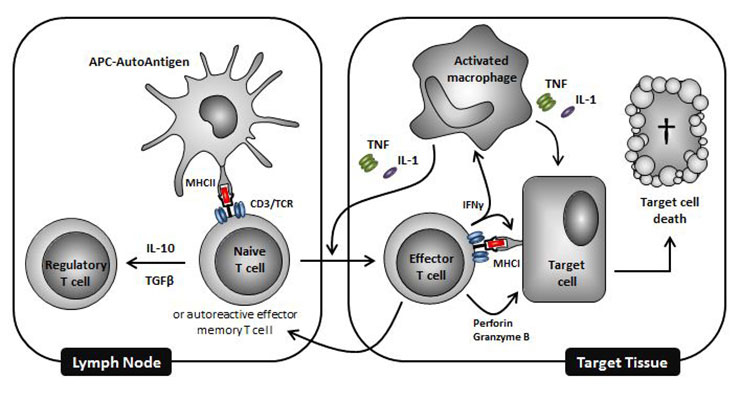
Figure 1
Schematic representation of some key immune mediators involved in the pathogenesis of autoimmune diseases. Combinations of drugs targeting distinct pathways in this process represent promising ways of curing autoimmune diseases.
DOI: https://doi.org/10.4414/smw.2012.13711
As T cells play a key role in the process of transplant rejection through recognition of foreign antigens presented by MHC molecules expressed on antigen presenting cells (APC), the first therapeutic monoclonal antibody (mAb) to be approved by the FDA was an anti-T cell antibody for the treatment of solid-organ transplantation in 1986. Muromonab (Orthoclone OKT3®) is a mouse IgG2a directed against the epsilon chain of the CD3-TCR complex, which is expressed on mature T cells. Muromomab exerts potent immunosuppressive effects via two mechanisms, (1.) T cells are transiently eliminated from the circulation as a result of cell margination and cell death, while a fraction of T cells are actually depleted in tissues, and (2.) remaining and re-appearing T cells transiently persist as CD3-TCR negative (a phenomenon termed antigenic modulation) and thus are unable to form an immune synapse with cells presenting antigenic peptides. The outcome of antigenic modulation is that T cells can no longer be triggered by the transplanted alloantigens. T cell function returns to normal within days after cessation of antibody administration. Randomised clinical trials have shown that muromonab effectively treats acute renal allograft rejection [1]. In addition, it has also been used successfully to treat acute rejection in liver and heart transplant recipients [1].
Anti-CD3 mAbs are administered clinically once a day for several consecutive days. Muromomab is of murine origin and highly immunogenic in humans. Most patients produce high titre anti-mouse antibodies following a course of treatment. In addition, muromonab is associated with a wide spectrum of side effects which occur almost immediately after administration of only the first doses [2]. These include flu-like symptoms such as fever, chills, nausea, vomiting and headaches. This first-dose response is called the “cytokine release syndrome” and occurs in almost all patients. Approximately 5% of patients experience more serious reactions, such as cardiopulmonary distress, seizures, encephalopathy, meningitis, renal insufficiency and graft thrombosis. The severity of these side effects diminishes with successive doses, due to antigenic modulation and T cell depletion. These unwanted effects of muromonab are a consequence of T cell activation, which results in the release of numerous cytokines into the systemic circulation.
To gain further insight into the mechanism of action of anti-CD3 therapies, the hamster antibody 145-2C11, directed against the epsilon chain of the murine CD3/TCR complex, was developed [3]. Like muromonab, 145-2C11 induces a first dose reaction in vivo associated with the transient release of cytokines including tumour necrosis factor (TNF), interferon-gamma (IFN-γ) and interleukin-2 (IL-2) [4, 5]. In addition, in mice, 145-2C11 induces transient antigenic modulation, elimination of T cells from the circulation and T cell depletion in tissues, and prevents acute rejection of fully mismatched allograft [5]. These findings show that 145-2C11 is a surrogate molecule for muromonab, and two major pharmacodynamic discoveries were in fact made using 145-2C11 in vivo. First, anti-CD3 mAb treatment reverses an ongoing autoimmune process and promotes long-term disease remission. Beyond their immunosuppressive properties, anti-CD3 mAbs exert potent immunoregulatory effects via preferential killing of activated effector T cells and/or induction of transforming growth factor-beta (TGF-β)-dependent adaptive regulatory T cells (Tregs) [6, 7]. Second, the ‘anti-CD3-induced first dose reaction’, associated with cytokine release, is mainly due to binding of the crystallizable fragment (Fc) to Fc gamma receptor (FcγR)-bearing leukocytes, since anti-CD3 mAbs without Fc or bearing a Fc with reduced capacity to bind FcγRs remain immunosuppressive and capable of inducing long lasting tolerance [8].
Due to its efficacy and although it provokes severe side effects, muromonab has been used extensively in the field of transplantation. In addition to allograft rejection, T cells play a key role in the pathogenesis of many autoimmune diseases including multiple sclerosis, type-1 diabetes (T1D), inflammatory bowel disease (IBD), psoriasis and rheumatoid arthritis. Hence in the early nineties muromonab was also administered to patients with severe multiple sclerosis [9]. However, due to its toxicity, development for the treatment of autoimmune diseases with this anti-CD3 mAb was stopped.
To alter the risk-to-benefit ratio and thus allow assessment of CD3-directed therapies for the treatment of autoimmune diseases, several Fc-modified anti-human CD3ԑ mAbs were created, including humanised versions of rodent anti-human CD3 mAbs (visilizumab, teplizumab, otelixizumab) and fully human mAb (foralumab). These mAbs have a similar engineered element built into their Fc portion. Amino acid mutations are introduced into the second heavy constant domain in order to reduce FcγR binding and the resultant cytokine release associated with cross-linking of the CD3-TCR complex via an Fc-dependent mechanism. In vitro studies using human blood cells and clinical trials in solid-organ transplant recipients show that these mAbs have immunosuppressive properties similar to that of muromonab while they do not have its severe unwanted immune activating capacity. Furthermore, using otelixizumab in transgenic non-obese diabetic (NOD) mice expressing the epsilon chain of the human CD3/TCR complex, Kuhn and colleagues have investigated the therapeutic efficacy and mechanism of action of non-FcγR binding anti-CD3 therapies in T1D [10]. They showed that, similarly to that of 145-2C11 in normal mice, Fc-modified anti-human CD3 induce durable disease remission that is dependent on transferable T cell mediated tolerance [10]. In T1D, anti-CD3 mAbs appear to exert therapeutic effects in two consecutive phases. The first one, during drug exposure, involves alteration in the circulation of T cells and T cell unresponsiveness as a consequence of antigenic modulation as well as killing of a fraction of T cells by apoptosis. The second phase starts when the antibody has cleared from the circulation; it involves re-expression of CD3/TCR complexes on remaining T cells with enrichment for protective TGF-β dependent adaptive Tregs. Tregs appear more resistant to apoptosis induced by non-FcγR binding anti-CD3 antibodies compared with both recently activated effector and naive T cells [11]. Non-FcγR binding anti-CD3 therapies are being developed for the treatment of various autoimmune diseases (table 1).
