNA methylation in common disorders like rheumatoid arthritis is becoming more and more evident [19]. However, in the next few sections, we will focus on the role of DNA methylation in cancer and in so-called imprinting disorders, as the epigenotype-phenotype relationship is (at least partly) well established in these disorders, and as DNA methylation-based routine diagnostic tests are available.
|
Table 1: Disorders associated with aberrant DNA methylation. Importantly, aberrant DNA methylation often comes along with alterations in other gene regulators (e.g. chromatin remodeling, histone modification). The table is not exhaustive. |
| |
Imprinting disorders
|
Cancer
|
X-chromosomal recessive disorders
|
Some trinucleotide repeat disorders
|
Defects in gene expression regulation machinery
|
|
Type of aberrant methylation
|
Presence of only methylated or only unmethylated alleles (instead of one methylated and one unmethylated allele) |
Promoter hyper- / hypo-methylation
Altered methylation in gene body |
Preferential silencing of healthy allele in female carriers of X-linked recessive disorders due to nonrandom X-inactivation |
Promoter hypermethylation |
Partly hypomethylation (ICF syndrome)
Various types of aberrant methylation (ATRX syndrome)
Loss of a transcription factor that binds methylated CpGs (Rett syndrome) |
|
Cause of aberrant methylation
|
Lack of paternal or maternal allele (due to deletion or uniparental disomy)
Imprinting centre defect |
Errors caused by epigenetic machinery (difficult to prove)
Somatic, tumourigenic genetic mutation
Cellular adaptation (not directly involved in tumourigenesis) |
Stochastic effects
Selective advantage of cells expressing mutated allele
Genetic alteration in X-inactivation machinery |
Expansion of a trinucleotide repeat in noncoding regions of a specific gene |
Mutations in DNMT3B(ICF syndrome)
Mutations in ATRX (ATRX syndrome)
Mutations in MECP2 (Rett syndrome) |
|
Affected tissues
|
All body cells |
Only cancerous tissue
Rare: constitutional hypermethylation leading to cancer predisposition |
All body cells in which healthy allele is not expressed |
All body cells |
All body cells |
|
Consequence
|
Aberrant gene expression (e.g. silencing of essential genes) |
Aberrant gene expression (e.g. silencing of tumour suppressor genes) in affected cells |
Mutated allele expressed in >>50% body cells |
Silencing of the corresponding gene (?) |
Aberrant gene expression |
|
Examples
|
Angelman syndrome
Prader-Willi syndrome
Beckwith-Wiedemann syndrome
Silver-Russell syndrome |
All cancer types |
Duchenne’s muscular dystrophy
Haemophilia B |
Fragile X syndrome
Friedreich ataxia |
ICF syndrome
ATRX syndrome
Rett syndrome |
|
Association with laboratory medicine
|
Detection of aberrant methylation important for diagnosis |
Used as diagnostic and prognostic marker routinely (only few tests available) and experimental cancer testing |
Rarely tested |
Detection of repeat elongation |
Detection of genetic mutations in DNMT3B,
ATRX,orMECP2
|
| ATRX = α-thalassaemia, mental retardation, X-linked; ICF = immunodeficiency-centromeric instability-facial anomalies |
Imprinting disorders
As mentioned above, genomic imprinting is characterised by the silencing of either the maternal or the paternal allele of specific loci, resulting in parent-of-origin-specific expression of the corresponding genes. Their expression will accordingly fail if only paternal or maternal alleles are present. This is the case when one allele is deleted, or when either two paternal or two maternal copies of the locus are present (the latter being called uniparental disomy [UPD]). If these genetic aberrations are present in all body cells (i.e. are constitutional) or most cells (in the case of mosaicism), the affected individuals manifest so-called imprinting disorders.
Prader-Willi syndrome (PWS) and Angelman syndrome (AS) are distinct neurogenetic disorders that arise from the loss of either the paternal (PWS) or the maternal (AS) allele at 15q11-q13. PWS is characterised by severe hypotonia and feeding difficulties in early infancy, followed by obesity due to excessive eating in late infancy. Besides delayed development, individuals show cognitive impairment, specific behavioural characteristics like tantrums and stubbornness, and hypogonadism. AS in turn is characterised by severe developmental delay, absent or limited speech, gait ataxia, and a specific behaviour that includes frequent laughter [20].
The 15q11-q13 region contains several genes that are expressed either from the paternal or the maternal allele only, including the small nucleolar RNA 116 (SNOD116; paternally expressed) and E6-AP ubiquitin-protein ligase (UBE3A; maternally expressed). Lack of expression of SNOD116 and UBE3Aare nowadays assumed to be the main causes of PWS and AS, respectively. The methylation status of another locus at 15q11-q13, the promoter of the small nuclear ribonucleoprotein-associated protein N (SNRPN), is a well established molecular marker for PWS and AS: normal individuals present with a methylated (maternal) and an unmethylated (paternal) allele and thus 50% methylation at this locus, whereas PWS and AS are characterised by 100% and 0% methylation of SNRPN due to lack of either the paternal or maternal allele [20].
Two other imprinting disorders are Silver-Russell-syndrome (SRS) and Beckwith-Wiedemann syndrome (BWS).
BWS is associated with overgrowth, tumour predisposition and congenital malformation, and has a reported incidence of about one in 13,700 live births. Considering the high clinical variability of the disorder with mild phenotypes, the actual incidence might be higher. The molecular aetiology is highly complex and involves dysregulation of imprinted growth regulatory genes on chromosome 11p15.5. The region can be divided in two imprinting control regions or imprinting centres (ICs), with IC1 (also called H19DMR) containing IGF2, a paternally expressed cytokine, and H19, a maternally expressed untranslated RNAPolII transcript. IC2 (also called KvDMR1) contains, among other genes, KCNQ1OT1, a paternally expressed gene whose untranslated transcript is suspected to regulate negatively the expression of several other imprinted genes of the region, including maternally expressed CDKN1C,a negative regulator of cell proliferation. The parent-of-origin-specific expression of these genes is controlled by differential methylation, which is disturbed in BWS owing to either IC1 hypermethylation, IC2 hypomethylation or UPD at 11p15.5. [21, 22].
SRS can be regarded as the epigenetical and clinical opposite of BWS. Clinically, it is characterised by severe intrauterine and postnatal growth retardation, and relative macrocephaly, whereas epigenetically it is often associated with IC1 hypomethylation, which results in downregulation of IGF2. However, SRS has also been associated with maternal UPD of chromosome 7 [21, 23].
DNA methylation analysis in cancer
In oncology, DNA methylation is currently making important steps from bench to bedside: on the one hand, demethylating agents are now routinely used in several malignancies (discussed below); on the other hand, the aberrant methylation of several gene promoters is routinely used as a diagnostic and prognostic marker in laboratory medicine.
In contrast to imprinting disorders, where the aberrant methylation can often be clearly related to a specific genetic aberration, the reasons for aberrant methylation patterns in cancer are less well-established. The most obvious cause is that aberrant methylation arises upon the advent of specific genetic alterations. The altered methylation pattern then significantly contributes to tumourigenisis, for example, by silencing cell cycle control genes [9]. Alternatively, it has been proposed that aberrant methylation might arise without genetic alteration because of random errors of the epigenetic machinery (so-called epimutations) [24], and it might also be induced by environmental factors like diet [25]. Recent research has established that altered methylation states spread across the genome, that is, that hypermethylation of a specific gene promoter increases the probability that promoters of adjacent genes are also hypermethylated [26]. Importantly, changed methylation patterns might simply be due to the physiological adaptions that the cancer cells undergo during tumour formation, without being directly involved in tumourigenesis. Next generation sequencing (NGS, a technique allowing the parallel sequencing of thousands or even millions of target sequences) will significantly advance the elucidation of the pathways involved in the advent of aberrant methylation in cancer. However, it is likely that the question “who is leader, who is follower in tumourigenesis?” will have complex and partly ambiguous answers.
