From “magic bullets” to specific cancer immunotherapy
DOI: https://doi.org/10.4414/smw.2013.13734
Carsten
Riether, Christian
Schürch, Adrian
Ochsenbein
Summary
The immune system is able to specifically target antigen-expressing cancer cells. The promise of immunotherapy was to eliminate cancer cells without harming normal tissue and, therefore, with no or very few side effects. Immunotherapy approaches have, for several decades, been tested against several tumours, most often against malignant melanoma. However, although detectable immune responses have regularly been induced, the clinical outcome has often been disappointing. The development of molecular methods and an improved understanding of tumour immunosurveillance led to novel immunotherapy approaches in the last few years. First randomised phase III trials proved that immunotherapy can prolong survival of patients with metastatic melanoma or prostate cancer. The development in the field is very rapid and various molecules (mainly monoclonal antibodies) that activate the immune system are currently being tested in clinical trials and will possibly change our treatment of cancer. The ultimate goal of any cancer therapy and also immunotherapy is to cure cancer. However, this depends on the elimination of the disease originating cancer stem cells. Unfortunately, cancer stem cells seem resistant to most available treatment options. Recent developments in immunotherapy may allow targeting these cancer stem cells specifically in the future. In this review, we summarise the current state of immunotherapy in clinical routine and the expected developments in the near future.
Immunosurveillance of cancer
The concept that elements of the immune system contribute to cancer control was already proposed more than 100 years ago by the German pioneer in immunology and Nobel Prize winner Paul Ehrlich. Although details of the different effector mechanisms of the immune system were unknown at that time, he proposed that soluble factors that he called ‘magic bullets’ are responsible for cancer control [1]. In the 1950s, Burnet and Thomas formulated the immunosurveillance hypothesis [2]. They proposed that the development of genetic alterations and early cancer are very frequent events, but these early tumours never become clinically apparent because they are destroyed by a very efficient immune system. Their hypothesis was challenged by experimental data and clinical observations in the 1970s. Patients that were treated with immunosuppressive medication due to organ transplantations did not develop solid tumours such as breast, colon or lung cancer at a higher incidence than controls [3]. However, immunosuppressed patients had a highly increased incidence of aggressive lymphomas associated with Epstein-Barr virus infection [4].
Although the immune system does not play the central role in the immunosurveillance as proposed by Burnet and Thomas, more recent clinical studies provide evidence that cellular immunity actually contributes to the control of carcinomas. Oncogenesis is a consequence of different genetic alterations leading to uncontrolled cell proliferation and metastasis. These genetic alterations encode proteins that are quantitatively and/or qualitatively different to the proteins expressed in the cell of origin. Therefore, these proteins can serve as antigens and can be recognised by the immune system. Tumour antigens that are selectively expressed by the tumour and are not expressed on normal healthy tissues are ideal antigens for immunotherapy. Cancer testis antigens (CTA) are important representatives of this group [5]. CTAs are only expressed on cancer cells and in the testis. Since the testis is an immunologically privileged organ and does not express major histocompatibility class I (MHC class I) molecules that allow antigen presentation, the cellular immune response is selective against the cancer cells.
The immune response against cancer involves different effector mechanisms of adaptive and innate immunity, including cytotoxic CD8+ T cells (CTLs), antibodies, natural killer (NK) cells and also granulocytes and macrophages. Experimental evidence, mainly from murine tumour models, supports the view that anti-tumoural immunity is largely mediated by CTLs [6]. Indeed, clinical studies in patients suffering from malignant melanoma, ovarian cancer or colon cancer support this view. In all these different cancer entities, the infiltration of CD8+ T cells in the tumour correlates with improved prognosis [7].
Despite these effector mechanisms, the immune system often fails to control the disease. Clinically documented spontaneous remissions are very rare, but sometimes occur in malignant melanoma. Why then is the immune control of cancer often insufficient? There is a variety of different very well defined escape mechanisms among tumours. First, anti-tumoural immune responses select for cancer cells with low immunogenicity by favouring loss or down-regulation of specific antigens or antigen presenting molecules, a process known as cancer immunoediting [8]. Second, tumour cells may express cell-bound or soluble immunosuppressive factors. Third, tumour blood vessels lacking adhesion molecules, such as intercellular adhesion molecule-1 (ICAM-1), may limit extravasation and infiltration of the tumour by CTLs [9]. Fourth, anti-tumoural immunity may be hampered by immunoregulatory mechanisms, such as regulatory T cells (Tregs), myeloid derived suppressor cells and cytokines. The main function of Tregs in healthy individuals is the suppression of an uncontrolled immune activation to self-antigens, preventing autoimmunity. In cancer patients, however, the frequency of these Tregs in the circulation and especially at the tumour site, is massively increased. Within the tumour environment of most cancer types, Tregs suppress anti-tumoural immunity. For example, the frequency of tumour infiltrating Tregs in patients with ovarian cancer stage III and IV is a negative prognostic factor [10]. In contrast, in some gastrointestinal tumours, intratumoural Tregs seem to correlate with an improved prognosis [11]. Therefore, determining the number of tumour infiltrating Tregs may not be sufficient to predict disease outcome for all tumour entities.
Immunotherapy in clinical routine
Immunotherapeutic strategies have been used for decades in the field of oncology, although the mode of action has not been completely solved yet. For example, Bacillus Calmette-Guérin (BCG) instillations are used to treat non-muscle invasive bladder cancer after surgical ablation [12]. It is assumed that BCG locally activates innate immunity, decreasing the relapse rate. In addition, allogeneic haematopoietic stem cell transplantation (alloSCT) and the resulting graft versus leukaemia effect (GvL) is mediated by effectors of the adaptive immune system, such as T cells and possibly NK cells. Moreover, part of the anti-tumour effect of certain cytotoxic drugs may actually be mediated by the immune system. Although apoptotic cells are often removed by phagocytes without inducing an immune response, certain cytotoxic drugs induce cell death that elicits anti-tumour immune responses. This “immunogenic” cell death has been described for anthracyclines and oxaliplatin [13]. The activation of dendritic cells and immune effector mechanisms depends on the exposure of cell death associated molecular patterns (CDAMPs). Some of the molecular pathways involved have been characterised recently and include the exposure of calreticulin or the release of high-mobility group box 1 (HMGB1) from dying cells [14, 15].