| Table 1: CD3-specific antibodies in clinical development for the treatment of autoimmune diseases. | ||||
| INN | Other names | Format | Fcmutations | Main clinical trials in autoimmune diseases (Phase, Clinical Trials.gov NCT number) |
| Teplizumab | MGA031, hOKT3γ1(Ala-Ala) | Humanised IgG1 | L234/A and L235/A | Type-1 diabetes (Phase 3, 00920582) Psoriasis (Phase 1/2, 00954915) Psoriatic arthritis (Phase 2, 00239720) |
| Otelixizumab | TRX4, ChAglyCD3, GSK2136525 | Chimaeric/ Humanised IgG1 | N297/A | Type-1 diabetes (Phase 3, 01123083) Thyroid eye disease (Phase 1, 01114503) Rheumatoid arthritis (Phase 1, 01101555) |
| Visilizumab | Nuvion, HuM291 | Humanised IgG2 | V234/A and V237/A | Ulcerative colitis (Phase 2/3, 00279422) Crohn’s disease (Phase 2, 00267722) |
| Foralumab | 28F11-AE, NI-0401 | Human IgG1 | L234/A and L235/E | Crohn’s disease (Phase 1/2, 00630643) |
Phase 1/2 clinical trials have been conducted to investigate the capacity of teplizumab and otelixizumab to suppress the autoimmune process and preserve the residual β-cell function in patients with new onset T1D. The first antibody, teplizumab, was investigated in 24 patients in whom the disease had been diagnosed within the previous six weeks [12]. In this open label study, patients were randomised to receive a single 14-day course of daily infusions with teplizumab (34 mg cumulative dose for a patient weighing 70 kg) or no antibody treatment, and were followed for one year. Nine of the twelve patients, treated with the therapeutic mAb, maintained or increased insulin production in response to a mix meal after one year, while only two out of twelve control patients had a sustained response. The most frequent side effects of teplizumab were mild to moderate fever and anaemia (9/12 patients) and a pruritic urticarial rash that developed on the hands and occasionally the trunk and feet (7/12 patients). An extension of this phase 1/2 trial was conducted to continue evaluation of these patients as well as to include additional subjects [13]. In total, 42 patients were randomised for prolonged assessment of the safety and efficacy of teplizumab. After two years the effects of teplizumab were still significant in terms of insulin production and exogenous insulin intake compared with the control group. In fact, Herold and colleagues reported notable maintenance of insulin production for up to 5 years in three patients treated with a single 12-day course of teplizumab [14]. No evidence of long term toxic effects (i.e. up to two years after antibody treatment) was observed.
More recently these results were confirmed in another investigator-sponsored Phase 2 clinical trial (AbATE) coordinated by the immune tolerance network (ITN). This placebo-controlled randomised study was conducted in 83 patients in whom the disease had been diagnosed within the previous 8 weeks. Teplizumab was administered intravenously daily for 14 days using a dose escalation course (from 51 µg/m2 on day 1, to 826 µg/m2 on days 5–14, corresponding to a cumulative dose of approximately 17 mg for a patient with a body surface area of 1.9 m2) at study entry, with the possibility of a second course after 12 months [15]. At 12 and 24 months post first course of anti-CD3 treatment, increased insulin production was observed in response to a mix meal [16].
The second humanised anti-CD3 mAb, otelixizumab, also produced very promising results in a double-blind phase 2 study in T1D. In this study, 80 patients who had been treated with insulin for less than 4 weeks were equally randomised to receive a single 6-day course of daily infusions with otelixizumab (48 mg cumulative dose) or placebo [17]. At 6, 12 and 18 months, residual β-cell function was best maintained in patients treated with otelixizumab versus the placebo group. In addition, daily insulin intake increased in the placebo group but not in the drug-treated group. Interestingly, this effect was more pronounced in patients with baseline β-cell function at or above the 50th percentile of the 80 patients. In this subgroup, 12 of 16 patients who received otelixizumab (75%) needed minimal doses of insulin (≤0.25 IU/kg per day) compared with none of the 21 patients who received placebo. Infusion-related reactions were similar to those observed with teplizumab, i.e. fever, headache, gastrointestinal symptoms, arthralgia, myalgia and rash. In addition, 30 of the patients in the treatment group had a syndrome similar to acute mononucleosis with sore throat, fever, cervical adenopathy or all of these starting between days 16 and 21 after the first infusion and resolving within 7 to 12 days. In 35 of 37 patients who were healthy carriers of the virus at the time of inclusion this syndrome was associated with an increase in circulating plasma Epstein-Barr virus (EBV) DNA copies which normalised at 6–12 weeks [18]. This symptomatic reactivation of EBV was not reported with teplizumab therapy. Keymeulen and colleagues showed that treatment with otelixizumab delayed the rise in insulin requirements of patients with recent-onset T1D, and reduced its amplitude over 48 months [19]. The therapeutic effect was better in patients with higher baseline residual β-cell function and a younger age. Also, importantly, no long-term adverse events were observed. Taken together, these studies with humanized and Fc mutated anti-CD3 mAbs suggest that a single short treatment course of anti-CD3 therapy attenuates the decline of β-cell function for at least two years and possibly four to five years in patients with recent onset T1D.
These encouraging results obtained from pilot clinical studies prompted confirmatory large trials with teplizumab and otelixizumab to assess their efficacy in T1D. Phase 3 randomised, double-blind, placebo-controlled clinical trials with teplizumab and otelixizumab have been conducted to assess their safety and efficacy in recent onset T1D. The PROTÉGÉ trial evaluated teplizumab in 513 patients aged 8–35, who had been diagnosed with T1D for 12 weeks or less. In contrast to previous studies, this clinical trial was conducted with two courses of teplizumab 6 months apart. Similarly, the DEFEND-1 trial evaluated otelixizumab in patients aged 12–45 with newly diagnosed T1D at over 100 study centres throughout North America and Europe. The study drug was administered not more than 90 days after the initial disease diagnosis. However, it was announced in 2010 and 2011 that the primary efficacy endpoint of both studies was not met. Interestingly, exploratory, post-hoc analyses suggested that, at 1 year, teplizumab does prevent the decline of β-cell function and can provide glycaemic control at reduced doses of insulin in children when used early after diagnosis of T1D [20]. However, the data showed that repeating anti-CD3 injections 6 months apart might not be sufficient to prolong anti-CD3 treatment efficacy. Also to be noted is that a high incidence of patients (76.6%) treated with teplizumab developed anti-drug antibodies [20]. A similar incidence of anti-drug antibodies (77.5%) was observed in the phase 2 trial with otelixizumab [21]. These findings suggest that immunogenicity might prevent repeated treatment with teplizumab and otelixizumab.