The best established DNA methylation aberration in cancer is promoter hypermethylation, which usually silences expression of the corresponding gene. However, hypomethylation of promoters and methylation alterations within the gene body rather than in the promoter and hemimethylation (methylation of single DNA strands) have also been shown to be associated with carcinogenesis [27]. As promoter hypermethylation is associated with gene silencing, its effect resembles that of a loss-of-function mutation in the given gene. The affected genes are commonly associated with tumour suppression [28] and DNA damage repair [5], and will abrogate proper cell cycle control and increase mutation rates when their functionality is lost. However, genes of other cellular pathways have also been found to be hypermethylated in cancer. Interestingly, there are examples where promoter hypermethylation is not only observed in the cancerous tissue, but in most (if not all) normal somatic cells of an individual. For example, constitutional hypermethylation of MLH1, a mismatch repair gene, has been suggested to mediate a predisposition to cancer, just as a constitutional genetic MLH1 mutation would [29, 30].
Dozens of genes have been found to show altered methylation states in many different cancer types; however, only very few have yet found their way into clinical diagnostics. To our knowledge, commercial kits are currently available for only four cancer methylation markers, namely SEPT9, MGMT, MLH1 and SHOX2.
Septin-9 (SEPT9)
SEPT9, a gene involved in cytokinesis and cell cycle control, was identified as a potential biomarker for blood-based colorectal cancer (CRC) screening [31]: in contrast to healthy controls, CRC-affected patients showed SEPT9 promoter methylation in DNA derived from blood plasma. A study by the renowned ARUP laboratories reported a sensitivity in CRC detection of 90% (95% confidence interval [CI] 77.4%‒96.3%; sensitivity for stage I and II CRC 86.8%) and specificity of 88% (95% CI 79.6%‒93.7%), when using a modified HeavyMethyl assay (see below) testing for SEPT9 methylation [32]. However, these data were challenged by a study [33] comparing the serum-based SEPT9 assay with the so-called “sDNA test” (which investigates methylated BMP3, NDRG4, VIM and TFPI2,as well as mutant KRAS and haemoglobin in stool samples [34]). The sensitivity of the SEPT9 test was only 60% (95% CI 41%–77%), as compared with 87% (95% CI 69%–96%) in the sDNA test.
When considering these results, one should bear in mind (1.) that CRC-screening is a multi-million market, and (2.) that the various studies of (methylation-based) CRC detection were presumably at least partly supported or initiated by competing test suppliers. It is thus likely that the controversy over the test best-suited for CRC screening will continue for a while. An important feature of an ideal test is the positive predictive value, which was 3.61% for ARUP’s SEPT9 test, meaning that only a small number of individuals with a positive test result will indeed suffer from CRC (negative predictive value 99.94%) [32]. Even though these values are, to our knowledge, not available for the sDNA test, we assume that they are in a similar range. A relatively huge number of SEPT9- and sDNA-positive patients will thus have to undergo colonoscopy to be free from the suspicion of CRC.
Short stature homeobox 2 (SHOX2)
SHOX2 was found to be hypermethylated in lung cancer when lung tumour tissues were compared with normal lung tissue in genome-wide methylation profiling. SHOX2, a transcription factor of the homeobox gene family, had previously not been associated with carcinogenesis [35]. Its usefulness as diagnostic marker was validated by assessing SHOX2 methylation in Saccomanno-fixed bronchial lavage samples from 125 patients and 125 controls. The HeavyMethyl assay used gave valid measurements in 82% of the samples (100 cases, 104 controls). This relatively low number was mainly attributed to the long storage of some of the samples, which was necessary to ensure that the control patients remained free of lung cancer. Of the cancer patients, 78 tested positive, whereas 100 of the controls tested negative, giving a sensitivity of 78% (95% CI 69%–86%) and a specificity of 96% (90%–99%) [36].
O6-methylguanine-DNA methyltransferase (MGMT)
The DNA repair protein O6-methylguanine-DNA methyltransferase (MGMT) removes O6-alkyl-guanine from DNA, a lesion induced by alkylating mutagens. In 2000, it was reported that MGMT methylation predicts the outcome of glioma patients treated with the alkylating agent carmustine. Methylation of the MGMT promoter was associated with regression of the tumour, and prolonged overall and disease-free survival, which can be attributed to increased carmustine-sensitivity of cancer cells due to silencing of MGMT upon methylation [37]. The same relationship between MGMT promoter methylation and treatment effectiveness was also reported for the treatment of glioblastoma with temozolomide [37]. MGMT methylation can thus be considered the classical example of a predictive methylation marker.
MutL homolog 1, colon cancer, nonpolyposis type 2 (Escherichia coli) (MLH1)
MLH1 is a DNA mismatch repair (MMR) gene with a tight yet complex association with cancer: heterozygous constitutional genetic mutations are associated with hereditary forms of CRC, namely nonpolyposis colorectal cancer (HNPCC; alias Lynch syndrome) [38]. Other MMR genes have been found to be mutated in HNPCC, including MSH2, MSH6 and PMS2. Characteristic of HNPCC tumours is the elongation of microsatellites (repeating sequences of two to six bases) due to defective MMR, the so-called microsatellite instability.
In CRC with microsatellite instability, but no identified MMR gene mutation, the cancer might be due to somatic promoter hypermethylation of MLH1. The hypermethylation silences MLH1 expression, with a mutation-like effect on MLH1 function [39]. In contrast to MMR mutations, such MLH1 methylation is a sporadic event, which is evidently an important point when counselling CRC patients and their relatives. Apart from MLH1 methylation, sporadic microsatellite-unstable CRC might be due to the BRAF mutation V600E. Testing microsatellite-unstable CRC first for MLH1 methylation and the BRAF V600E mutation allows sporadic CRC to be indentified, and negative samples can then be screened for MMR gene mutation in order to test for HNPCC.
An important aspect of MLH1 methylation analysis (and of DNA methylation analysis in general) is the exact genomic region to be investigated. Even though a study from 1999 showed that loss of MLH1 expression is correlated with the methylation pattern of the proximal, but not the distal, part of the MLH1 promoter, more than half of the MLH1 studies published in the subsequent 7 years analysed nonspecific regions of the MLH1 promoter [40]. This, together with the differing sensitivities and specificities of the techniques employed, might explain the partly poor reproducibility of methylation tests.
