Hematopoietic stem cell transplantation: a review and recommendations for follow-up care for the general practitioner
DOI: https://doi.org/10.4414/smw.2012.13696
Jakob R
Passweg, Joerg
Halter, Christoph
Bucher, Sabine
Gerull, Dominik
Heim, Alicia
Rovó, Andreas
Buser, Martin
Stern, André
Tichelli
Summary
The first hematopoietic stem cell transplantation (HSCT), the replacement of the hematopoietic system, by hematopoietic stem cells from the patient (autologous HSCT) or from another person (allogeneic HSCT), was performed almost 45 years ago. Today autologous HSCT is used to bridge hematopoietic failure after high dose chemotherapy for the treatment of selected hematopoietic and non-hematopoietic tumours. Allogeneic HSCT is used to treat congenital or acquired marrow failure, and, more commonly, to exploit the graft versus tumour effect of allogeneic cells against high risk hematologic malignancies. In 2010, 30,000 patients were treated with HSCT (12,000 allogeneic and 18,000 autologous HSCT) in Europe.
Substantial progress has been made in the field of allogeneic HSCT in the last decade. First the article describes advances in patient and donor selection, the current concepts of choosing the optimal stem cell source and the most appropriate preparative regimen. Furthermore, recent advances in supportive care are described. We describe how these innovations have allowed indications for allogeneic HSCT to be expanded. Finally, prospects for future developments will be outlined.
Introduction
Replacement of the hematopoietic system, either in the form of hematopoietic stem cells from the patient themself (autologous hematopoietic stem cell transplantation; HSCT) or from another person (allogeneic HSCT), is a procedure that was used for the first time in man almost 45 years ago. Autologous HSCT is used to bridge hematopoietic failure during high dose chemotherapy for the treatment of tumours of the hematopoietic system that are adequately sensitive to this treatment. In this sense autologous stem cell support is not a "trans-plant" but the term autologous HSCT is commonly used. Results of both of these procedures are often reported together as they use similar technology. Allogeneic HSCT is used to replace the hematopoietic system in patients with acquired or congenital failure, and more commonly to exploit the graft versus tumour effect of allogeneic cells [1–2]. According to the European Group for Blood and Marrow Transplantation 2010 annual survey, 33,362 transplants were performed [3]. Of these 33,362 transplants, 13,345 were allogeneic and 20,017 autologous. Figure 1 shows the evolution of transplant numbers with a continuous increase from 1990 to 2010. Two major changes in clinical practice are mirrored in the curves displayed: The fist was the introduction of Imatinib (1999) for the treatment of chronic myeloid leukaemia (CML) resulting in a transient slowing of transplant numbers in the early 2000s. The second is the discontinuation of autologous HSCT for breast cancer after the publication of negative data.

Figure 1
Autologous and Allogeneic HSCT in Europe 1990–2010 (reprinted from: Passweg JR, Baldomero H, Gratwohl A, Bregni M, Cesaro S, Dreger P, et al.; for the European Group for Blood and Marrow Transplantation (EBMT). The EBMT activity survey: 1990–2010. Bone Marrow Transplant. 2012;47(7):906-23 [3]).
Indications
Indications for autologous and allogeneic HSCT are shown in table 1, and the distribution of its use in Europe for the year 2010 are shown in figure 2A and 2B. The vast majority of allogeneic HSCT (70%) are done for hematologic malignancies, in particular: acute myeloid leukaemia (AML, 32%) and acute lymphoblastic leukaemia (ALL 16%), myelodysplastic syndromes (MDS 16%) and non Hodgkin lymphoma (NHL) (9%) [4–10]. Beside these frequent indications, these is a host of less frequent indications including many malignant and non malignant diseases (fig. 2).
The most frequent indications for autologous HSCT in Europe are plasma cell myeloma (PCM), non Hodgkin lymphoma and Hodgkin lymphoma (HL). Whereas most patients with PCM receive autologous HSCT as part of the upfront treatment strategy, the majority of patients with NHL and in particular HL receive this treatment once they have relapsed after chemotherapy. Smaller numbers of autologous transplants are performed to treat solid tumours (germ cell tumours and solid tumours of childhood, such as neuroblastoma or Ewing sarcoma). Finally, about 150 autologous transplants are performed each year for autoimmune disorders [11–16].