Visilizumab and foralumab were both tested in IBD. An open-label phase 1 study was conducted with visilizumab in 32 patients with severe steroid-refractory ulcerative colitis [22]. Eight patients received visilizumab at a dose of 15 µg/kg/day intravenously on 2 consecutive days. Dose-limiting toxicity due to prolonged lymphopenia (T cell recovery >30 days) occurred in 2 of the first 8 patients. The dose was therefore reduced to 10 µg/kg in the next 24 patients. On day 30, 84% of patients showed a clinical response, 41% achieved clinical remission and 44% achieved endoscopic remission. 45% of patients did not require salvage therapies or colectomy during the first year postdose. Mild to moderate symptoms of cytokine release occurred in 100% and 83% of patients in the 15- and 10-µg/kg dose groups respectively. Plevy and colleagues concluded that visilizumab had an acceptable safety profile at the 10 µg/kg dose level and may be clinically beneficial in patients with severe intravenous corticosteroid-refractory ulcerative colitis. However, a confirmatory randomised, double-blind, placebo-controlled trial failed to demonstrate that visilizumab was effective for the treatment of IBD. Sandborn and colleagues conducted a randomised, double blind, placebo-controlled trial of visilizumab in intravenous corticosteroid-refractory ulcerative colitis with a planned recruitment of 150 patients [23]. This trial was discontinued prematurely by the data safety monitoring board after 127 patients had been randomised when an interim analysis showed that the study drug was neither safe nor effective in treating severe intravenous corticosteroid-refractory ulcerative colitis. Treatment with visilizumab at a cumulative dose of 0.7 mg (for a patient weighing 70 kg) was associated with increased rates of infection, cytokine release syndrome, cardiac and vascular disorders. It is unclear why the tolerability of visilizumab is significantly inferior to that of the other Fc modified anti-human CD3 mAbs. This may be linked to its unique capacity to address preferentially activated T cells. Partial activation of T cells is a prerequisite for therapeutic efficacy with anti-CD3 therapy and visilizumab was selected on the basis of its ability to induce partial agonism of the CD3/TCR in order to kill preferentially activated T cells by activation-induced cell death (AICD) in vitro [24]. Thus visilizumab’s propensity to induce T cell activation as compared to the other Fc modified anti-CD3 therapies is probably responsible for the poor tolerability of visilizumab in vivo. In view of its unfavourable benefit-to-risk profile the clinical development of visilizumab was stopped.
Foralumab was assessed in patients with moderate to severe active Crohn’s disease [25]. In this double-blind placebo-controlled, dose-escalation study, foralumab was administered intravenously as a single 5-day treatment course. The primary endpoints of the trial were safety and the ability to modulate the CD3/TCR complex. Secondary objectives included the therapeutic response to foralumab over time, defined as either clinical remission or clinical response, and the effect of foralumab on mucosal repair. Foralumab was tolerated at doses ≤1 mg with manageable side effects. No significant improvement of Crohn’s disease activity index was observed, but a statistically significant improvement in the Crohn’s disease endoscopic index score was observed in the 1 mg dose group compared to placebo. With only 7 patients recruited in the placebo group and 11 in the 1 mg dose group, the study was not powered to assess clinical efficacy.
Several reasons may explain the disappointing results of some of these clinical trials in T1D and IBD. Firstly, for example, in the PROTÉGÉ trial a composite endpoint of reduced insulin requirement with maintenance of glycaemic control was used [20]. This endpoint had not been validated as a surrogate outcome measure for assessment of therapeutic efficacy. Using glycated haemoglobin (HbA1c) as a primary endpoint in this trial may have been inappropriate, as most patients already had low levels of HbA1c at study entry. In this context the preservation of insulin production or the need for exogenous insulin would perhaps have been better markers of treatment efficacy [20]. Secondly, for safety reasons the dose of the anti-CD3 mAbs used in the phase 3 trials was reduced as compared with that used in the pilot studies. Despite 2 dosing cycles of teplizumab in the PROTÉGÉ trial while the original phase 2 trial consisted of only one cycle, the cumulative doses were reduced. PROTÉGÉ had 3 dose groups of 17 mg (14-day full dose group), 5.6 mg (14-day low dose group) and 4.6 mg (6-day full dose group) equivalent for a patient with a body surface area of 1.9 m2, which corresponds to a reduction of approximately two, six and seven times the dose used in the pilot study respectively [12, 20]. Also, the cumulative dose of otelixizumab used for the phase 3 DEFEND-1 trial was reduced to 3.1 mg per patient, corresponding to a dose about 15 times lower than that which proved effective in the phase 2 trial, which was 48 mg [17]. Similarly, the equivalent cumulative dose of visilizumab used in the phase 3 study was 0.7 mg for a patient weighing 70 kg [23], which corresponds to half the dose used in the phase 2 study of visilizumab [22].
Figure 1
Schematic representation of some key immune mediators involved in the pathogenesis of autoimmune diseases. Combinations of drugs targeting distinct pathways in this process represent promising ways of curing autoimmune diseases.
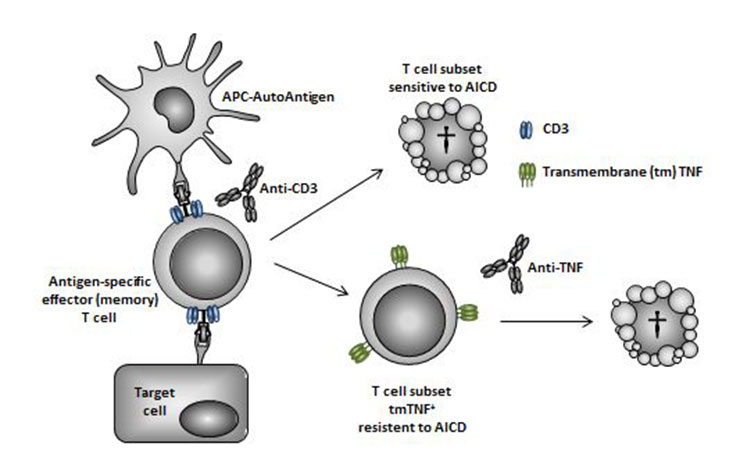
Figure 2
Schematic representation of a possible complementary mechanism of action for anti-CD3 and anti-TNF combination therapy on pathogenic T cells. In response to antigen presentation and exposure to anti-CD3 mAb in the secondary lymphoid and target organs, antigen-specific effector and/or effector memory T cells either undergo apoptosis through the process of activation-induced cell death (AICD) or survive and express several activation markers, including transmembrane (tm) TNF. An anti-TNF mAb can then deplete by ADCC (and/or reverse signalling) pathogenic T cells resistant to anti-CD3 induced AICD.
Thus, despite the creation of engineered humanised/human antibody alternatives to muromonab, treating large numbers of patients in a manner that ensures efficacy while minimising side effects remains a challenge. Interestingly, a therapeutic effect of teplizumab in the PROTÉGÉ trial (post-hoc exploratory analysis) was observed with the highest dose [20]. Because of adverse events observed in the phase 2 trials, the dose of anti-CD3 antibodies in phase 3 confirmatory trials had to be reduced and was therefore probably insufficient. Thus, going forward, what strategies can be envisaged that will widen the therapeutic window when dosing patients with anti-CD3 mAbs? Combining anti-CD3 mAbs with other drugs may be the most effective way to reduce toxicity while allowing significant therapeutic benefit to occur. Importantly, the choice and protocol may be disease-specific.