DNA demethylating drugs
The reversible nature of DNA methylation makes it an attractive target for cancer therapies. This reversibility is exploited by the DNA demethylating drugs azacitidine [41] and decitabine [42], which act by inhibiting DNMTs. This does not lead to active demethylation; rather, these agents exert their effects by inhibiting maintenance of the acquired tumourigenic DNA methylation pattern during cellular division. DNA demethylation then leads to re-expression of genes that counteract the tumour phenotype, such as tumour suppressor genes. Alternatively, histone deacetylase inhibitors are used to re-establish nontumourigenic gene expression patterns [43]. DNMT inhibitors have been proposed for, or are currently being tested in, several disorders, with the main focus on various cancer types [43]. Earlier, DNMT inhibitors have been tested in the hereditary disorder sickle cell anaemia, where azacitidine was shown to induce re-expression of foetal haemoglobin [44]. Currently, the main use of these drugs is in myelodysplastic syndromes (MDSs), with decibatine and azacitidine being FDA-approved for these disorders. A low dosage of one of the drugs is a common intervention in high-risk MDS, resulting in increased overall survival rates as well as haematological improvements and improvement in quality of life [43].
Analysis of DNA methylation
Assays for DNA methylation analysis must be able to detect this small chemical modification in a sequence-specific manner, a task that is hampered by the fact that DNA methylation is not replicated (and thus amplified) during the process of the polymerase chain-reaction (PCR). Techniques for the analysis of DNA methylation can be classified by the methylation detection system and the number of loci investigated, with detection systems being divided into three categories: (1.) antibodies raised against methylated cytosines; (2.) bisulphite treatment converting unmethylated cytosines (but not methylated cytosines) into uracil; and (3.) endonucleases (or restriction enzymes) cutting specifically in the presence or absence of methylation. In terms of the number of loci analysed, everything from single cytosines to whole methylomes (i.e. the methylation status of all loci throughout the entire genome) is feasible.
5mC-antibodies
Antibodies raised against methylcytosines, first described in the eighties [45], enable detection of DNA methylation by both immunofluorescence [46] and immunoprecipitation. The latter enriches methylated DNA molecules from the DNA under investigation, which can then be detected by various methods. Commonly used methods include DNA microarrays [47] and NGS [48], both of which allow analysis of entire methylomes, whereas testing of single genomic loci is usually achieved with bisulphite- or endonuclease-based methods (see below).
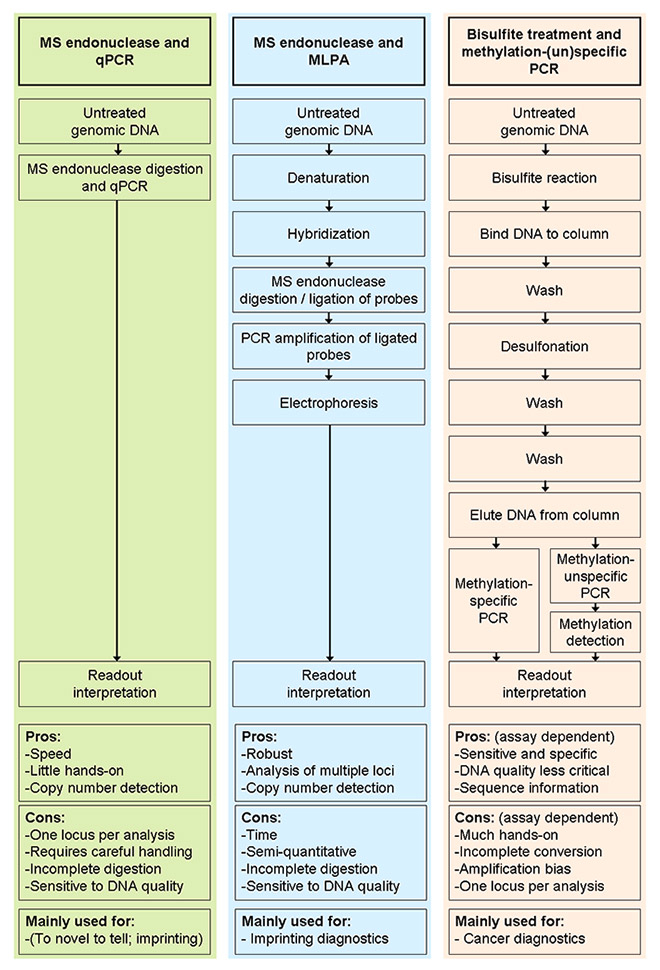
Figure 1
Comparison of combinations of methylation-sensitive DNA restriction plus (A) qPCR (performed as a one-step reaction) or (B) MLPA, and of(C) bisulphite-based approaches. Each arrow indicates a pipetting step. The number of pipetting steps for the bisulphite treatment depends on the protocol employed, as it does in assays combining bisulphite and methylation-unspecific primers. Technicians' hands-on times can be reduced by using pipetting robots.
DNA = deoxyribonucleic acid; MLPA = multiplex ligation-dependent probe amplification; MS = methylation sensitive; PCR = polymerase chain-reaction; qPCR = quantitive PCR.
Bisulphite treatment of DNA
Treating DNA with sodium bisulphite deaminates unmethylated cytosines to uracils, while methylated cytosines are chemically protected and remain methylated cytosines. Frommer and colleagues [49] were the first to exploit the chemical properties of (un)methylated cytosines for DNA methylation analysis, simply by PCR-amplifying and Sanger sequencing bisulphite-treated DNA: methylated cytosine residues are displayed as cytosines when sequencing bisulphite-treated DNA, whereas unmethylated cytosines are displayed as thymines (uracil is replicated as thymine in PCR). Nowadays, a plethora of alternative methods have joined Sanger sequencing in detecting cytosine conversion.
As with immunoprecipitated DNA, NGS can be used to characterise DNA methylation patterns at a genome-wide level when applied to bisulphite-treated DNA [50]. Methods for the analysis of specific genomic loci in turn are usually based on PCR amplification of the region of interest. In these approaches, DNA methylation analysis can be achieved in two ways. With methylation-specific primers, only methylated molecules are amplified. Accordingly, detection of PCR amplification is evidence that the locus under investigation is (at least partly) methylated, whereas no amplification indicates absence of methylation. In contrast, methylation-nonspecific primers amplify both methylated and unmethylated molecules, and methylation status is then assigned in a second, independent reaction [51]. When proper calibrators and/or standards are included, all assays described below (with the exception of Sanger sequencing) can be used for relative quantification (i.e. they are capable of assessing the proportion of methylated and unmethylated sequences in a sample). However, their abilities to detect sensitively and specifically small numbers of methylated molecules in a sample vary considerably.
Assays based on methylation-specific primers
Several methods have been proposed for detection of amplification in assays using methylation-specific primers. In the traditional methylation-specific PCR (MSP), PCR products are detected using gel electrophoresis and, accordingly, no specialised equipment is required [52]. However, MSP is hampered by its nonquantitative nature: as it is an endpoint measurement, no conclusions on the exact number of methylated molecules in the sample can be drawn. In addition, being a two-step procedure (first PCR, then electrophoresis), traditional MSP is relatively labour-intensive and there is an increased risk of mixing up samples.
This is overcome by quantitative MSP assays, in which the amplification of target molecules is monitored in real-time by adding a fluorescent reporter. These quantitative assays require equipment for real-time PCR, such as the LightCycler instrument. In the MethyLight assay [53], an analogue of the well-known TaqMan real-time PCR assay, the reporter is an oligonucleotide probe tha binds within the amplicon sequence and is labelled with a fluorescent dye and a quencher. During amplification, the probe is destroyed, releasing the dye from its quencher and thus resulting in increased fluorescence. Alternatively, amplification is monitored using SYBR Green I, an agent that becomes fluorescent when intercalating into double-stranded DNA molecules such as PCR products [54, 55]. This type of fluorescence generation circumvents the need for fluorescent probes, but might be compromised by reduced assay specificity due to signals generated by amplification of off-target sequences.