For autologous HSCT, risks include the short and long term toxicities of the high dose treatment, called in this instance the preparative regimen, constituting of combination chemotherapy with or without total body irradiation. Consequences may include organ damage and the risks of infection and bleeding associated with the marrow aplasia. This period is generally short as hematopoietic stem cells previously cryopreserved are thawed and infused after the end of the preparative regimen. Relapse remains the main issue after autologous HSCT but treatment-related late effects are increasingly recognised, and so long-term follow up is currently recommended for all patients after autologous HSCT. Risks are different for allogeneic HSCT in spite of using similar technology. In allogeneic HSCT the consequences of the immune mediated phenomena, graft versus tumour effects or graft versus host disease (GvHD) in its acute or chronic form predominate.
|
Table 1: Common indications for HSCT. |
|
Autologous
|
| Malignancy |
Plasma cell myeloma |
| Non Hodgkin lymphoma |
| Hodgkin lymphoma |
| Acute myeloid leukaemia |
| Neuroblastoma |
| Ewing sarcoma |
| Other rare cancers of childhood |
| Germ-cell tumours |
| Non malignant disorders |
Autoimmune diseases |
|
Allogeneic
|
| Malignancy |
Acute myeloid leukaemia |
| Acute lymphoblastic leukaemia |
| Chronic myeloid leukaemia (refractory to tyrosine kinase inhibitors) |
| Myelodysplastic syndromes (high risk) |
| Myeloproliferative neoplasia (high risk) |
| Non Hodgkin lymphoma |
| Hodgkin lymphoma |
| Plasma cell myeloma |
| Chronic lymphocytic leukaemia |
|
Non malignant disorders
|
| Acquired |
Severe aplastic anaemia |
| Paroxysmal nocturnal hemoglobinuria (not eligible for complement inhibition treatment) |
| Congenital |
Fanconi anaemia and other marrow failure syndromes |
| Thalassemia |
| Sickle cell disease |
| Congenital immunodeficiency syndromes |
| Inborn errors of metabolism |
Stem cell source
Bone Marrow (BM): Traditionally stem cells were harvested by repeated aspirations from the iliac crest. However, this procedure requires an operation theatre, two physicians and two technicians. Post-harvest hip pain in donors is common and the need of red blood cell transfusion is sometimes necessary. Time to recovery of marrow function in the patient after HSCT of BM is approximately 17–20 days.

Figure 2A
Indications for allogeneic HSCT in Europe in 2010 (reprinted from: Passweg JR, Baldomero H, Gratwohl A, Bregni M, Cesaro S, Dreger P, et al.; for the European Group for Blood and Marrow Transplantation (EBMT). The EBMT activity survey: 1990–2010. Bone Marrow Transplant. 2012;47(7):906-23 [3]).
Abbr: AML Acute Myeloid Leukaemia, ALL Acute Lymphoblastic Leukaemia, CML Chronic Myeloid Leukaemia; MDS/MPS Myelodysplastic Syndromes/Myeloproliferative Neoplasias, CLL Chronic Lymphocytic Leukaemia, PCD plasma cell disorders, HD Hodgkin Lymphoma, NHL Non Hodgkin Lymphoma, Hemo/thal Hemoglobinopathies/Thalassemias, BMF Bone Marrow Failure, PID Primary Immunodeficiencies, IDM Inborn Errors of Metabolism, AID Autoimmune Disorders
Peripheral Blood Stem Cells (PBSC): Hematopoietic stem cells can be mobilised into the peripheral blood by using granulocyte colony-stimulating factor (G-CSF). G-CSF mobilised stem cells collected by continuous large volume (15–25 l) apheresis are the preferred stem cell source for HSCT today. As higher doses of stem cells can be harvested by apheresis compared to BM collection, time to engraftment is shorter. In autologous HSCT, PBSC have completely replaced marrow as a stem cell source. PBSC contain 10–100 times more T lymphocytes than marrow, explaining the increased incidence of chronic graft versus host disease (GvHD) after allogeneic HSCT with PBSC compared to BM, associated with better disease free survival in sibling donor HSCT in patients with advanced disease stages [17]. Whereas a recently published randomised clinical study in unrelated donor transplantation showed increased chronic GvHD without impact on other outcomes [18].
Mobilisation of PBSC is increased when G-CSF is given after chemotherapy, feasible only in recipients of autologous HSCT. Some patients fail to mobilise an adequate number of stem cells, risk factors are pre-treatment with many lines of chemotherapy and in particular with certain alkylating agents such as melphalan. Plerixafor is an inhibitor of CXC chemokine receptor 4 (CXCR4) on stem cells mediating adhesion and is useful in mobilisation failure [19–20]. For autologous HSCT, stem cells are cryo-preserved, thawed and re-infused after the patient has completed the preparative regimen. In allogeneic HSCT, apheresis is usually performed on the day of transplantation so that the stem cells can be transplanted without the need for cryopreservation.