To reduce infusion-related reactions, pre- and co-medication is already a practise sometimes invoked. Indeed, pre-treatment with corticosteroids represents the most widely used strategy. Early on it was recognised in preclinical models that hydrocortisone [26] and methylprednisolone [27] inhibit release of TNF, IL-2 and IL-6, and thus symptoms such as hypothermia and diarrhoea. This effect translates to the clinical setting as corticosteroids alone [28], or in association with pentoxyfylline [29] or indomethacin [30], reduce the release of cytokines and their associated symptoms after the first infusion of muromonab.
As TNF plays a substantial role in anti-CD3 mediated side effects, an alternative is selective inhibition with anti-TNF blockers. In animals, pre-treatment with an anti-TNF mAb, reverses the anti-CD3-induced hypothermia, diarrhoea and hypomobility [31]. With human kidney allograft recipients a single dose of 0.4 or 2 mg/kg anti-TNF mAb one hour before the first muromonab administration is effective in preventing the first dose reaction [32].
Importantly, in both animal models and clinical practice, inhibiting cytokine release by corticosteroids or anti-TNF mAb has no impact on the therapeutic efficacy of anti-CD3 in transplantation. Similarly, in the murine model of multiple sclerosis, experimental allergic encephalomyelitis (EAE), pre-treatment with ciclosporin inhibits anti-CD3 mAb-mediated cytokine release without affecting clinical efficacy [33]. This is in contrast to use in NOD mice where ciclosporin blocks the ability of anti-CD3 mAbs to reverse the development of spontaneous T1D [34]. Interestingly, cyclophosphamide [34] and rapamycin [35] have also been shown to exert detrimental effect on the therapeutic efficacy of anti-CD3 mAb in NOD mice.
In EAE, protection from clinical disease progression induced by non-FcγR binding anti-CD3 mAbs is associated with an increase in the frequency of CD4+CD25+ T cells, although neither depletion of Tregs nor anti-TGF-β treatment abrogated the treatment’s efficacy [36]. In contrast, in NOD mice CD3-specific antibodies induce transferable tolerance involving CD4+CD25+ T cells. Also, remission of T1D is abrogated by co-administration of a neutralising anti-TGF-β antibody [37]. The fact that calcineurin and mTOR inhibitors block the anti-CD3-induced tolerogenic effects in NOD mice suggests that effective CD3/TCR intracellular signalling is required for tolerance induction via TGF-β. Combining immunosuppressive drugs with anti-CD3 mAbs to diminish their side effects should therefore be envisaged with extreme caution. Thus, preclinical testing should be performed in an experimental model that allows assessment of potential impact on all anti-CD3 induced immunoregulatory properties that may be involved in their therapeutic efficacy in the clinic.
A more attractive strategy to improve the clinical outcome with anti-CD3 mAbs is combination treatment aiming at improving clinical efficacy. This strategy is currently the subject of intensive investigation for the treatment of autoimmune diseases such as T1D and rheumatoid arthritis, using different approaches. In T1D the therapeutic interventions aim at (1.) controlling the autoimmune process to inhibit the destruction of β-cells in the pancreas, or (2.) restoring insulin secretion by either stimulation/expansion of residual β-cells or replacement of β-cells. In patients with no or too few residual β-cells, therapies replacing the destroyed β-cells can reduce the risks of life-impacting hypoglycaemia. Combination of anti-CD3 mAb treatment, to eliminate autoreactive T cell responses, with intraportal allogeneic islet infusion to restore endogenous insulin secretion, represents a promising approach. Hering and colleagues have used teplizumab during pancreatic islet transplantation with 4 of 6 subjects becoming insulin independent for 1 year [38]. Recently, a larger study reported that induction therapy with teplizumab promotes long-term islet graft survival and function [39]. Remarkably, 50 percent of diabetic recipients were insulin-independent 5 years after the last allo-islet infusion. In NOD mice, anti-CD3 mAb treatment and transplantation of embryonic pancreatic precursor cells induce long-term remission of T1D [40].
In patients with residual functional β-cell mass, an approach to halting the autoimmune process consists in using a drug that prevents tissue damage or even accelerates tissue regeneration/repair in combination with an anti-CD3 mAb. To this end, Sherry et al. tested the hypothesis that exendin-4 would enhance remission of T1D in NOD mice treated with 145-2C11 [41]. Exendin-4 is a glucagon-like peptide-1 receptor agonist that was shown to reduce insulitis scores, enhance β-cell mass and improve glucose tolerance in NOD mice [42]. The combination of exendin-4 and anti-CD3 mAb therapy enhanced remission of T1D in NOD mice by enhancing the recovery of the residual islets. Such a combination has also been proposed using teplizumab in combination with exenatide, a synthetic version of exendin-4 marketed as Byetta and Bydureon, in new onset T1D. The strategy is for teplizumab to turn off the autoimmune process while exenatide would reduce the rate of apoptosis resulting in an increase in β-cell mass and function [43]. Another strategy consists in combining anti-CD3 mAb therapy with other immunomodulating agents with a view to more efficiently, and perhaps more specifically, halting the autoimmune process. T1D is the result of an imbalance between autoaggressive and Treg subsets. To complement the capacity of anti-CD3 mAbs to deplete autoaggressive T cells and promote the induction of Tregs, antigen-specific immunotherapy represents an attractive strategy. Combination treatment with anti-CD3 mAb and intranasal delivery of proinsulin peptide has already been shown to reverse disease in NOD mice and a virus-induced diabetic mouse model with much greater efficacy than with the anti-CD3 mAb or peptide monotherapies [44]. Efficacy is associated with expansion of CD25+ Foxp3+ and insulin specific Tregs which produce regulatory cytokines (IL-10, TGF-β, and IL-4). Remarkably, these cells can transfer tolerance to immunocompetent recipient mice with recent onset T1D and suppress heterologous autoaggressive CD8+ T cell responses. Combined anti-CD3 mAb treatment and antigen-based intervention also provides a positive outcome when using glutamic acid decarboxylase of 65 kd (GAD65)-expressing plasmid to express the protein [45]. Synergism was observed using GAD65 treatment and a suboptimal dose of non-FcγR binding anti-CD3 and shown to be associated with expansion of GAD65-specific Tregs secreting IL-10, TGF-β, and IFN-γ. Interestingly, anti-CD3 and GAD65 vaccine synergistically reversed T1D in a virus-induced diabetic mouse model (C57BL/6 background) but not in NOD mice [45]. This finding suggests that the therapeutic efficacy of combined anti-CD3 and antigen-based therapy is dependent on the genetic background. This may have implications for the successful translation of this strategy into the clinical setting.