In contrast, despite testing different approaches in phase I and II studies during the past decades, active immunotherapy has only reached clinical routine in the last few years. We now focus on the successful implementation of immunotherapies and their daily clinical use.
Passive immunotherapy
Passive immunotherapy describes the ex vivo generation of effector molecules (e.g., antibodies) or effector cells (e.g., CTLs) that are then transferred to a patient. For approximately ten years, monoclonal antibodies (mAbs) have been an integral part of the treatment of lymphoma and solid tumours. mAbs may block important growth factors or growth factor receptors on cancer cells or directly induce apoptosis of the cancer cell. In addition, mAbs activate parts of the immune system via the Fc-region of the antibody. Therefore, a main effector mechanism of these mAbs is the antibody dependent cell-mediated cytotoxicity (ADCC) that is executed by macrophages, NK cells and probably granulocytes. Experimental evidence with Fc gamma receptor-deficient mice supports the view that at least part of the anti-tumoural effect of clinically relevant antibodies, such as Rituximab (MabThera®), Trastuzumab (Herceptin®) and Cetuximab (Erbitux®) is mediated via ADCC [16]. In contrast to mAbs, adoptive T cell therapy (ACT) did not reach clinical routine yet.
Active immunotherapy
Active immunotherapy describes the process of vaccination with a tumour antigen and the consequent activation of the patient’s immune response. Different possibilities to provide tumour antigen have been tested in preclinical models and in clinical studies. These include injection of purified tumour antigens, defined peptide fragments of specific antigens or the expression of the antigens by viral vectors. Protein antigens and peptides have to be injected together with adjuvants to improve the processing and presentation of the antigen by dendritic cells (DCs) and to provide a depot effect. Alternatively, DCs are loaded with the antigen ex vivo prior to injection. So far, this method seems most promising. Recently, in a large randomised phase III study, men suffering from metastatic, castration-resistant prostate cancer were treated with autologous DCs generated from peripheral blood mononuclear cells natured in vitro using a recombinant fusion protein consisting of GM-CSF and the prostate-specific antigen prostate acid phosphatase (PA2024). This cellular immunotherapy, called Sipuleucel-T (Provenge®), induced specific immune responses in these patients, reduced the risk of death by 22% and improved median overall survival by 4.1 months compared to the placebo group [17].
Immunomodulation
In addition to the antigen-specific immunotherapies described above, the immune system may also be activated non-specifically. This can be achieved by modulation of co-stimulatory and co-inhibitory molecules. The interaction of B7 molecules (CD80, CD86) with cytotoxic T lymphocyte antigen 4 (CTLA-4) leads to suppression of T cells. Targeting CTLA-4 with a blocking mAb (e.g., Ipilimumab, Yervoy®) will force B7 molecules to interact with the co-stimulatory molecule CD28 leading to T cell activation. In two large phase III trials, treatment of metastatic melanoma patients with Ipilimumab prolonged overall survival by approximately four months. Of note, Ipilimumab was the first therapy for metastatic melanoma that significantly improved overall survival [18]. Interestingly, Ipilimumab rarely induces objective clinical responses according to response evaluation criteria in solid tumours (RECIST). In some patients, tumour size even increased early after therapy. This may be due to tumour infiltration by immune cells or by oedema. Importantly, the immune system led to a long-term stabilisation of the disease. However, the unspecific immune activation did not only induce anti-tumoural immunity but also autoimmunity, leading to diarrhoea, hepatitis, skin toxicity and hypophysitis. The potential of Ipilimumab is now under investigation in clinical phase II/III studies for different solid tumours.
Novel developments
Modulating T cell activity by interfering with co-stimulatory or co-inhibitory pathways
The hallmark study with Ipilimumab in advanced melanoma [18] created interest in the evaluation of further co-inhibitory proteins, such as T cell immunoglobulin and mucin domain-containing protein 3 (TIM-3), lymphocyte activation gene 3 (LAG-3) and programmed death (PD) family members. PD-1 is primarily expressed on activated T and B cells. Similar to CTLA-4, the interaction of PD-1 with its ligands PD-L1 or PD-L2 limits T cell activation to constrain autoimmune reactions. Constitutive signalling via PD-1 on antigen-specific T cells was documented to induce T cell anergy and exhaustion in chronic viral infections and cancer, resulting in a failure to control the disease [19].