Independent of the detection format, MSP assays may be prone to false-positives when the primers amplify unmethylated (and thus converted) on-target sequences, owing to mispriming. Such unspecificity can be overcome by using more stringent reaction conditions, for example, by increasing the annealing temperature. However, it is possible that this will not only increase the specificity of the assay, but also decrease its sensitivity. As the MethyLight assay may employ not only methylation-specific primers, but also methylation-specific probes, the problem of nonspecificity is less acute there; accordingly, detection of methylated alleles in the presence of a 10,000-fold excess of unmethylated alleles has been reported [53].
Assays based on methylation-nonspecific primers
Methylation-nonspecific primers amplify both methylated and unmethylated alleles, and detect the methylation status in a second reaction. This second reaction basically aims to distinguish cytosines (= methylated cytosines) from thymines (= unmethylated cytosines).
A commonly used approach is the aforementioned Sanger sequencing (based on incorporation of differentially labelled dideoxynucleotides), in which the entire amplified sequence can be analysed [49]. This is attractive in exploratory projects, but Sanger sequencing has a limited sensitivity of around 20% for detecting methylated molecules [56]. Sensitivity is increased to 5% in pyrosequencing [57], which determines short sequence stretches with a reaction that is based on the pyrophosphate released upon incorporation of a deoxy-nucleotide during strand elongation. Finally, if one is interested in the base-specific methylation patterns of single DNA molecules, Sanger sequencing of cloned PCR products was for a long time the method of choice, although it is now being replaced by NGS.
The conversion of unmethylated cytosines to thymines during bisulphite treatment will influence the melting behaviour of PCR products, with methylated DNA having a higher melting temperature owing to the higher GC content. This can be exploited in melting curve analysis, in which the complementary strands of PCR products are melted in the presence of SYBR Green I [58]. At low temperatures, SYBR Green I will be fluorescent as it intercalates into the double-stranded PCR products. When the temperature is increased, the complementary strands of the PCR products will dissociate at a specific temperature, resulting in a sudden decrease of fluorescence due to release of SYBR Green I. The temperature-fluorescence-patterns thus obtained reflect GC content (and thus the methylation pattern) of the amplified sequence. The detection limit of this technique is about 5% methylated DNA in an excess of unmethylated DNA [51]. However, melting curves may become hard to interpret when DNA methylation patterns are highly heterogeneous.
Finally, HeavyMethyl [59] is a hybrid of methylation-specific and -nonspecific primers, as its primers are basically methylation-nonspecific, but amplification of unmethylated molecules is blocked by blockers (oligonucleotides that cannot be elongated by the polymerase) that bind specifically to unmethylated molecules. Amplification is detected with a TaqMan probe (see above) specific for methylated DNA. In summary, the HeavyMethyl PCR is basically methylation-nonspecific, but is made methylation-specific by the presence of the blockers and by the methylation-specific TaqMan probe (fig. 2). Compared with MSP-based approaches, HeavyMethyl has the advantage that false-positive rates are very low, because the blockers provide methylation-specificity at every cycle, whereas a single mispriming event in an MSP-based assay will result in a positive signal [51]. It is thus not astonishing that HeavyMethyl is able to generate a positive signal in a mix of 1 methylated molecule in 1,600 unmethylated molecules, but no amplification occurs in the same mix without a methylated molecule [59].
Several other approaches based on methylation-nonspecific primers have been proposed for analysis of single bases. However, these techniques are, in part, so complex that introducing them would be beyond the scope of this review. We thus refer to the review by Kristensen and Hansen [51] on detection of DNA methylation by means of bisulphite-based approaches.
Use of bisulphite-based assays in diagnostics
Owing to their high sensitivity and specificity, bisulphite-based protocols have become the gold standard for the analysis of DNA methylation in cancer. Ideally, an assay should have the following features: to exclude the possibility that absence of amplification signals is not simply due to absence of DNA, assays should include a control PCR, which can also be used to correct for variable DNA input; in addition, an ideal assay should include a reaction checking for complete conversion of unmethylated cytosines to uracils [60], as bisulphite reactions have repeatedly been reported to be incomplete [61]. Evidently, these unmethylated and unconverted cytosines mimic methylated sequences, leading to false positive results.
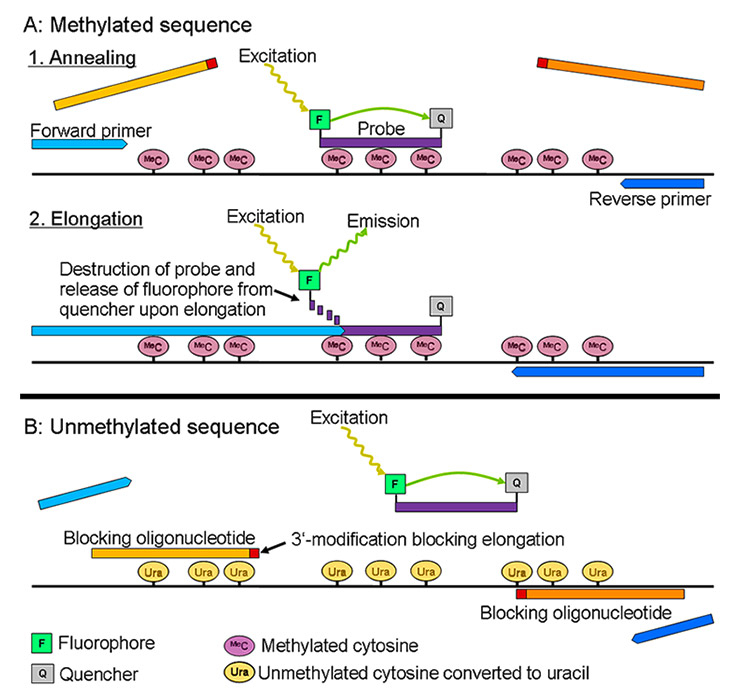
Figure 2
Principle of the HeavyMethyl assay. (A) With methylated DNA, the blocking oligonucleotides (amber) cannot bind their target sequence, leaving space for the methylation-nonspecific primers (blue) to anneal. Upon elongation, the methylation-specific probe (that has bound to the target sequence) is destroyed by the 5'-3' exonuclease activity of the Taq polymerase, releasing the fluorochrome F from its quencher Q (Taqman principle). The fluorochrome becomes fluorescent upon this release, which is detected by the qPCR instrument. (B) In unmethylated sequences, the blockers bind their target sequences, preventing the annealing and elongation of the primers. In addition, the methylation-specific probe will not bind its target; accordingly, it will not be destroyed, and there will be no increase in fluorescence.
DNA = deoxyribonucleic acid; PCR = polymerase chain-reaction; qPCR = quantitive PCR.
Besides possible incomplete conversion, the complex multistep nature of bisulphite-based assays is a major drawback, demanding much hands-on time and increasing the risk of mixing up samples. Additionally, it may be a challenge to design appropriate primers and probes for bisulphite assays: as most cytosines are converted to uracils (behaving like thymines in PCR), the complexity of the genomic sequences is reduced, which increases the risk that the primer binding sites are not unique. Finally, PCR might preferentially amplify either methylated or unmethylated (and bisulphite converted) DNA [57, 61], introducing a bias that has to be corrected for by including appropriate standards in each reaction setup. These drawbacks have led to the development of alternative assays based on enzymatic rather than chemical detection of DNA methylation. Such assays based on methylation-sensitive endonucleases are introduced in the next section.