Takiishi and colleagues developed a more sophisticated strategy for tolerance restoration in T1D using mucosal delivery of Lactococcus lactis genetically modified to secrete the whole proinsulin autoantigen along with the immunomodulatory cytokine IL-10 [46]. Combination therapy with their bacterial construct administered orally and a suboptimal intravenous dose of 145-2C11 in NOD mice with established T1D induced autoantigen-specific long-term tolerance. The authors showed that the frequency of Tregs in the pancreatic islets was increased and that these cells suppressed the autoimmune response in an autoantigen-specific manner. Interestingly, higher autoantibody levels at diagnosis can distinguish responders from non-responders among recipients of combined anti-CD3 and insulin immunotherapy [47]. Co-administration of oral insulin was shown to improve and prolong the therapeutic efficacy of anti-CD3 therapy. Long-term protection was achieved by maintaining elevated insulin-specific Treg numbers that efficiently lowered diabetogenic effector memory T cells. This study suggests that pre-existing anti-insulin autoantibody levels can be used as biomarkers to distinguish future responders from non-responders among recipients of combined anti-CD3/oral insulin treatments. The levels of anti-serpinB13 autoantibodies have recently been shown to inversely correlate with the levels of anti-insulin autoantibodies and thus could also be used as a biomarker [48]. Interestingly, exposure to anti-serpinB13 mAb decreased islet inflammation and, further, co-administration of this reagent and a suboptimal dose of anti-CD3 mAb accelerated recovery from T1D in NOD mice [48].
Very recently, combination therapy with a bioactive vitamin D3 analogue (TX527), ciclosporin and anti-CD3 mAb, all used at sub-therapeutic doses, was shown to reduce synergistically recurrent autoimmune responses to a grafted islet mass in NOD mice [49]. The combination therapy surpassed anti-CD3 monotherapy in reducing pro-inflammatory cytokine responses and increasing the frequency of Tregs. The authors of the study concluded that individual agents of the combination therapy cooperate to enhance their individual potency, thereby offering an interesting strategy circumventing the dose-related side effects of anti-CD3 mAbs currently encountered in the treatment of autoimmune diseases.
Another strategy consists in combining anti-CD3 mAb therapy with neutralisation of pro-inflammatory cytokines participating in the T cell subset imbalance and tissue damage (fig. 1). Using the classical NOD model, Ablamunits and colleagues have recently demonstrated that combination of anti-CD3 mAb therapy with IL-1 blockade synergistically induces persistent remission of islet inflammation [50]. Ex vivo investigations suggest that complementary mechanisms involving depletion of pathogenic T cells and increased regulatory function of T cells and splenocytes resulted in synergistic therapeutic effects. Similarly, a short course treatment regime involving a combination of anti-CD3 and anti-TNF mAbs synergistically and dose dependently inhibits the progression of established collagen-induced arthritis (CIA) [51, 52]. The impact was remarkable as the effect was maintained for over 3 weeks after the end of the 5-day treatment period. When animals were administered an antibody that would bind and neutralise the anti-CD3 mAb, and thus reverse the generalised immunosuppression, the therapeutic effect was maintained, suggesting induction of tolerance [51]. However, the mechanism did not involve expansion of CD25+Foxp3+ Tregs or depend on TGF-β or programmed death-ligand 1. Anti-CD3 and anti-TNF combination therapy efficiently depleted Th1 and Th17 pathogenic T cells in the periphery. In the joints, treatment with the combination therapy was associated with a reduced number of CD4+ T cells. It is unclear from these experiments whether T cells are directly depleted in the joints or whether their migration into the joints is inhibited. However, since it has been shown that using an anti-TNF mAb in CIA inhibits migration of Th1 and Th17 cells to the joints [53], we speculate that pathogenic T cells are killed in the periphery, e.g. in secondary lymphoid tissues such as the draining lymph nodes and spleen. Putting the results of this research into a clinical context is supported by the experience reported recently by Reinke and colleagues [54]. Using a combination of muromonab and infliximab, they have observed improved clinical success in kidney transplant recipients receiving a second or third time organ. The combination therapy involved selective depletion of donor-specific effector memory T cells. Thus, considering recent literature together with our recently published data, we would propose the following mechanism for the anti-CD3 and anti-TNF combination therapy (fig. 2). During an autoimmune response, self-antigens are presented by APC which derive pathogenic effector/memory T cell populations. In the presence of an anti-CD3 mAb, a subset of this population will become “immuno-blind” (CD3/TCR negative) and partially activated, and thus sensitive to undergo apoptosis through the process of AICD [55]. In CIA, non-FcγR binding anti-CD3 mAbs do not sufficiently deplete pathogenic T cells, and thus their efficacy is limited. In the context of our investigation it is conceivable that the subset of pathogenic T cells that escape anti-CD3 induced AICD are activated and consequently express transmembrane TNF, the precursor form of soluble TNF. As a one-two punch, an anti-TNF may therefore deplete by CDC, ADCC or even reverse signalling pathogenic T cells resistant to anti-CD3-mediated AICD via the mechanism of engaging transmembrane TNF [56].
Although counterintuitive, extensive preclinical data show that mucosal (both oral and nasal) delivered anti-CD3 mAbs can exert potent immunoregulatory functions and ameliorate experimental autoimmune and inflammatory diseases [57]. Prophylactic treatment with anti-CD3 significantly attenuated the development of CIA via nasal administration as well as via oral administration, although not to a statistically significant degree [58]. In contrast, no effect was observed when the anti-CD3 was administered therapeutically. Remarkably, oral or nasal therapeutic co-administration of anti-CD3 with an emulsome adjuvant that enhances Th2 responses resulted in suppression of ongoing disease with less joint damage, a decrease in TNF and IFN-γ mRNA expression in the joints, and a reduction in anti-collagen antibodies. These results show that mucosal anti-CD3 therapy may be useful for the treatment of arthritis in combination with an emulsome-based adjuvant to enhance its therapeutic efficacy [58].
Using an anti-CD3 targeting therapy remains one of the only strategies where induction of tolerance and immunomodulation translates from laboratory testing to clinical trials. Thus, using anti-CD3 therapeutic mAbs remains an attractive mechanism for a physician to have in the pharmacopoeia. Since anti-CD3 mAbs cannot be administered in the clinic at a therapeutic dose without evoking cytokine-related reactions and/or EBV reactivation, strategies are under intensive investigation in an effort to successfully translate the unique immunoregulatory properties of anti-CD3 mAbs in the clinic. One consists of administering the drug orally. The upside of oral treatment with anti-CD3 mAbs is that there is no systemic drug exposure and thus no generalised immunosuppression associated with EBV reactivation, and no side effects related to cytokine release [57]. It would therefore be ideal for chronic treatment, which may be required for the treatment of most autoimmune diseases. A clinical study to evaluate the clinical efficacy of oral anti-CD3 in IBD is ongoing [59]. Another attractive strategy to strengthen clinical efficacy of low dose anti-CD3 strategy consists of combining anti-CD3 mAbs with other drugs to induce and sustain clinical remission. Monotherapy has its limits, ranging from targeting a single arm of the immune process to more adverse events due to higher dose requirements. High variability in disease amongst patients with autoimmune disorders, especially those to be enrolled in phase 3 trials and even more so when a drug is prescribed as a marketed product, constitutes a challenge to be addressed with mono-immunotherapies. Indeed, in T1D, no mono- immunotherapy has reported long term disease remission [60]. Thus, combination immunotherapies are increasingly considered to improve the therapeutic efficacy of anti-CD3 mAbs. It is reasonable to anticipate that identifying the appropriate combination (drug and treatment regimen including the dose of individual compounds) will provide a variety of synergistic effects in the clinical setting, as demonstrated in animal model systems (table 2). Recommendations have been made for the development of combination immunotherapies for the treatment of T1D. Recently the T1D combination therapy assessment group with representatives from the ITN and Juvenile Diabetes Research Foundation (JDRF) indicated a preference for combination therapies using anti-CD3 and either antigen (such as oral insulin, GAD65 or proinsulin) or IL-1 blockade for the treatment of recent onset T1D [61]. While in patients newly diagnosed with rheumatoid arthritis, combining anti-CD3 and anti-TNF therapies represents an attractive strategy for induction of clinical remission [51]. These approaches may also be the basis for broader applications in other chronic inflammatory and autoimmune diseases with high unmet medical needs such as IBD and multiple sclerosis.