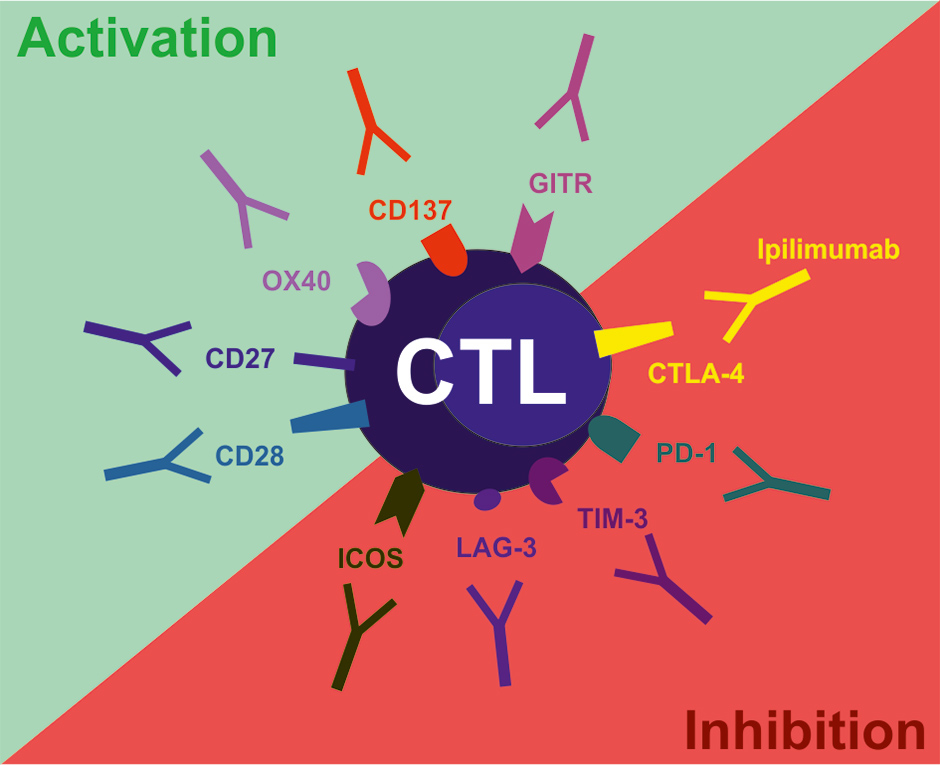
Figure 1
Examples of co-stimulatory and co-inhibitory molecules on T cells suitable for targeting with agonistic and blocking antibodies, respectively. CTLA-4, cytotoxic T lymphocyte antigen-4; PD-1, programmed cell death 1; TIM-3, T cell immunoglobulin and mucin domain-containing protein 3; LAG-3, lymphocyte activation gene 3; ICOS, inducible T cell co-stimulator; GITR, glucocorticoid-induced TNF family related gene; CD, cluster of differentiation.
We, and others, recently demonstrated that blocking PD-1 signalling results in improved tumour control in pre-clinical mouse models [20]. In 2006, the first human anti-PD-1 antibody was tested in a phase I trial with patients suffering from refractory solid tumours [21]. Even though patients with advanced disease resistant to conventional treatment were included in this phase I study, tumour regression was observed in some cases (one durable complete response and two partial responses in a total of 39 patients). Objective responses correlated with lymphocyte infiltration in metastatic tumours. In addition, lower toxicity has been reported compared to Ipilimumab. This correlates well with a more moderate autoimmunity phenotype of PD-1-/- mice compared to CTLA-4-/- mice [19]. These promising results led to phase I trials with anti-PD1 and anti-PD-L1 antibodies in advanced clear-cell renal cell carcinoma, melanoma, non-small cell lung cancer (NSCLC), prostate cancer and colorectal cancer. Objective tumour responses were observed with either antibody at a frequency of about 10-20%. Importantly, some of the treated patients had a prolonged disease stabilisation rate. At least for the activity of the anti-PD-1 antibody, the expression of its ligand PD-L1 on tumour cells seems to be a predictive factor for response [22, 23]. Currently, four pharmaceutical companies develop antibodies that block the PD-1 signalling pathway.
Furthermore, comparable promising results have been shown for blocking antibodies or small molecule inhibitors against LAG-3, TIM-3 and B and T lymphocyte attenuator (BTLA). Combination treatment regimens, e.g. parallel blockade of CTLA-4 and PD-1, act synergistically in the activation of anti-tumoural immunity and might be superior to monotherapy in terms of efficacy and side effects [24].
In addition, T cell co-stimulatory receptors such as CD28 and the TNF receptor family members CD137, OX40, GITR and CD27 may serve as potential targets for agonistic mAbs in order to activate anti-tumoural T cell responses (fig. 1). Unfortunately, data from a phase I clinical trial on an agonistic CD28 mAb demonstrated severe treatment-related toxicities due to a cytokine-release syndrome [25]. In contrast, the TNF receptor family members seem promising targets for improving anti-tumoural immunity in pre-clinical models. Administration of agonistic anti-mouse CD137 mAb resulted in reduced tumour growth even in poorly immunogenic tumours. Re-challenge experiments in anti-CD137-treated mice demonstrated that these mice developed memory cells specific for tumour-antigens and were subsequently protected from further tumour growth. Administration of agonistic anti-CD137 mAb even prevented recurrence of primary tumours after resection and prevented metastasis formation [26]. In addition, the combination of agonistic anti-CD137 mAb and Trastuzumab in a HER-2 positive breast cancer model resulted in an enhanced activation of the immune system and improved tumour control compared to Trastuzumab alone [27].
T cell-mediated anti-tumoural immunity can be induced by administration of agonistic anti-OX40 mAbs or OX40-ligand-IgG fusion proteins, as shown in several pre-clinical studies. Agonistic anti-OX40 mAb had the most potent anti-cancer effect in combination with chemotherapy and irradiation [28]. More recently, it was demonstrated that dual treatment using agonistic anti-OX40 mAb and IL-2 resulted in increased anti-tumoural immunity against several types of cancer [29]. These promising results led to the development of human agonistic anti-OX40 mAbs that are currently being tested in early clinical trials. Furthermore, agonistic anti-GITR mAbs reduced Treg cell activity and enhanced tumour rejection and in parallel reduced Treg cell activity in pre-clinical tumour models [30].
Current evidence supports the view that CD27-signalling improves anti-tumoural immunity as well. However, these findings are controversial. Some pre-clinical studies demonstrate that agonistic anti-CD27 mAbs effectively activate immune cells to control or eliminate lymphomas, leukemia and solid tumours. Consequently, the CD27-targeting therapeutic strategy is currently evaluated in a phase I trial for patients with haematological malignancies and selected solid tumours. In contrast, we could recently document in a murine tumour model that activation of CD27 induces progression of solid tumours by inducing regulatory Tregs [31].
In summary, many pre-clinical studies have demonstrated that targeting co-stimulatory or co-inhibitory pathways alone or in combination with conventional therapy increases anti-tumoural immunity and may even eradicate established tumours in some situations. Therefore, it is expected that in addition to the already approved mAb Ipilimumab, many new immunomodulating antibodies will be used to treat cancer in the future.