Methylation-sensitive endonucleases
In order to destroy selectively the unmethylated DNA of bacteriophages, while leaving methylated “self” DNA intact, bacteria have developed a set of endonucleases cutting (or “digesting”) only unmethylated DNA. This property can be exploited for DNA methylation analysis, with commonly used methylation-sensitive (MS) endonucleases being HpaII (selectively cutting unmethylated CCGG sequences) and HhaI (selectively cutting unmethylated GCGC). First tests detected (non)digestion with Southern blots, which is, however, a time-consuming procedure requiring large quantities of DNA.
Combining methylation-sensitive endonuclease and qPCR
An alternative to Southern blotting is PCR, in which the DNA under investigation is first digested with an MS endonuclease, followed by PCR with primers that are designed to span the locus under investigation (the latter must evidently contain the recognition sequence of the MS endonuclease used). After digestion, only methylated (= uncuttable) sequences are still amplifiable, with amplification being detected by means of gel electrophoresis. These assays are not quantitative, a drawback that can be overcome by quantifying the number of undigested molecules with probe- or SYBR Green I-based qPCR [62, 63]. Simultaneous testing of undigested DNA corrects for varying DNA input, making these assays quite accurate. Investigating an unmethylated control locus additionally allows a check for the presence of endonuclease inhibitors. Last but not least, by neatly combining the information obtained from the various reactions, one can not only measure DNA methylation, but also check for copy number changes of the investigated sequence [64]. This last feature makes these assays particularly useful for imprinting disorder diagnostics, as the aberrant methylation pattern observed in these disorders is often caused by deletions.
Combination of endonuclease digestion and qPCR in a single reaction has recently been proposed [64]: DNA, MS endonuclease and qPCR are mixed in a single tube and then submitted to a temperature regimen that enables first DNA digestion and then qPCR. This procedure allowed diagnosis of 35 samples with PWS and AS, reducing the analysis time to 90 minutes after DNA extraction. To our knowledge, this is the only methylation assay requiring only a single pipetting step and using untreated genomic DNA as template. Because only one step is needed, the hands-on time and the risk of mixing up samples are reduced, making the assay well-suited for both diagnostic purposes and high-throughput investigations (fig. 3).
A major drawback of qPCR-based assays is errors introduced by temperature differences in the qPCR instruments [65, 66]. As DNA methylation is often analysed in GC-rich loci (which usually denature poorly during PCR), qPCR results may be biased by slightly different reaction-well temperatures – a cumbersome feature observable in all qPCR instruments. However, these errors can be eliminated by ensuring thorough DNA denaturation during the PCR process by, for example, adding DMSO to the reaction.
Methylation-specific multiplex ligation-dependent probe amplification
A commonly used alternative to qPCR for quantification of uncut (= methylated) DNA molecules is multiplex ligation-dependent probe amplification (MLPA, or MS-MLPA when combined with MS endonuclease treatment) [67]. In MLPA, a left and a right oligonucleotide probe hybridise adjacently on the sequence of interest, allowing ligation of the probes in a subsequent reaction step. As hybridisation and ligation can only occur in the presence of the target sequence, the quantity of the target sequence is reflected by the number of ligated probe pairs. Ligated probe pairs are PCR-amplified with fluorescently labelled primers, and PCR products are detected with sequencing-type electrophoresis instruments (so-called fragment analysis). Quantifying the peak size of PCR products finally allows conclusions on the number of target DNA molecules to be drawn (fig. 4).
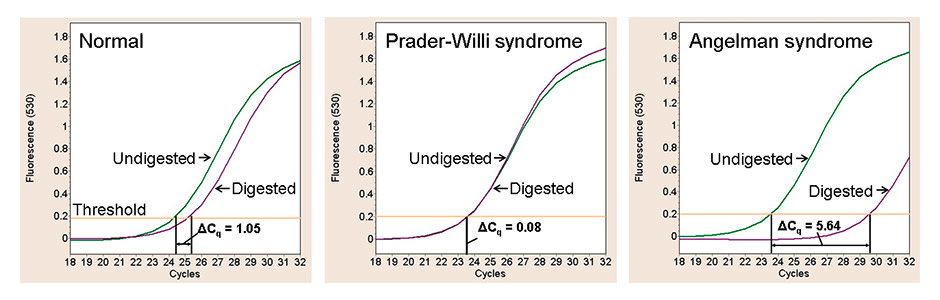
Figure 3
SNRPN-qPCR results of three DNA samples digested with the methylation-sensitive endonuclease HpaII (purple lines) or left undigested (green lines). In healthy individuals, 50% of the SNRPN molecules are unmethylated; they are digested and thus not amplifiable. Accordingly, it takes one cycle more than with the undigested sample to obtain a certain fluorescence signal. In Prader-Willi syndrome, SNRPN is fully methylated, and the SNPRN molecules are not digestible – amplification will thus be same in the digested and undigested sample. In Angelman syndrome, finally, SNRPN is unmethylated and thus fully digestible, and there will be no or only minor amplification signals.
DNA = deoxyribonucleic acid; PCR = polymerase chain-reaction; qPCR = quantitive PCR.
The ingenious feature of MLPA is that several probe pairs detecting different targets can be multiplexed in a single reaction, making the investigation of up to fifty loci in a single reaction feasible (distinguishing the signal of different probe pairs is achieved by their differing lengths) (fig. 4). Evidently, this is attractive for diagnostics and compensates for the drawbacks of MLPA, which are its semiquantitative nature, the elevated hands-on time (five pipetting steps) and reaction time (>18 h), and its relative inflexibility when used to analyse new loci. However, as compared with qPCR-based assays, MLPA can be considered to be more robust: firstly, it analyses multiple loci, and, secondly, it requires less pipetting experience as it is relatively insensitive to small changes in reaction composition. In addition, apart from a sequencing-type electrophoresis instrument, MLPA requires no special equipment.
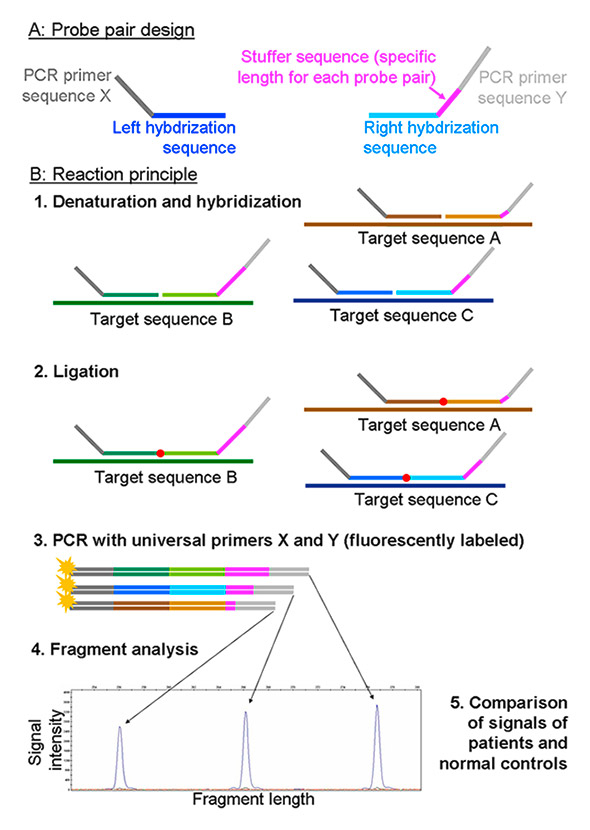
Figure 4
Reaction principle of MLPA. In methylation-sensitive MLPA, probes are designed such that they target a restriction site of a methylation-sensitive endonuclease. During ligation, the MS endonuclease cuts those probes that hybridised to an unmethylated target strand, while those hybridised to a methylated strand are left intact and can be PCR amplified.