| Table 2: Synergism between CD3-specific antibodies and various therapeutic interventions in established experimental autoimmunity. | ||||||||
| Anti-CD3 therapy* | Treatment combined with anti-CD3* | Animal model | Outcome; Reported mode of action | Ref. | ||||
| 145-2C11 format | Route | Dose | Drug | Route | Dose | |||
| Hamster IgG (FcγR binding) | i.p. | 100 μg/d 2×, 5 days apart | Anti-TNF | i.p. | 300 μg/d 2×, 5 days apart | CIA | Long-lasting (>8 weeks) disease remission; not determined | [52] |
| Chimaeric Fc-mutated IgG (non-FcγR binding) | i.p. | 50 μg/d for 5 consecutive days | Anti-TNF | i.p. | 100 μg once | CIA | Long-lasting (>4 weeks) disease remission; anti-inflammatory effects and depletion of pathogenic T cells | [51] |
| F(ab’)2 (non-FcγR binding) | Oral | 5 μg/d for 5 consecutive days | Emulsome | Oral | Anti-CD3 administered emulsified in 5% emulsome | CIA | Long-lasting (>14 weeks) inhibition of disease progression; anti-inflammatory effects (reduced TNF and IFN-γ mRNA expression in the joints) and induction of LAP+ Tregs | [58] |
| Nasal | 0.5 μg 3× every other day | Nasal | ||||||
| F(ab’)2 | i.p. | 50 μg/d for 5 consecutive days | IL-1RA | i.p. | 10 mg/d for 5 consecutive days | NOD | Accelerates and improves rate of disease remission; anti-inflammatory effects, elimination of pathogenic T cells and increased regulatory activity of innate and adaptive immune cells | [50] |
| Anti-IL-1β | i.p. | 75 μg/d 3×, 2 days apart | ||||||
| Hamster IgG | i.v. | 2.5 μg/d for 5 consecutive days | Vitamin D3 analogue (TX527) andCiclosporin | i.p. per os | 100 μg/kg every 2d for 60 days 5 mg/kg/d for 30 days | Islet transplanted NOD | Prolonged graft acceptance; anti-inflammatory effects, elimination of pathogenic T cells and increased Foxp3+ Tregs | [49] |
| F(ab’)2 | i.v. | 40 μg/d for 5 consecutive days | Proinsulin | i.n. | 40 μg/d 4×, 2–5 days apart | NOD and H2dRIP-LCM V-NP | Enhances rate of disease remission; expansion of Ag-specific Foxp3+ Tregs | [44] |
| Hamster IgG | i.v. | 2.5 μg/d for 5 consecutive days | Lactococcus lactis expressing IL-10 and proinsulin | i.g. | 2×109 CFU 5×/week for 6 weeks | NOD | Enhances rate of long-lasting (>14 weeks) disease remission; expansion of Ag-specific Foxp3+ Tregs | [46] |
| F(ab’)2 | i.v. | 5–25 μg/d for 3 consecutive days | Insulin | i.g. | 0.5–1 mg 2×/week for 5 weeks | NOD | Long-lasting (>16 weeks) remission of severe disease (high levels of anti-insulin autoantibodies at disease onset) with reduced disease recurrence rate; Depletion of pathogenic T cells and expansion of Ag-specific Foxp3+ Tregs | [47] |
| F(ab’)2 | i.v. | 40 μg/d for 4 consecutive days | GAD65 expressing plasmid | i.m. | 100 μg/leg/d 3×, 5–7 days apart | H2bRIP-LCM V-GP | Enhances rate of disease remission in appropriate genetic background; expansion of Ag-specific Foxp3+ Tregs | [44, 45] |
| Hamster IgG | 10 μg/d 4× | Anti-serpinB13 | 100 μg/d 4× | NOD | Accelerates disease remission; not determined | [48] | ||
| Hamster IgG | i.v. | 10 μg/d for 5 consecutive days | Exendin-4 | i.p. | 75 ng/d for 10 consecutive days | NOD | Enhances rate of disease remission; inhibition of autoimmune destruction and enhancement of functional recovery of residual β-cells | [41] |
| Hamster IgG | i.v. | 10 μg/d for 5 consecutive days | Histocompatible embryonic pancreatic precursor cells | SRC | 6–10 anlagen | NOD | Long-lasting (>6 weeks) disease remission; inhibition of autoimmune destruction of β-cells and restoration of β-cell mass and insulin production | [40] |
| F(ab’)2 | i.v. | 50 μg/d for 5 consecutive days | ||||||
| i.v. = intravenous; i.p. = intraperitoneal; i.g. = intragastric; i.n. = intranasal; i.m. = intramuscular; SRC = subrenal capsule; CFU = colony-forming unit. *Drugs were administered in sick animals i.e. after diabetes or arthritis had been diagnosed. | ||||||||
Acknowledgement:We thank Dr C. De Min and D. Slack for critical reading of the manuscript.
Funding / potential competing interests: The work was funded by NovImmune SA. Y. Dean and M. Kosco-Vilbois are shareholders in NovImmune SA, the discoverer and developer of NI-0401/Foralumab. M. Kosco-Vilbois is an employee at NovImmune SA. Y. Dean, F. Dépis and M. Kosco-Vilbois are inventors on patents filed by NovImmune SA relating to composition of matter as well as the clinical use of NI-0401/Foralumab.
1 Hooks MA, Wade CS, Millikan WJ, Jr. Muromonab CD-3: a review of its pharmacology, pharmacokinetics, and clinical use in transplantation. Pharmacotherapy. 1991;11(1):26–37.
2 Norman DJ, Chatenoud L, Cohen D, Goldman M, Shield CF, III. Consensus statement regarding OKT3-induced cytokine-release syndrome and human antimouse antibodies. Transplant Proc. 1993;25(2 Suppl 1):89–92.
3 Leo O, Foo M, Sachs DH, Samelson LE, Bluestone JA. Identification of a monoclonal antibody specific for a murine T3 polypeptide. Proc Natl Acad Sci U S A 1987;84(5):1374–8.
4 Ferran C, Sheehan K, Dy M, Schreiber R, Merite S, Landais P, et al. Cytokine-related syndrome following injection of anti-CD3 monoclonal antibody: further evidence for transient in vivo T cell activation. Eur J Immunol. 1990;20(3):509–15.
5 Hirsch R, Eckhaus M, Auchincloss H, Jr., Sachs DH, Bluestone JA. Effects of in vivo administration of anti-T3 monoclonal antibody on T cell function in mice. I. Immunosuppression of transplantation responses. J Immunol. 1988;140(11):3766–72.