Adoptive T cell therapy
ACT employs the transfusion of large numbers of autologous or allogeneic T cells with high avidity for tumour antigens. The source of tumour-specific T cells is either naturally-occurring autologous T cells from the tumour microenvironment (e.g., tumour-infiltrating lymphocytes) or blood or genetically engineered T cells expressing high affinity tumour-specific T cell receptors (TCRs) [32]. Main problems in the clinical development of ACT were the labour-intensive development of specific T cell clones or T cell lines in vitro, their short half-life after transfer into the patient and the need of an individual development of T cells due to HLA-restriction. The in vivo half-life of the transferred T cells could be increased after lymphodepletion before adoptive transfer. Lymphodepletion with chemotherapy or irradiation may reduce immunosuppressive cells and generate space in lymphoid organs to allow a more efficient engraftment of the transferred cells. Adoptive T cell therapy after lymphodepletion showed objective responses in heavily pre-treated melanoma patients [32, 33].
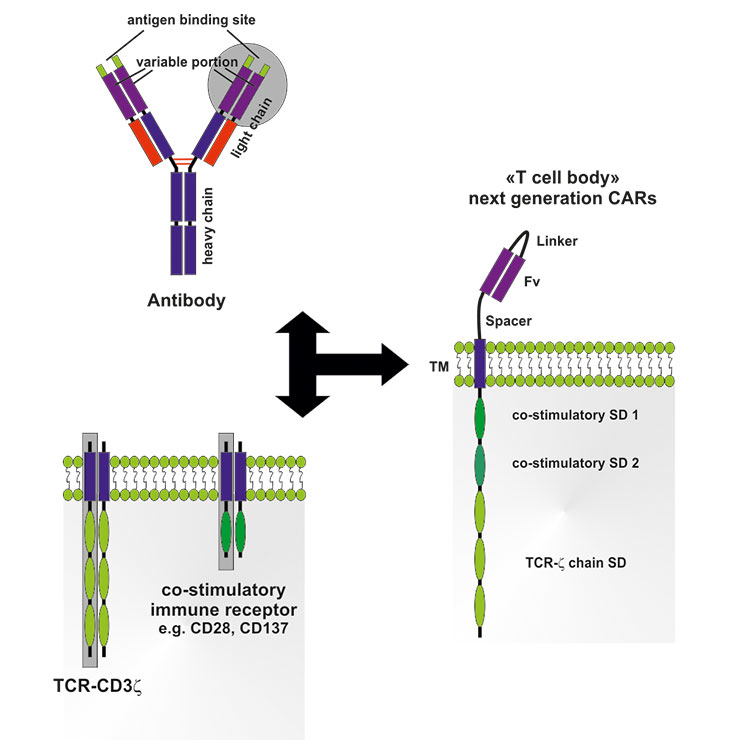
Figure 2
Structure and assembly of next-generation chimeric antigen receptors (CARs).
CARs are comprised of the variable portion (Fv) from an antibody with a defined specificity for a tumour antigen linked via a transmembrane domain (TM) to intracellular signaling domains (SDs) from the T cell receptor (TCR)-complex (CD3ζ chain), as well as one or several SDs from co-stimulatory receptors (e.g. CD28, OX40, CD137). Consequently, T cells expressing genetically engineered CARs can be activated independent of antigen-presenting cells; recognition of tumour antigen by the Fv-portion results in full activation of T cells.
Advances in T cell engineering using lentiviral and retroviral vectors carrying genetically engineered TCRs expanded the opportunities for ACT. Genetically engineered T cells were shown to recognise and destroy solid tumours expressing the cognate antigens. This resulted in clinical studies testing the role of ACT using genetically engineered T cells in tumours other than melanoma such as neuroblastoma, synovial cell sarcoma, leukaemia and lymphoma [34]. The high affinity TCRs expressed on genetically engineered T cells are either of human or murine origin. For example, TCR DNA sequences originating from a tumour-reactive T cell of a cancer patient can be isolated and cloned into autologous T cells from HLA-compatible donors. Furthermore, human T cells can be engineered to express mouse TCRs. T cells in human HLA transgenic mice immunised with human melanoma antigens generated TCRs with high avidity against tumour antigens. These TCRs were cloned into autologous human T cells, which were injected into melanoma patients and elicited a complete response in 19% of the cases [35].
Another promising possibility to direct T cells to a given tumour is the genetic engineering of a chimeric antigen receptor (CAR). CAR T cells express a so called “T-body”, a receptor consisting of a recognition domain and a signalling domain from the humoral and cellular adaptive immune system, respectively. In the majority of the cases, the CAR consists of a single chain variable fragment (Fv) of an mAb specific for a tumour antigen and an activating immune receptor, such as the TCR-associated CD3ζ chain. Next-generation CARs additionally contain co-stimulatory sequences, e.g. the cytoplasmic domain of CD28 or OX40, to allow full activation of the T cell (fig. 2). Upon binding of the Fv to the tumour antigen, the T cell is activated and elicits its effector function. Thereby, CAR T cells bypass the need of functional antigen processing and expression on MHC class I or II molecules on the surface of tumour cells. In pre-clinical tumour models, first-generation CAR T cells showed successful anti-tumoural activity, resulting in tumour regression [36]. The safety of CAR ACT could be demonstrated in phase I clinical trials, however, the outcomes regarding tumour control were rather disappointing. CAR T cells did not efficiently migrate to tumour sites and showed limited persistence, activation and effector function in vivo. In contrast, long-term anti-tumoural activity of CAR T cells was demonstrated in one clinical trial in which 19 patients with high-risk neuroblastoma were treated using ACT with T cells engineered to express CARs directed against the GD2 antigen. In 3 of 11 patients, a complete remission was observed that was associated with persistence of CAR T cells and improved survival [37]. The positive outcomes concerning safety and feasibility and the promising objective responses in this clinical trial resulted in the development of next-generation CARs that are currently under investigation [38, 39]. For example, in B cell lymphoma patients, T cells containing a second-generation CAR equipped with a co-stimulatory CD28 domain showed an improved in vivo-persistence compared to T cells containing first-generation CARs only targeting the CD19 antigen [39]. Subsequently, these next-generation CARs preferentially targeting lymphoma and leukaemia antigens were used in phase I and II clinical trials. Third-generation CARs containing two co-stimulatory signalling domains from CD28 and CD137 demonstrated safety, long-term persistence and anti-tumour activity in patients with lymphoma [40]. In a recent study, chronic lymphocytic leukaemia (CLL) patients treated with a low number of CAR T cells (1x105 CAR T cells / kg body weight) targeting CD19 and containing the co-stimulatory signalling domain of CD137 showed a complete remission. Furthermore, CAR-expressing T cells persisted more than 6 months, expanded more than 1000-fold and showed a CD19-specific immune response in the blood and bone marrow (killing of >1000 CLL cells per CAR cell). Most importantly, however, some of these CAR-T cells developed an effector memory phenotype allowing them to potentially expand upon secondary encounter with CLL cells and prevent relapse [41].