For more than 20 years [21] it has been known that cord blood harvested shortly after birth is very rich in potent hematopoietic stem cells. Therefore, cord blood banks have been established to provide stem cell products for allogeneic HSCT. Subsequently cord blood units (CBU) from unrelated donors have rapidly become a common stem cell source. In Europe, about 900 cord blood transplants are performed annually [3]. Cord blood units contain fewer hematopoetic stem cells than marrow or PBSC products. As a consequence, risk of non-engraftment is higher. Due to immunologic immaturity of co-transplanted T-cells, a greater number of HLA mismatches can be tolerated. By the infusing of two CBU into a single recipient (double-cord HSCT), the problem of low stem cell dose and risk of non-engraftment can be circumvented. To shorten time to engraftment and to lower engraftment failure, in vitro cord blood expansion and co-infusion of non-stem-cells facilitating engraftment are investigated intensively [22].
Patient selection
Disease stage at transplant remains the major determinant of survival after autologous HSCT, and to a lesser degree after allogeneic HSCT. HSCT is better tolerated in general if performed early in the disease course. Based on studies in the 1980s and 1990s, autologous HSCT are either done as part of the first line treatment as in PCM or in early relapse (e.g. for high risk NHL and HL). For allogeneic HSCT, disease stage is of major importance because of higher relapse risks and higher risks of toxicity, but also the main immunologic complication: GvHD occurs less frequently if the treatment is done at early disease stage. Assuming future improvement in transplant technology and in supportive care, the risk/benefit ratio has to be continuously assessed in prospective studies. Currently HSCT for acute leukaemias are done in first or second complete remission, whereas many patients with lymphoid neoplasias receive allogeneic HSCT after having failed several lines of treatment often including autologous HSCT. For patients with myelodysplastic syndromes, studies have shown that waiting for disease progression to the stage of blast excess is beneficial as compared to early transplantation, if maximisation of life expectancy is considered [23]. Conversely in CML, patients are increasingly transplanted in advanced stages of the disease compared to transplants in chronic phase before the introduction of tyrosine kinase inhibitors as patients will only come to transplant after having failed multiple drugs [3]. This results in an unfavourable shift towards higher TRM and relapse when compared to historic data, which was mainly generated with patients in chronic phase [24].

Figure 2B
Indications for autologous HSCT in Europe in 2010 (reprinted from: Passweg JR, Baldomero H, Gratwohl A, Bregni M, Cesaro S, Dreger P, et al.; for the European Group for Blood and Marrow Transplantation (EBMT). The EBMT activity survey: 1990–2010. Bone Marrow Transplant. 2012;47(7):906-23 [3]).
Abbr: PCD: plasma cell disorders, HD Hodgkin Lymphoma, NHL Non Hodgkin Lymphoma, AID Autoimmune Disorders
Due to the significant procedure related morbidity and mortality of allogeneic HSCT, only diseases with poor prognosis (<50% survival at 5yrs with conventional treatment) are currently indications for allogeneic transplantation. Scores have been established incorporating risk factors for transplantation, such as disease stage, type of donor (matched sibling donor versus unrelated donor), patient age and disease duration [25], and in addition scoring of co-morbidities has been shown to be useful [26]. Such scores are helpful for individual patient counselling, in so far as we learn more and more about disease risks such as genetic alterations reflecting the malignant potential of leukaemia biology [27]. Thus, patients with high risk disease and low transplant risk scores may be preferentially counselled to undergo HSCT, whereas patients with lower risk disease and a high transplant risk score may prefer alternative treatment strategies.