MLPA = multiplex ligation-dependent probe amplification; PCR = polymerase chain-reaction.
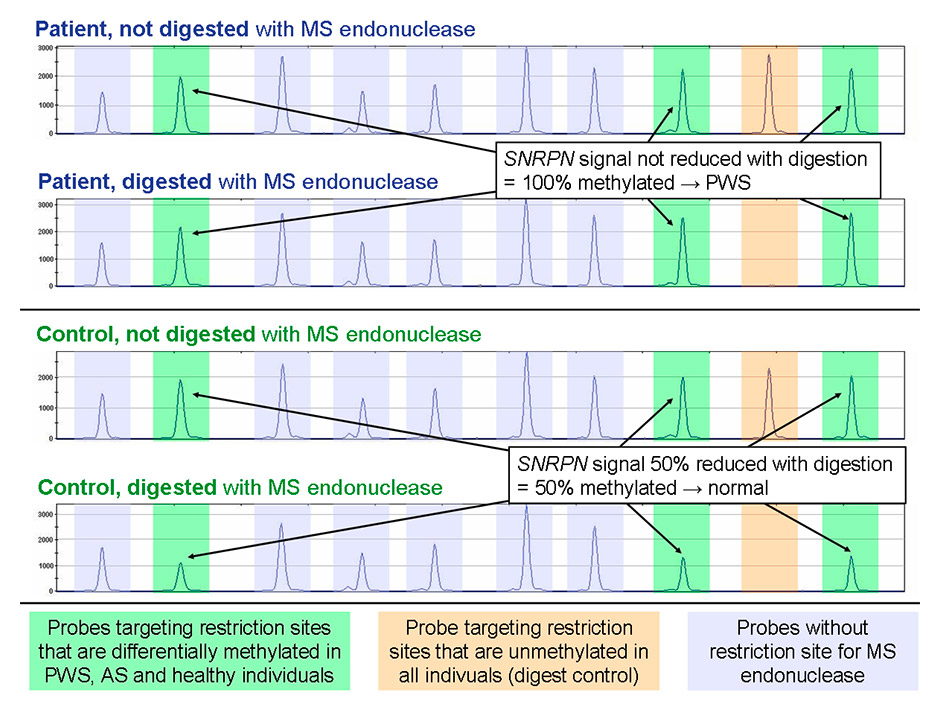
Figure 5
Detail of typical MS-MLPA results in the diagnosis of Prader-Willi syndrome and Angelman syndrome. Each peak represents the signal of a probe pair testing a specific locus. Amber highlights the peaks of an unmethylated locus that is used as digest control, while green peaks interrogate the methylation status of the SNRPN locus. No signal is visible at the control locus with digested DNA, so the methylation-sensitive restriction of the DNA was successful. SNRPN in turn is not digestible in the Prader-Willi syndrome sample due to full SNRPN methylation; accordingly, there is no reduction in peak intensity. In the healthy individual, the unmethylated SNRPN molecules (50%) are digested, resulting in halving of the peak height. Purple peaks do not contain the recognition sequence of the MS endonuclease and are thus unaffected by digestion; they are used for copy number quantification of the SNRPN locus.
AS = Angelman syndrome; DNA = deoxyribonucleic acid; MPLA = multiplex ligation-dependent probe amplification; MS = methylation-sensitive; MS-MPLA = MPLA combined with MS endonuclease; PWS = Prader-Willi syndrome.
Like qPCR-based assays, MS-MLPA takes advantage of MS endonucleases to distinguish methylated from unmethylated strands. Added to the ligation reaction, the MS endonuclease will cut hybrids of unmethylated target sequence strands and the corresponding probes, impeding subsequent PCR amplification of the ligated probes. On the other hand, hybrids of methylated target sequence strands and the corresponding probes are not cut and thus are amplifiable after ligation. Apart from targeting restriction sites of interest, MS-MLPA usually interrogates unmethylated control loci to check for complete digestion. In addition, probes containing no restriction sites allow copy number changes to be detected. A typical MS-MLPA reaction setup measures both the sample of interest and a control sample with and without MS endonuclease. By comparing signals obtained with and without digestion, one can determine the DNA methylation levels (fig. 5); copy number changes are detected by comparing undigested signals of the sample of interest and the control [68].
Use of MS endonuclease-based assays in diagnostics
Their ability to detect both DNA methylation and the copy numbers status of specific loci makes MS endonuclease-based assays of particular interest in the diagnosis of imprinting disorders: besides detecting the aberrant DNA methylation, these techniques will also tell if the aberrant methylation is due to a deletion. As MLPA has become the standard technique for detection of medium- to large-sized deletions in specific genes and is thus a commonly used method in diagnostic laboratories, MS-MLPA is presumably more commonly used than qPCR-assays.
Even though MS endonuclease-based strategies have been used to identify possible methylation biomarkers [31], their application in routine cancer diagnostics is hampered by the fact that they may not be able to digest all unmethylated DNA molecules present in a sample, leading to false-positive signals. This is not a problem when analysing imprinting disorders (where only methylation levels of 0%, 50% and 100% need to be differentiated), but becomes problematic in cancer diagnostics aiming to detect very low levels of DNA methylation. Incomplete digestion is worsened when analysing fragmented DNA (64), a feature typically encountered when extracting DNA from frozen or formalin-fixed paraffin-embedded tissues or from blood plasma, which are samples commonly investigated in cancer diagnostics. This problem may be overcome by increasing the amount of endonuclease in the reaction. However, one will nevertheless have to check thoroughly for complete DNA digestion.
It has been emphasised that assays for methylation detection in cancer should be truly quantitative [69]; this might (together with the above-mentioned problem of incomplete digestion) explain the poor performance of MS-MLPA in a recent study comparing different methylation assays in cancer diagnostics [70]. Assays combining MS endonucleases and qPCR (which is considered to be “more quantitative” than MLPA) might thus have a higher potential for cancer diagnostics. However, to our knowledge there is currently no data to support this hypothesis.
Conclusion
Over the last few years, DNA methylation analysis has become the first-step procedure in the diagnosis of imprinting disorders. This can be explained by the more or less straightforward epigenotype-phenotype relationship in this domain, and the fact that that the methylation tests employed require a relatively low sensitivity and specificity. In addition, imprinting disorders are usually detectable in all tissues, making analysis of blood DNA (which can be considered the “Rolls-Royce” among the available analytes for genetic and epigenetic assays) feasible.
In contrast, DNA methylation analysis is still awaiting the final breakthrough in cancer diagnostics and prognostics. Apart from the presumably less straightforward epigenotype-phenotype relationship in cancer, as compared with imprinting disorders, cancer assays are hampered by the fact that they usually must be able to detect DNA methylation with high specificity and sensitivity – and this often with low quality DNA retrieved from body fluids. In addition, investigation of different loci using various techniques within the same gene has led to conflicting results, which evidently abolishes the credibility of methylation biomarkers. Accordingly, there is a huge need for standardisation of both the methods used and the loci investigated [69].