One of the major limitations of CAR ACT is that lymphodepletion with chemotherapy or irradiation and their associated side effects has to precede the transfer of CAR T cells. Pre-clinical studies addressing this issue recently demonstrated that CD19-specific CARs that constitutively secrete IL-12 eradicate lymphoma even in the absence of a pre-conditioning regimen [42].
Targeting cancer stem cells
Cancer stem cells (CSCs) are a subpopulation of cancer cells that are thought to drive the growth of tumours, similar to somatic stem cells that drive the growth of healthy proliferative tissues (e.g., bone marrow, epithelia). CSCs possess stem cell characteristics, i.e. dormancy/quiescence, self-renewal and unlimited proliferative potential, and have the ability to generate all the distinct malignant cell types within the tumour. In addition, they are thought to be capable of seeding to distant sites to initiate metastases. Depending on the origin of the tumour, CSC frequencies range from <1% to the majority of cells in the tumour. Leukaemia CSCs, so-called leukaemia stem cells (LSCs), were the first CSCs to be characterised and are among the most well-defined CSCs. Subsequently, CSCs were found in solid tumours as well, such as breast, ovarian, colon and brain cancer [43, 44].
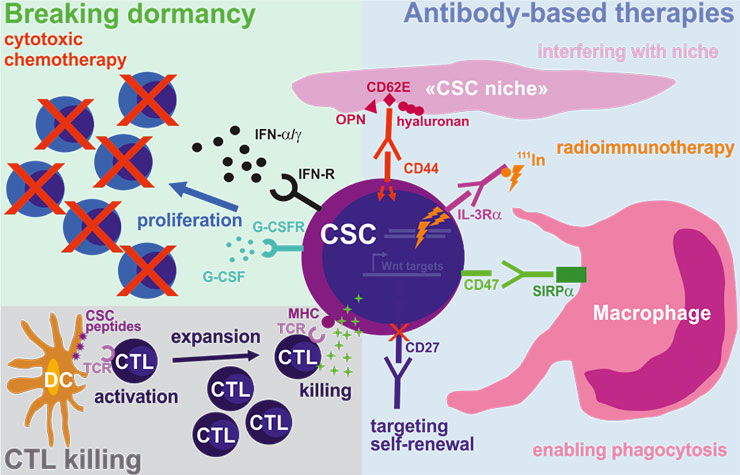
Figure 3
Targeting cancer stem cells (CSCs) using immunotherapy.
As compared to the bulk of tumour cells, CSCs may also be targeted using antibody-based therapies by blocking pathways important for homing and engraftment (CD44), self-renewal (CD27), protection against phagocytosis (CD47) and by delivering radioactive compounds (IL-3Rα). In addition, strongly activated and expanded cytotoxic CD8+ T cells (CTLs) specific for CSC antigens are promising candidates for a potent and long-lasting elimination of CSCs. Finally, forcing CSCs into the cell-cycle by breaking their dormancy using proliferation-activating molecules, followed by conventional cytotoxic chemotherapy, may represent an attractive two-step strategy to eradicate CSCs.
Cure of cancer implies the elimination of CSCs. However, CSCs display increased resistance against chemotherapy, irradiation and even targeted therapy. Several factors favour CSC resistance, such as the expression of high levels of ABC pumps that expel small molecules and the localisation in hypoxic niches, preventing the accumulation of reactive oxygen species after radiotherapy. Therefore, new therapies that selectively destroy CSCs are needed (fig. 3). Immunotherapy may be an attractive strategy to directly attack CSCs, but prior to developing new treatments, a more detailed understanding of the biology of CSCs and the distinctions from normal somatic stem cells is needed [45, 46].
First, methods need to be developed that allow prospective isolation of live CSCs from bulk tumour mass as well as isolation of somatic stem cells as their cognate healthy counterparts. Surface molecules that distinguish CSCs from bulk tumour cells have been found for various haematological and solid tumours (table 1). However, these molecules are rarely exclusively expressed by the CSC but are markers for normal somatic stem cells as well, highlighting the necessity to define more specific molecules or to use combinations.
Secondly, we need models that allow analysing and understanding differences in the biology of CSCs and somatic stem cells with regards to cell survival, proliferation, differentiation, responses to injury and drug resistance. Without this knowledge, it will be impossible to design drugs targeting pathways critical for CSC maintenance that are non-toxic to normal somatic stem cells. Today, the most widely used models for CSC propagation are murine xenograft assays analysing the repopulation of and serial transplantation in immunodeficient mice (NOD/SCID mice; NSG mice), as well as in vitro clonogenicity assays (long-term cultures; spheroids). However, some caveats should be kept in mind when analysing CSCs using these models, such as a) plasticity of the CSC state (CSC targeting is only an option if the CSC phenotype is stable), b) the experimental procedures (species barrier, transplantation setting and cellular stress) and c) the use of bona fideCSC markers (most CSC markers such as CD133 are also widely expressed in healthy tissues). These issues are comprehensively reviewed in [47].