Donor selection
In allogeneic HSCT, HLA compatibility is of greater importance than in organ transplantation, as rejection occurs not only in the host versus graft direction, but also as graft versus host disease (see below). Given that all genes encoding HLA antigens are located on chromosome 6, the probability that any given siblings are HLA identical is 25%. Approximately 15–30% of patients referred for allogeneic HCT have at least one suitable HLA-identical sibling donor. For patients without a suited family donor, a search for an HLA-matched unrelated donor is undertaken. International donor registries have grown and now include nearly 20 million HLA typed donors. In addition there are about 500,000 units of cord blood available in cord blood banks all over the world ( http://www.bmdw.org ). Figure 3 shows the remarkable increase in the use of unrelated donors over time in Europe. The likelihood of identifying an HLA-matched unrelated donor varies according to patient HLA alleles and ethnicity, which influences HLA diversity. Caucasians have an approximate 60% chance of finding an appropriately matched donor for class I and class II HLA loci. Other options include HLA-mismatched unrelated donors, umbilical cord blood where mismatches are more easily tolerated, and HLA-haploidentical (half matched) family members [28, 29].
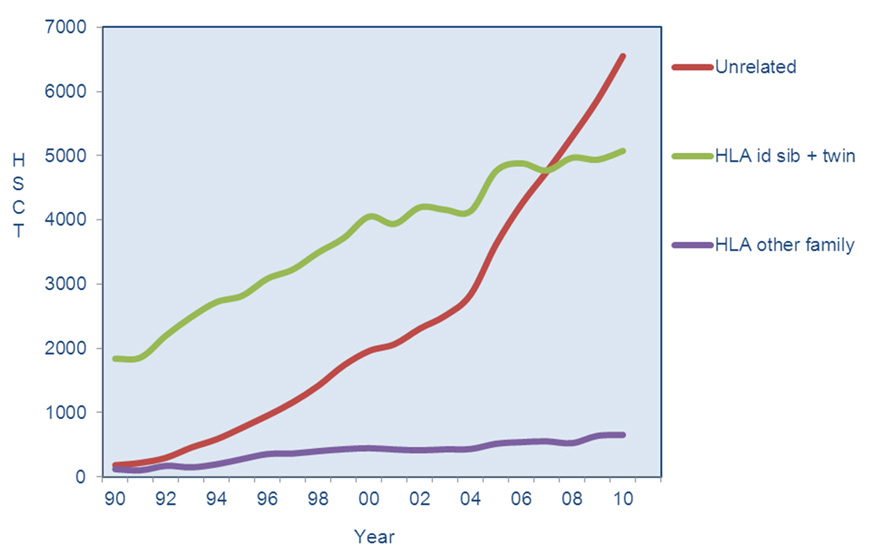
Figure 3
Use of different donor types for allogeneic HSCT in Europe 1990–2010 (reprinted from: Passweg JR, Baldomero H, Gratwohl A, Bregni M, Cesaro S, Dreger P, et al.; for the European Group for Blood and Marrow Transplantation (EBMT). The EBMT activity survey: 1990–2010. Bone Marrow Transplant. 2012;47(7):906-23 [3]).
Evidence suggests that certain alleles may be more ‘permissive’, which means they may be less of a barrier to successful transplantation. This is further complicated by the fact that polymorphisms outside of HLA may play a role as well [30] in deciding whether a particular donor, recipient combination will result in tolerance or in immunologic activation, and other cellular components will be of influence as is exemplified by natural killer cell reactivity which may exert a strong graft versus tumour effect [31].
For patients without a HLA matched donor, using a half matched (haploidentical, typically a parent or another relative) donor is an option. Due to HLA disparity, stringent T-cell depletion of the allograft is necessary. Problems include graft rejection, infection and disease relapse because of reduced graft-versus-tumour (GvT) activity after depletion of donor lymphocytes and poor immune reconstitution. GvT effects after haploidentical HSCT rely in part on NK-cell alloreactivity. NK cells interact with target cells through a complex system of activating and inhibitory signals. We have used this system to transfer purified donor NK cells to exert anti-leukaemia activity [32] and are currently studying the use of in vitro expanded donor derived NK cells as adoptive treatment.
Advances in the availability of HLA typed unrelated donors as well as development of alternative donor transplantation – including cord blood transplantation and HSCT from haploidentical donors – have improved access to transplantation for patients who were previously unable to undergo this treatment because of lack of a suitable donor. Satisfactory outcomes have been reported in uncontrolled studies. However, risks and benefits of alternative donor transplantation remain to be elucidated.
Preparative (conditioning) regimens
Preparative regimens in autologous HSCT carry the goal to administer chemotherapy toxin to the tumour, without any compromises in relation to marrow toxicity, but whilst preserving non-hematopoietic organ function. Administering HSC serves to restore bone marrow function and to shorten aplasia.