NGS (i.e. high-throughput sequencing) of bisulphite-treated DNA is currently dramatically increasing the volume of available data on DNA methylation, and it is likely that this information will result in the discovery of new biomarkers for various disease conditions. In addition, the decreasing costs of NGS will presumably soon allow routine analysis of multiple methylated loci in a single patient – a task that will be further facilitated by third-generation sequencing technologies, which will enable methylation of single DNA molecules to be detected without bisulphite conversion and/or PCR amplification [71, 72]. However, just as it is becoming more and more evident for genetic NGS results, interpretation of such huge epigenetic data sets will be a major challenge. The task will be facilitated by recent advances in the elucidation of functional genetic and epigenetic elements throughout the genome by the ENCODE project [73], which will allow focus on sequences that are likely to be functionally relevant. However, disease-associated changes in DNA methylation will still have to be validated by thoroughly comparing diseased and nondiseased samples of the same tissue, ideally combined with experimental investigation of the functional effects of the methylation changes. In addition, investigation of corresponding genetic data will allow genetic causes of epigenetic aberrations to be identified.
References
1 Wyatt GR. The purine and pyrimidine composition of deoxypentose nucleic acids. Biochem J. 1951;48(5):584–90.
2 Jeltsch A. Beyond Watson and Crick: DNA methylation and molecular enzymology of DNA methyltransferases. Chembiochem. 2002;3(4):274–93.
3 Casadesús J, Low D. Epigenetic gene regulation in the bacterial world. Microbiol Mol Biol Rev. 2006;70(3):830–56.
4 Fatemi M, Pao MM, Jeong S, Gal-Yam EN, Egger G, Weisenberger DJ, et al. Footprinting of mammalian promoters: use of a CpG DNA methyltransferase revealing nucleosome positions at a single molecule level. Nucleic Acids Res. 2005;33(20):e176.
5 Jin B, Robertson KD. DNA methyltransferases, DNA damage repair, and cancer. Adv Exp Med Biol. 2013;754:3–29.
6 Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14(3):204–20.
7 Meister P, Mango SE, Gasser SM. Locking the genome: nuclear organization and cell fate. Curr Opin Genet Dev. 2011;21(2):167–74.
8 Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–97.
9 Baylin SB, Jones PA. A decade of exploring the cancer epigenome – biological and translational implications. Nat Rev Cancer. 2011;11(10):726–34.
10 Füllgrabe J, Kavanagh E, Joseph B. Histone onco-modifications. Oncogene. 2011;30(31):3391–403.
11 Ferguson-Smith AC. Genomic imprinting: the emergence of an epigenetic paradigm. Nat Rev Genet. 2011;12(8):565–75.
12 Chow JC, Yen Z, Ziesche SM, Brown CJ. Silencing of the mammalian X chromosome. Annu Rev Genomics Hum Genet. 2005;6:69–92.
13 Maksakova IA, Mager DL, Reiss D. Keeping active endogenous retroviral-like elements in check: the epigenetic perspective. Cell Mol Life Sci. 2008;65(21):3329–47.
14 Puck JM, Willard HF. X inactivation in females with X-linked disease. N Engl J Med. 1998;338(5):325–8.
15 Gibbons RJ, McDowell TL, Raman S, O’Rourke DM, Garrick D, Ayyub H, et al. Mutations in ATRX, encoding a SWI/SNF-like protein, cause diverse changes in the pattern of DNA methylation. Nat Genet. 2000;24(4):368–71.
16 Jin B, Tao Q, Peng J, Soo HM, Wu W, Ying J, et al. DNA methyltransferase 3B (DNMT3B) mutations in ICF syndrome lead to altered epigenetic modifications and aberrant expression of genes regulating development, neurogenesis and immune function. Hum Mol Genet. 2008;17(5):690–709.
17 Greene E, Mahishi L, Entezam A, Kumari D, Usdin K. Repeat-induced epigenetic changes in intron 1 of the frataxin gene and its consequences in Friedreich ataxia. Nucleic Acids Res. 2007;35(10):3383–90.
18 McLennan Y, Polussa J, Tassone F, Hagerman R. Fragile x syndrome. Curr Genomics. 2011;12(3):216–24.
19 Liu Y, Aryee MJ, Padyukov L, Fallin MD, Hesselberg E, Runarsson A, et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol. 2013;31(2):142–7.
20 Ramsden SC, Clayton-Smith J, Birch R, Buiting K. Practice guidelines for the molecular analysis of Prader-Willi and Angelman syndromes. BMC Med Genet. 2010;11:70.
21 Priolo M, Sparago A, Mammì C, Cerrato F, Laganà C, Riccio A. MS-MLPA is a specific and sensitive technique for detecting all chromosome 11p15.5 imprinting defects of BWS and SRS in a single-tube experiment. Eur J Hum Genet. 2008;16(5):565–71.
22 Choufani S, Shuman C, Weksberg R. Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet. 2010;154C(3):343–54.
23 Eggermann T. Russell-Silver syndrome. Am J Med Genet C Semin Med Genet. 2010;154C(3):355–64.
24 Martin DIK, Cropley JE, Suter CM. Epigenetics in disease: leader or follower? Epigenetics. 2011;6(7):843–8.
25 Waterland RA, Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol. 2003;23(15):5293–300.
26 De S, Shaknovich R, Riester M, Elemento O, Geng H, Kormaksson M, et al. Aberration in DNA methylation in B-cell lymphomas has a complex origin and increases with disease severity. PLoS Genet. 2013;9(1):e1003137.
27 Ehrlich M, Lacey M. DNA hypomethylation and hemimethylation in cancer. Adv Exp Med Biol. 2013;754:31–56.
28 Esteller M. CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene. 2002;21(35):5427–40.
29 Dobrovic A, Kristensen LS. DNA methylation, epimutations and cancer predisposition. Int J Biochem. Cell Biol. 2009;41(1):34–9.
30 Gazzoli I, Loda M, Garber J, Syngal S, Kolodner RD. A hereditary nonpolyposis colorectal carcinoma case associated with hypermethylation of the MLH1 gene in normal tissue and loss of heterozygosity of the unmethylated allele in the resulting microsatellite instability-high tumor. Cancer Res. 2002;62(14):3925–8.
31 Lofton-Day C, Model F, DeVos T, Tetzner R, Distler J, Schuster M, et al. DNA Methylation Biomarkers for Blood-Based Colorectal Cancer Screening. Clinical Chemistry. 2008;54(2):414–23.
32 Warren JD, Xiong W, Bunker AM, Vaughn CP, Furtado LV, Roberts WL, et al. Septin 9 methylated DNA is a sensitive and specific blood test for colorectal cancer. BMC Med. 2011;9:133.
33 Ahlquist DA, Taylor WR, Mahoney DW, Zou H, Domanico M, Thibodeau SN, et al. The stool DNA test is more accurate than the plasma septin 9 test in detecting colorectal neoplasia. Clin Gastroenterol Hepatol. 2012;10(3):272–277.e1.
34 Ahlquist DA, Zou H, Domanico M, Mahoney DW, Yab TC, Taylor WR, et al. Next-generation stool DNA test accurately detects colorectal cancer and large adenomas. Gastroenterology. 2012;142(2):248–256; quiz e25–26.