Finally, we need to understand why most chemotherapeutics effectively eradicate bulk tumour cells without affecting CSCs. Patients achieving clinical remission from their cancer may harbour dormant, drug-resistant CSCs that persist and cause disease relapse years or even decades later [48]. Therefore, developing methods to detect and quantify these residual CSCs is essential to design more comprehensive combinatorial or sequential treatment regimens.
Antibody targeting of cancer stem cells
mAbs against CD44, an adhesion molecule and E-selectin ligand, markedly reduced human acute myeloid leukaemia (AML) repopulation and led to absence of leukaemia in serially xenotransplanted NOD/SCID mice. Anti-CD44 mAbs directly interfered with (LSC-niche interactions and altered the LSC fate [49]. In a murine chronic myeloid leukaemia (CML) model, the expression of CD44 on LSCs was increased compared to haematopoietic stem cells (HSCs), and CD44-deficient LSCs and LSCs pre-treated with a blocking anti-CD44 mAb were unable to home to and engraft in recipient bone marrow [50]. Several anti-CD44 mAbs are now tested for human AML, breast cancer, head and neck cancer and melanoma [51].
CD47, that interacts with signal-regulatory protein α (SIRPα) on macrophages and inhibits macrophage-induced phagocytosis, is normally expressed on circulating HSCs to avoid phagocytosis. Weissman and colleagues demonstrated that CD47 is overexpressed on mouse and human leukaemia cells and LSCs to evade macrophage killing. CD47 is an adverse prognostic factor in human AML and a possible drug target on human AML LSCs [52]. Furthermore, CD47 was not only overexpressed in myeloid leukaemias but also in acute lymphocytic leukaemia (ALL), and targeting CD47 using mAbs in an ALL xenograft model eliminated the leukaemia, suggesting the elimination of ALL LSCs. In addition, these authors showed that CD47 is expressed on a variety of human solid tumours and CSCs, e.g. bladder, ovarian, brain, breast, colon, hepatocellular and prostate cancer, and that its expression is higher on tumour cells than on normal tissue cells [53].
Recent work from our laboratory demonstrates that LSCs may be targeted by blocking CD27 signalling. The TNF-receptor family member CD27 is expressed on CML LSC and CD27 signalling activated the canonical Wnt pathway, induced LSC proliferation, increased differentiation to malignant granulocytes and promoted disease progression. Blocking CD27 signalling by transplanting CD27-deficient leukaemias or by mAb treatment reduced accumulation of nuclear β-catenin in LSCs, delayed disease progression and prolonged survival [54].
Another promising approach to target CSCs is the coupling of mAbs to radioisotopes. Reilly and colleagues used a mouse mAb specific for the IL-3Rα chain expressed on AML LSCs, which was modified with a nuclear localization signal and 111Indium. This mAb was internalised upon binding to IL-3Rα and caused DNA double strand breaks in AML cell lines and primary human AML cells [55].
Further promising strategies to directly attack CSCs using mAbs aim at the interruption of signalling pathways essential for stem cell self-renewal and maintenance, such as the Wnt/Frizzled, Delta/Notch and Hedgehog/Patched pathways [51].
Cellular therapy against cancer stem cells
AalloSCT and donor lymphocyte infusions (DLIs) are immunotherapies for high-risk leukaemia patients, which potentially lead to cure of the disease, implying the elimination of LSCs. The mechanisms underlying this phenomenon have been termed graft versus leukaemia effect (GvL). GvL is most probably mediated by minor histocompatibility-specific CTLs as well as NK cells [56]. In a murine xenograft model of human AML, Bonnet and colleagues were able to demonstrate that minor histocompatibility-specific CTLs eliminate AML LSCs, providing evidence that CTLs may represent a therapeutic strategy to eliminate CSCs [57]. These findings were supported by a recent study in a murine CML model showing that DLIs are able to block LSC engraftment and eradicate leukaemia in combination with imatinib [58].
For solid tumours, several attempts to eradicate CSCs are being investigated, including vaccination using CSCs fused to DCs or CSC peptide-pulsed DCs to prime CTL responses [59], the induction of γδ T cells that eliminate CSCs directly or via the secretion of IL-17 [60] and NK cell-mediated CSC killing [61].
Breaking CSC dormancy using immunomodulation
Dormancy and quiescence are hallmarks of stem cells, preserving self-renewal capability and preventing stem cell exhaustion. Upon tissue injury, stem cells leave their dormant state and begin to proliferate, giving rise to transit-amplifying daughter cells as well as new stem cells (asymmetric division). Since proliferating cells are more sensitive to DNA damage-inducing chemotherapeutics and drugs affecting the mitotic spindle, it was proposed to imply a two-step therapeutic strategy by priming dormant CSCs with proliferation-inducing compounds prior to conventional or targeted chemotherapy. Today, several molecules are known to induce proliferation of HSCs, such as IFN-α, IFN-γ, G-CSF and arsenic trioxide [46, 62, 63]. Indeed, until 2001, IFN-α was the standard therapy for newly diagnosed CML, leading to long-term remissions in a significant number of CML patients. The introduction of imatinib dramatically changed treatment of CML, however, it has to be taken life-long, and CML often relapses after drug discontinuation. Interestingly, in a French CML trial, the only patients experiencing long-term remission after discontinuation of imatinib had been treated with IFN-α before [46]. These findings indicate that combination treatment protocols may be a promising concept to eradicate dormant CSCs.