6 You S, Candon S, Kuhn C, Bach JF, Chatenoud L. CD3 antibodies as unique tools to restore self-tolerance in established autoimmunity, their mode of action and clinical application in type 1 diabetes. Adv Immunol. 2008;100:13–37.
7 Chatenoud L. Immune therapy for type 1 diabetes mellitus-what is unique about anti-CD3 antibodies? Nat Rev Endocrinol. 2010;6(3):149–57.
8 Chatenoud L. CD3-specific antibody-induced active tolerance: from bench to bedside. Nat Rev Immunol. 2003;3(2):123–32.
9 Weinshenker BG, Bass B, Karlik S, Ebers GC, Rice GP. An open trial of OKT3 in patients with multiple sclerosis. Neurology. 1991;41(7):1047–52.
10 Kuhn C, You S, Valette F, Hale G, van EP, Bach JF, et al. Human CD3 transgenic mice: preclinical testing of antibodies promoting immune tolerance. Sci Transl Med. 2011;3(68):68ra10.
11 Penaranda C, Tang Q, Bluestone JA. Anti-CD3 therapy promotes tolerance by selectively depleting pathogenic cells while preserving regulatory T cells. J Immunol. 2011;187(4):2015–22.
12 Herold KC, Hagopian W, Auger JA, Poumian-Ruiz E, Taylor L, Donaldson D, et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med. 2002;346(22):1692–8.
13 Herold KC, Gitelman SE, Masharani U, Hagopian W, Bisikirska B, Donaldson D, et al. A single course of anti-CD3 monoclonal antibody hOKT3gamma1(Ala-Ala) results in improvement in C-peptide responses and clinical parameters for at least 2 years after onset of type 1 diabetes. Diabetes. 2005;54(6):1763–9.
14 Herold KC, Gitelman S, Greenbaum C, Puck J, Hagopian W, Gottlieb P et al. Treatment of patients with new onset type 1 diabetes with a single course of anti-CD3 mAb Teplizumab preserves insulin production for up to 5 years. Clin Immunol. 2009;132(2):166–73.
15 ClinicalTrials.gov. U.S. National Institutes of Health [updated 2009 May 13; cited 2012 Sep 5]. Available from: http://clinicaltrials.gov/ct2/show/NCT00129259.
16 Sprangers B, Van der Schueren B, Gillard P, Mathieu C. Otelixizumab in the treatment of type 1 diabetes mellitus. Immunotherapy. 2011;3(11):1303–16.
17 Keymeulen B, Vandemeulebroucke E, Ziegler AG, Mathieu C, Kaufman L, Hale G, et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med. 2005;352(25):2598–608.
18 Keymeulen B, Candon S, Fafi-Kremer S, Ziegler A, Leruez-Ville M, Mathieu C, et al. Transient Epstein-Barr virus reactivation in CD3 monoclonal antibody-treated patients. Blood. 2010;115(6):1145–55.
19 Keymeulen B, Walter M, Mathieu C, Kaufman L, Gorus F, Hilbrands R, et al. Four-year metabolic outcome of a randomised controlled CD3-antibody trial in recent-onset type 1 diabetic patients depends on their age and baseline residual beta cell mass. Diabetologia. 2010;53(4):614–23.
20 Sherry N, Hagopian W, Ludvigsson J, Jain SM, Wahlen J, Ferry RJ, Jr. et al. Teplizumab for treatment of type 1 diabetes (Protege study): 1-year results from a randomised, placebo-controlled trial. Lancet. 2011;378(9790):487–97.
21 Hale G, Rebello P, Al B, I, Bolam E, Wiczling P, Jusko WJ, et al. Pharmacokinetics and antibody responses to the CD3 antibody otelixizumab used in the treatment of type 1 diabetes. J Clin Pharmacol. 2010;50(11):1238–48.
22 Plevy S, Salzberg B, Van AG, Regueiro M, Hommes D, Sandborn W, et al. A phase I study of visilizumab, a humanized anti-CD3 monoclonal antibody, in severe steroid-refractory ulcerative colitis. Gastroenterology. 2007;133(5):1414–22.
23 Sandborn WJ, Colombel JF, Frankel M, Hommes D, Lowder JN, Mayer L, et al. Anti-CD3 antibody visilizumab is not effective in patients with intravenous corticosteroid-refractory ulcerative colitis. Gut. 2010;59(11):1485–92.
24 Carpenter PA, Pavlovic S, Tso JY, Press OW, Gooley T, Yu XZ, et al. Non-Fc receptor-binding humanized anti-CD3 antibodies induce apoptosis of activated human T cells. J Immunol. 2000;165(11):6205–13.
25 van der Woude CJ, Stokkers P, van Bodegraven AA, Van AG, Hebzda Z, Paradowski L, et al. Phase I, double-blind, randomized, placebo-controlled, dose-escalation study of NI-0401 (a fully human anti-CD3 monoclonal antibody) in patients with moderate to severe active Crohn’s disease. Inflamm Bowel Dis. 2010;16(10):1708–16.
26 Ferran C, Dy M, Merite S, Sheehan K, Schreiber R, Leboulenger F, et al. Reduction of morbidity and cytokine release in anti-CD3 MoAb-treated mice by corticosteroids. Transplantation. 1990;50(4):642–8.
27 Alegre ML, Vandenabeele P, Depierreux M, Florquin S, Deschodt-Lanckman M, Flamand V, et al. Cytokine release syndrome induced by the 145-2C11 anti-CD3 monoclonal antibody in mice: prevention by high doses of methylprednisolone. J Immunol. 1991;146(4):1184–91.
28 Chatenoud L, Legendre C, Ferran C, Bach JF, Kreis H. Corticosteroid inhibition of the OKT3-induced cytokine-related syndrome – dosage and kinetics prerequisites. Transplantation. 1991;51(2):334–8.
29 Alegre ML, Gastaldello K, Abramowicz D, Kinnaert P, Vereerstraeten P, De PL, et al. Evidence that pentoxifylline reduces anti-CD3 monoclonal antibody-induced cytokine release syndrome. Transplantation. 1991;52(4):674–9.
30 Shield CF, III, Kahana L, Pirsch J, Vergne-Marini P, First MR, Schroeder TJ, et al. Use of indomethacin to minimize the adverse reactions associated with orthoclone OKT3 treatment of kidney allograft rejection. Transplantation. 1992;54(1):164–6.
31 Ferran C, Dy M, Sheehan K, Schreiber R, Grau G, Bluestone J, et al. Cascade modulation by anti-tumor necrosis factor monoclonal antibody of interferon-gamma, interleukin 3 and interleukin 6 release after triggering of the CD3/T cell receptor activation pathway. Eur J Immunol. 1991;21(10):2349–53.
32 Charpentier B, Hiesse C, Lantz O, Ferran C, Stephens S, O’Shaugnessy D, et al. Evidence that antihuman tumor necrosis factor monoclonal antibody prevents OKT3-induced acute syndrome. Transplantation. 1992; 54(6):997–1002.
33 Belmar NA, Lombardo JR, Chao DT, Li O, Ma X, Pong-Afar M, et al. Dissociation of efficacy and cytokine release mediated by an Fc-modified anti-CD3 mAb in a chronic experimental autoimmune encephalomyelitis model. J Neuroimmunol. 2009;212(1-2):65–73.