35 Schmidt B, Liebenberg V, Dietrich D, Schlegel T, Kneip C, Seegebarth A, et al. SHOX2 DNA methylation is a biomarker for the diagnosis of lung cancer based on bronchial aspirates. BMC Cancer. 2010;10:600.
36 Dietrich D, Kneip C, Raji O, Liloglou T, Seegebarth A, Schlegel T, et al. Performance evaluation of the DNA methylation biomarker SHOX2 for the aid in diagnosis of lung cancer based on the analysis of bronchial aspirates. Int J Oncol. 2012;40(3):825–32.
37 Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343(19):1350–4.
38 Bronner CE, Baker SM, Morrison PT, Warren G, Smith LG, Lescoe MK, et al. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature. 1994;368(6468):258–61.
39 Parsons MT, Buchanan DD, Thompson B, Young JP, Spurdle AB. Correlation of tumour BRAF mutations and MLH1 methylation with germline mismatch repair (MMR) gene mutation status: a literature review assessing utility of tumour features for MMR variant classification. J Med Genet. 2012;49(3):151–7.
40 Capel E, Fléjou J-F, Hamelin R. Assessment of MLH1 promoter methylation in relation to gene expression requires specific analysis. Oncogene. 2007;26(54):7596–600.
41 Issa J-PJ, Kantarjian HM, Kirkpatrick P. Azacitidine. Nat Rev Drug Discov. 2005;4(4):275–6.
42 Gore SD, Jones C, Kirkpatrick P. Decitabine. Nat Rev Drug Discov. 2006;5(11):891–2.
43 Seidel C, Florean C, Schnekenburger M, Dicato M, Diederich M. Chromatin-modifying agents in anti-cancer therapy. Biochimie. 2012;94(11):2264–79.
44 Charache S, Dover G, Smith K, Talbot CC Jr, Moyer M, Boyer S. Treatment of sickle cell anemia with 5-azacytidine results in increased fetal hemoglobin production and is associated with nonrandom hypomethylation of DNA around the gamma-delta-beta-globin gene complex. Proc. Natl. Acad Sci USA. 1983;80(15):4842–6.
45 Adouard V, Dante R, Niveleau A, Delain E, Revet B, Ehrlich M. The accessibility of 5-methylcytosine to specific antibodies in double-stranded DNA of Xanthomonas phage XP12. Eur J Biochem. 1985;152(1):115–21.
46 Santos F, Hendrich B, Reik W, Dean W. Dynamic reprogramming of DNA methylation in the early mouse embryo. Dev Biol. 2002;241(1):172–82.
47 Mohn F, Weber M, Schübeler D, Roloff T-C. Methylated DNA immunoprecipitation (MeDIP). Methods Mol Biol. 2009;507:55–64.
48 Down TA, Rakyan VK, Turner DJ, Flicek P, Li H, Kulesha E, et al. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nat Biotechnol. 2008;26(7):779–85.
49 Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci USA. 1992;89(5):1827–31.
50 Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, et al. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008;452(7184):215–9.
51 Kristensen LS, Hansen LL. PCR-based methods for detecting single-locus DNA methylation biomarkers in cancer diagnostics, prognostics, and response to treatment. Clin Chem. 2009;55(8):1471–83.
52 Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93(18):9821–6.
53 Eads CA, Danenberg KD, Kawakami K, Saltz LB, Blake C, Shibata D, et al. MethyLight: a high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000;28(8):E32.
54 Arya M, Shergill IS, Williamson M, Gommersall L, Arya N, Patel HRH. Basic principles of real-time quantitative PCR. Expert Rev Mol Diagn. 2005;5(2):209–19.
55 Chan MWY, Chu ESH, To K-F, Leung WK. Quantitative detection of methylated SOCS-1, a tumor suppressor gene, by a modified protocol of quantitative real time methylation-specific PCR using SYBR green and its use in early gastric cancer detection. Biotechnol Lett. 2004;26(16):1289–93.
56 Shen S, Qin D. Pyrosequencing data analysis software: a useful tool for EGFR, KRAS, and BRAF mutation analysis. Diagn Pathol. 2012;7:56.
57 White HE, Durston VJ, Harvey JF, Cross NCP. Quantitative analysis of SNRPN(correction of SRNPN) gene methylation by pyrosequencing as a diagnostic test for Prader-Willi syndrome and Angelman syndrome. Clin Chem. 2006;52(6):1005–13.
58 Worm J, Aggerholm A, Guldberg P. In-tube DNA methylation profiling by fluorescence melting curve analysis. Clin Chem. 2001;47(7):1183–9.
59 Cottrell SE, Distler J, Goodman NS, Mooney SH, Kluth A, Olek A, et al. A real-time PCR assay for DNA-methylation using methylation-specific blockers. Nucleic Acids Res. 2004;32(1):e10.
60 Rand K, Qu W, Ho T, Clark SJ, Molloy P. Conversion-specific detection of DNA methylation using real-time polymerase chain reaction (ConLight-MSP) to avoid false positives. Methods. 2002;27(2):114–20.
61 Warnecke PM, Stirzaker C, Song J, Grunau C, Melki JR, Clark SJ. Identification and resolution of artifacts in bisulfite sequencing. Methods. 2002;27(2):101–7.
62 Oakes CC, La Salle S, Robaire B, Trasler JM. Evaluation of a quantitative DNA methylation analysis technique using methylation-sensitive/dependent restriction enzymes and real-time PCR. Epigenetics. 2006;1(3):146–52.
63 Bruce S, Hannula-Jouppi K, Lindgren CM, Lipsanen-Nyman M, Kere J. Restriction site-specific methylation studies of imprinted genes with quantitative real-time PCR. Clin Chem. 2008;54(3):491–9.
64 Von Kanel T, Gerber D, Schaller A, Baumer A, Wey E, Jackson CB, et al. Quantitative 1-step DNA methylation analysis with native genomic DNA as template. Clin Chem. 2010;56(7):1098–106.
65 Von Kanel T, Adolf F, Schneider M, Sanz J, Gallati S. Sample number and denaturation time are crucial for the accuracy of capillary-based LightCyclers. Clin Chem. 2007;53(7):1392–4.
66 Von Kanel T, Gerber D, Wittwer CT, Hermann M, Gallati S. Detecting and resolving position-dependent temperature effects in real-time quantitative polymerase chain reaction. Anal Biochem. 2011;419(2):161–7.
67 Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30(12):e57.
68 Nygren AOH, Ameziane N, Duarte HMB, Vijzelaar RNCP, Waisfisz Q, Hess CJ, et al. Methylation-specific MLPA (MS-MLPA): simultaneous detection of CpG methylation and copy number changes of up to 40 sequences. Nucleic Acids Res. 2005;33(14):e128.
69 Mikeska T, Bock C, Do H, Dobrovic A. DNA methylation biomarkers in cancer: progress towards clinical implementation. Expert Rev Mol Diagn. 2012;12(5):473–87.
70 Suijkerbuijk KPM, Pan X, Van der Wall E, Van Diest PJ, Vooijs M. Comparison of different promoter methylation assays in breast cancer. Anal Cell Pathol (Amst). 2010;33(3):133–41.
71 Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, et al. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods. 2010;7(6):461–5.
72 Clarke J, Wu H-C, Jayasinghe L, Patel A, Reid S, Bayley H. Continuous base identification for single-molecule nanopore DNA sequencing. Nat Nanotechnol. 2009;4(4):265–70.
73 Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74.