|
Table 1:Surface markers of cancer stem cells (adapted from Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev. Cancer. 2008;8:755–68 [44], and Krause DS, Van Etten RA. Right on target: eradicating leukemic stem cells. Trends Mol Med. 2007;13:470–81 [69]).B/A: BcR/ABL; bc: blast crisis; cp: chronic phase; CSC: cancer stem cell; EpCAM: epithelial cell adhesion molecule; M/A: MLL/AF9; N/H: NUP98/HOXA9. |
|
Tumour type
|
CSC (Immuno)phenotype
|
|
Leukemias
|
|
CML
|
| Human cpCML, B/A+
Mouse cpCML, B/A+
Human, bcCML, B/A+
Mouse, bcCML, B/A+ N/H+
|
lin- CD34+ CD38-
lin- Sca-1+ c-kithi CD44hi
lin- CD34+ CD38+ IL-3Ra+CD45RA+
lin-Sca-1+c-kitlowCD34+CD135+CD150-
|
|
AML, ALL
|
| Human AML
Human AML
Mouse AML, M/A+
Human B-ALL |
lin- CD34+ CD38- c-kit- CD90- IL-3Ra+ CD71- HLA-DR- CD47+
lin- CD34+ CD38- CD25+ CD32+ [64]
lin- Sca-1- c-kit+ CD34+ FcγR+ CD11b+
CD34+CD10- CD19- [65 ] |
|
Solid tumours (human)
|
|
Cell adhesion molecules
|
| Breast
Head and neck
Colon
Colon
Pancreas
Bladder
Prostate |
CD44+CD24-/low
CD44+
CD44+ CD24-/low
CD44+ EpCAMhi
CD44+ CD24+ EpCAM+
CD44+ [66]
CD44+ CD133+ CD49b/CD29hi [67] |
|
CD133
|
| Brain (glioblastoma, medulloblastoma)
Colon
Lung
Pancreas |
CD133+
CD133+
CD133+
CD133+
|
|
Miscellaneous
|
| Liver
Melanoma
Bladder
Brain
Mesenchymal |
CD90+
ABCB5+
CD47+ (66)
Autofluorescence (FL1+) (68)
Hoechst exclusion (side population) |
Concluding remarks
Immunotherapy is now part of clinical routine in the treatment of some tumours and novel immunotherapies against cancer will be available soon. New developments are expected in the field of modulating T cell activity by interfering with co-stimulatory or co-inhibitory pathways and in adoptive immunotherapy using CAR T cells. In recent years, clinical trials in oncology have well documented that only a small fraction of the treated patients profit from targeted therapy with newly developed drugs or, very comparably, from immunotherapy. Currently available data suggest that only a small fraction of melanoma patients benefit from Ipilimumab treatment, as indicated by long-term stabilisation of the disease. In addition to the establishment of novel therapeutic molecules, a main focus of investigation must therefore lay on the establishment of predictive biomarkers that allow selecting for patients who will respond to a defined immunotherapy. Unfortunately, current clinical developments in immunotherapy are not very focused and the efficacy of the novel molecules is tested in unselected patient populations in most or every tumour entity. However, since immunotherapy may be considered a prime example for targeted therapy, it is fundamental to characterise and define the requirements for successful tumour control, e.g. expression of tumour antigens on tumour cells, persistence and fate of transferred CAR T cells and others. The definition of predictive biomarkers will be crucial to define the role of the currently available and the novel immunotherapy strategies for each tumour in the future.
References
1 Ehrlich P. 1957. The Collected papers of Paul Ehrlich. Vol II. Pergamon Press: 8.
2 Burnet FM. Cancer: a biological approach. Br Med J. 1957;1:9.
3 Newstead CG. Assessment of risk of cancer after renal transplantation. Lancet. 1998;351:610–1.
4 Pietersma F, Piriou E, van Baarle D. Immune surveillance of EBV-infected B cells and the development of non-Hodgkin lymphomas in immunocompromised patients. Leuk Lymphoma. 2008;49:1028–41.
5 Suri A. Cancer testis antigens – their importance in immunotherapy and in the early detection of cancer. Expert Opin Biol Ther. 2006;6:379–89.
6 Ochsenbein AF, Sierro S, Odermatt B, Pericin M, Karrer U, Hermans J, et al. Roles of tumour localization, second signals and cross priming in cytotoxic T-cell induction. Nature. 2001;411:1058–64.
7 Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348:203–13.
8 Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–70.
9 Griffioen AW. Anti-angiogenesis: making the tumor vulnerable to the immune system. Cancer Immunol Immunother. 2008;57:1553–8.
10 Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–9.
11 Wilke CM, Wu K, Zhao E, Wang G, Zou W. Prognostic significance of regulatory T cells in tumor. International journal of cancer. Int J Cancer. 2010;127:748–58.
12 Brandau S, Suttmann H. Thirty years of BCG immunotherapy for non-muscle invasive bladder cancer: a success story with room for improvement. Biomed Pharmacother. 2007;61:299–305.
13 Locher C, Conforti R, Aymeric L, Ma Y, Yamazaki T, Rusakiewicz S, Tesniere A, et al. Desirable cell death during anticancer chemotherapy. Ann N Y Acad Sci. 2010;1209:99–108.
14 Apetoh L, Ghiringhelli F, Zitvogel L. Calreticulin dictates the immunogenicity of anti-cancer chemotherapy and radiotherapy. Medecine sciences: M/S 2007;23:257–8.
15 Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61.
16 Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6:443–6.
17 Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–22.
18 Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23.
19 Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704.
20 Mumprecht S, Schürch C, Schwaller J, Solenthaler M, Ochsenbein AF. Programmed death 1 signaling on chronic myeloid leukemia-specific T cells results in T-cell exhaustion and disease progression. Blood. 2009;114:1528–36.
21 Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28:3167–75.
22 Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54
23 Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–65.
24 Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64.
25 Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355:1018–28.
26 Narazaki H, Zhu Y, Luo L, Zhu G, Chen L. CD137 agonist antibody prevents cancer recurrence: contribution of CD137 on both hematopoietic and nonhematopoietic cells. Blood. 2010;115:1941–8.
27 Kohrt HE, Houot R, Weiskopf K, Goldstein MJ, Scheeren F, Czerwinski D, et al. Stimulation of natural killer cells with a CD137-specific antibody enhances trastuzumab efficacy in xenotransplant models of breast cancer. J Clin Invest. 2012;122:1066–75.
28 Croft M. Control of immunity by the TNFR-related molecule OX40 (CD134). Annu Rev Immunol. 2010;28:57–78.
29 Redmond WL, Triplett T, Floyd K, Weinberg AD. Dual anti-OX40/IL-2 therapy augments tumor immunotherapy via IL-2R-mediated regulation of OX40 expression. PloS ONE 2012;7:e34467.