34 Chatenoud L, Primo J, Bach JF. CD3 antibody-induced dominant self tolerance in overtly diabetic NOD mice. J Immunol. 1997;158(6):2947–54.
35 Valle A, Jofra T, Stabilini A, Atkinson M, Roncarolo MG, Battaglia M. Rapamycin prevents and breaks the anti-CD3-induced tolerance in NOD mice. Diabetes. 2009;58(4):875–81.
36 Kohm AP, Williams JS, Bickford AL, McMahon JS, Chatenoud L, Bach JF, et al. Treatment with nonmitogenic anti-CD3 monoclonal antibody induces CD4+ T cell unresponsiveness and functional reversal of established experimental autoimmune encephalomyelitis. J Immunol. 2005;174(8):4525–34.
37 Belghith M, Bluestone JA, Barriot S, Megret J, Bach JF, Chatenoud L. TGF-beta-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat Med. 2003;9(9):1202–8.
38 Hering BJ, Kandaswamy R, Harmon JV, Ansite JD, Clemmings SM, Sakai T, et al. Transplantation of cultured islets from two-layer preserved pancreases in type 1 diabetes with anti-CD3 antibody. Am J Transplant. 2004;4(3):390–401.
39 Bellin MD, Barton FB, Heitman A, Harmon J, Balamurugan AN, Kandaswamy R, et al. Potent induction immunotherapy promotes long-term insulin independence after islet transplantation in type 1 diabetes. Am J Transplant. 2012.
40 Begum S, Chen W, Herold KC, Papaioannou VE. Remission of type 1 diabetes after anti-CD3 antibody treatment and transplantation of embryonic pancreatic precursors. Endocrinology. 2009;150(10):4512–20.
41 Sherry NA, Chen W, Kushner JA, Glandt M, Tang Q, Tsai S, et al. Exendin-4 improves reversal of diabetes in NOD mice treated with anti-CD3 monoclonal antibody by enhancing recovery of beta-cells. Endocrinology. 2007;148(11):5136–44.
42 Hadjiyanni I, Baggio LL, Poussier P, Drucker DJ. Exendin-4 modulates diabetes onset in nonobese diabetic mice. Endocrinology. 2008;149(3):1338–49.
43 Masharani UB, Becker J. Teplizumab therapy for type 1 diabetes. Expert Opin Biol Ther. 2010;10(3):459–65.
44 Bresson D, Togher L, Rodrigo E, Chen Y, Bluestone JA, Herold KC, et al. Anti-CD3 and nasal proinsulin combination therapy enhances remission from recent-onset autoimmune diabetes by inducing Tregs. J Clin Invest. 2006;116(5):1371–81.
45 Bresson D, Fradkin M, Manenkova Y, Rottembourg D, von HM. Genetic-induced variations in the GAD65 T-cell repertoire governs efficacy of anti-CD3/GAD65 combination therapy in new-onset type 1 diabetes. Mol Ther. 2010;18(2):307–16.
46 Takiishi T, Korf H, Van Belle TL, Robert S, Grieco FA, Caluwaerts S, et al. Reversal of autoimmune diabetes by restoration of antigen-specific tolerance using genetically modified Lactococcus lactis in mice. J Clin Invest. 2012.
47 Mamchak AA, Manenkova Y, Leconet W, Zheng Y, Chan JR, Stokes CL, et al. Preexisting autoantibodies predict efficacy of oral insulin to cure autoimmune diabetes in combination with anti-CD3. Diabetes 2012.
48 Czyzyk J, Henegariu O, Preston-Hurlburt P, Baldzizhar R, Fedorchuk C, Esplugues E, et al. Enhanced anti-serpin antibody activity inhibits autoimmune inflammation in type 1 diabetes. J Immunol. 2012;188(12):6319–27.
49 Baeke F, Van Belle TL, Takiishi T, Ding L, Korf H, Laureys J, et al. Low doses of anti-CD3, ciclosporin A and the vitamin D analogue, TX527, synergise to delay recurrence of autoimmune diabetes in an islet-transplanted NOD mouse model of diabetes. Diabetologia. 2012.
50 Ablamunits V, Henegariu O, Hansen JB, Opare-Addo L, Preston-Hurlburt P, Santamaria P, et al. Synergistic reversal of type 1 diabetes in NOD mice with anti-CD3 and interleukin-1 blockade: evidence of improved immune regulation. Diabetes. 2012;61(1):145–54.
51 Depis F, Hatterer E, Lamacchia C, Waldburger JM, Gabay C, Reith W, et al. Long term amelioration of established collagen-induced arthritis achieved with short term therapy combining anti-CD3 and anti-TNF treatments. Arthritis Rheum. 2012.
52 Malfait AM, Williams RO, Malik AS, Maini RN, Feldmann M. Chronic relapsing homologous collagen-induced arthritis in DBA/1 mice as a model for testing disease-modifying and remission-inducing therapies. Arthritis Rheum. 2001;44(5):1215–24.
53 Notley CA, McCann FE, Inglis JJ, Williams RO. ANTI-CD3 therapy expands the numbers of CD4+ and CD8+ Treg cells and induces sustained amelioration of collagen-induced arthritis. Arthritis Rheum. 2010;62(1):171–8.
54 Baan CC, Gaston RS. Report of a joint ESOT and AST meeting: highlights in biologic agents and transplantation. Am J Transplant. 2011;11(4):681–6.
55 Yu XZ, Bidwell SJ, Martin PJ, Anasetti C. Anti-CD3 epsilon F(ab')2 prevents graft-versus-host disease by selectively depleting donor T cells activated by recipient alloantigens. J Immunol. 2001;166(9):5835–9.
56 Mitoma H, Horiuchi T, Tsukamoto H, Tamimoto Y, Kimoto Y, Uchino A, et al. Mechanisms for cytotoxic effects of anti-tumor necrosis factor agents on transmembrane tumor necrosis factor alpha-expressing cells: comparison among infliximab, etanercept, and adalimumab. Arthritis Rheum. 2008;58(5):1248–57.
57 da Cunha AP, Weiner HL. Induction of immunological tolerance by oral anti-CD3. Clin Dev Immunol 2012;2012:425021.
58 Wu HY, Maron R, Tukpah AM, Weiner HL. Mucosal anti-CD3 monoclonal antibody attenuates collagen-induced arthritis that is associated with induction of LAP+ regulatory T cells and is enhanced by administration of an emulsome-based Th2-skewing adjuvant. J Immunol. 2010;185(6):3401–7.
59 ClinicalTrials.gov. U.S. National Institutes of Health [updated 2011 Aug 27; cited 2012 Sep 30]. Available from: http://clinicaltrials.gov/ct2/show/NCT01287195.
60 Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature. 2010;464(7293):1293–300.
61 Matthews JB, Staeva TP, Bernstein PL, Peakman M, von HM. Developing combination immunotherapies for type 1 diabetes: recommendations from the ITN-JDRF Type 1 Diabetes Combination Therapy Assessment Group. Clin Exp Immunol. 2010;160(2):176–84.