30 Nocentini G, Ronchetti S, Petrillo MG, Riccardi C. Pharmacological modulation of GITRL/GITR system: therapeutic perspectives. Br J Pharmacol. 2012;165:2089–99.
31 Claus C, Riether C, Schurch C, Matter MS, Hilmenyuk T, Ochsenbein AF. CD27 signaling increases the frequency of regulatory T cells and promotes tumor growth. Cancer Res. 2012;72:3664–76.
32 Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012;12:269–81.
33 Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17:4550–7.
34 Schumacher TN, Restifo NP. Adoptive T cell therapy of cancer. Curr Opin Immunol. 2009;21:187–9.
35 Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–46.
36 Lipowska-Bhalla G, Gilham DE, Hawkins RE, Rothwell DG. Targeted immunotherapy of cancer with CAR T cells: achievements and challenges. Cancer Immunol Immunother. 2012;61:953–62.
37 Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118:6050–6.
38 Kohn DB, Dotti G, Brentjens R, Savoldo B, Jensen M, Cooper LJ, et al. CARs on track in the clinic. Mol Ther. 2011;19:432–8.
39 Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121:1822–6.
40 Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood. 2012;119:3940–50.
41 Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–33.
42 Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119:4133–41.
43 Huntly BJ, Gilliland DG. Leukaemia stem cells and the evolution of cancer-stem-cell research. Nat Rev Cancer. 2005;5:311–21.
44 Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–68.
45 Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Engl J Med. 2006;355:1253–61.
46 Essers MA, Trumpp A. Targeting leukemic stem cells by breaking their dormancy. Mol Oncol. 2010;4:443–50.
47 Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011;17:313–9.
48 Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer. 2007;7:834–46.
49 Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12:1167–74.
50 Krause DS, Lazarides K, von Andrian UH, Van Etten RA. Requirement for CD44 in homing and engraftment of BCR-ABL-expressing leukemic stem cells. Nat Med. 2006;12:1175–80.
51 Deonarain MP, Kousparou CA, Epenetos AA. Antibodies targeting cancer stem cells: a new paradigm in immunotherapy? mAbs 2009;1:12–25.
52 Ritchie DS, Smyth MJ. A new therapeutic target for leukemia comes to the surface. Cell. 2009;138:226–8.
53 Chao MP, Weissman IL, Majeti R. The CD47-SIRPalpha pathway in cancer immune evasion and potential therapeutic implications. Curr Opin Immunol. 2012;24:225–32.
54 Schürch C, Riether C, Matter MS, Tzankov A, Ochsenbein AF. CD27 signaling on chronic myelogenous leukemia stem cells activates Wnt target genes and promotes disease progression. J Clin Invest. 2012;122:624–38.
55 Leyton JV, Hu M, Gao C, Turner PV, Dick JE, Minden M, et al. Auger electron radioimmunotherapeutic agent specific for the CD123+/CD131- phenotype of the leukemia stem cell population. J Nucl Med. 2011;52:1465–73.
56 Weiden PL, Flournoy N, Thomas ED, Prentice R, Fefer A, Buckner CD, et al. Antileukemic effect of graft-versus-host disease in human recipients of allogeneic-marrow grafts. N Engl J Med. 1979;300:1068–73.
57 Bonnet D, Warren EH, Greenberg PD, Dick JE, Riddell SR. CD8(+) minor histocompatibility antigen-specific cytotoxic T lymphocyte clones eliminate human acute myeloid leukemia stem cells. Proc Nat Acad Sci USA. 1999;96:8639–44.
58 Lu YF, Gavrilescu LC, Betancur M, Lazarides K, Klingemann H, Van Etten RA. Distinct graft-versus-leukemic stem cell effects of early or delayed donor leukocyte infusions in a mouse chronic myeloid leukemia model. Blood. 2012;119:273–84.
59 Ning N, Pan Q, Zheng F, Teitz-Tennenbaum S, Egenti M, Yet J, et al. Cancer stem cell vaccination confers significant antitumor immunity. Cancer Res. 2012;72:1853–64.
60 Todaro M, D’Asaro M, Caccamo N, Iovino F, Francipane MG, Meraviglia S, et al. Efficient killing of human colon cancer stem cells by gammadelta T lymphocytes. J Immunol. 2009;182:7287–96.
61 Pietra G, Manzini C, Vitale M, Balsamo M, Ognio E, Boitano M, et al. Natural killer cells kill human melanoma cells with characteristics of cancer stem cells. Int Immunol. 2009;21:793–801.
62 Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature. 2010;465:793–7.
63 Trumpp A, Essers M, Wilson A. Awakening dormant haematopoietic stem cells. Nature reviews. Immunology. 2010;10:201–9.
64 Saito Y, Kitamura H, Hijikata A, Tomizawa-Murasawa M, Tanaka S, Takagi S, et al. Identification of therapeutic targets for quiescent, chemotherapy-resistant human leukemia stem cells. Sci Transl Med. 2010;2:17ra19.
65 Cox CV, Evely RS, Oakhill A, Pamphilon DH, Goulden NJ, Blair A. Characterization of acute lymphoblastic leukemia progenitor cells. Blood. 2004;104:2919–25.
66 Chan KS, Espinosa I, Chao M, Wong D, Ailles L, Diehn M, et al. Identification, molecular characterization, clinical prognosis, and therapeutic targeting of human bladder tumor-initiating cells. Proc Nat Acad Sci USA. 2009;106:14016–21.
67 Maitland NJ, Collins AT. Prostate cancer stem cells: a new target for therapy. J Clin Oncol. 2008;26:2862–70.
68 Clement V, Marino D, Cudalbu C, Hamou MF, Mlynarik V, de Tribolet N, et al. Marker-independent identification of glioma-initiating cells. Nat Meth. 2010;7:224–8.
69 Krause DS, Van Etten RA. Right on target: eradicating leukemic stem cells. Trends Mol Med. 2007;13:470–81.