In allogeneic HSCT, historically, marrow-ablative doses of chemotherapy and total-body irradiation (TBI) were thought to be necessary to eradicate malignancy; to provide immunosuppression to the recipient; and to create space in the stem cell niche, allowing engraftment of donor hematopoietic cells. A major development over the last 15 years was the introduction of reduced intensity regimens (RIC) removing myeloablative treatment and keeping treatment intensity just high enough to avoid graft rejection [33]. The goal was to promote engraftment and let the graft versus tumour effect eliminate tumour cells. Studies have shown this to be the case, as in patients with leukaemia relapse risks are somewhat higher if a reduced intensity regimen is given, but this increase is only around 10% and is offset by lower treatment related mortality [34]. The reduction in morbidity and mortality of RIC has made allogeneic HSCT available for older patients (i.e., patients aged 60 to 70 years), the age group that has the highest prevalence of most hematopoietic malignancies.
GvHD: prophylaxis, clinical manifestations, treatment
Graft-versus-host disease is the major complication of allogeneic HSCT. GvHD is initiated by donor T cells that respond to alloantigen presented on host antigen presenting cells. GvHD is more frequent and severe when HLA-mismatched grafts are used (major mismatch), but also occurs with a frequency of up to 60% when the donor and the recipient are fully HLA matched (minor mismatch). GvHD is either acute or chronic, affecting specific organ systems with distinctive clinical manifestations. Acute GvHD generally occurs in the first 100 days after HSCT but late onset acute GvHD may occur particularly after reduced intensity conditioning HSCT. Major targets are the skin, the gastrointestinal tract, and the liver, leading to exanthema of the skin (fig. 4A), enteritis and cholestatic hepatitis. Acute GvHD is graded according to a widely accepted system, where the degree of skin involvement, the amount of diarrhoea, the bilirubin level, and in severe forms the decrease in performance status is used. For GvHD prophylaxis, patients receive immunosuppressive drugs, most commonly a combination of cyclosporine and methotrexate or mycophenolic acid. In contrast to organ transplantation, this prophylaxis can be tapered off and stopped in patients not developing GvHD after 6 to 12 months. Patient age, disease stage and donor recipient sex mismatch (i.e. female donor for male recipients recognising antigens associated with the Y chromosome) are risk factors for GvHD. Polymorphisms and biomarkers allowing to predict GvHD are extensively studied as these would permit prediction on transplant associated risks and decisions on type and intensity of GvHD prophylaxis.
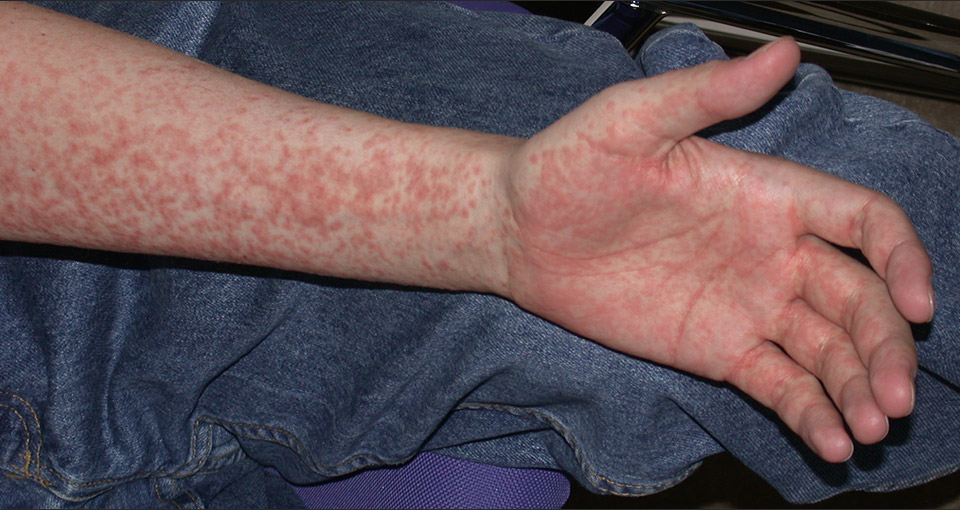
Figure 4A
Morbilliform exanthema of acute GvHD of the skin including the palma manus.

Figure 4B
Lichenoid features and irregular pigmentation of chronic GvHD of the skin.
Chronic GvHD generally occurs after day 100 post-transplant and is a chronic inflammatory condition characterised by sclerosis and multi-organ involvement (fig. 4B). The main risk factor of cGvHD is aGvHD, presumably by damaging the thymus and precluding functional positive and negative selection of donor lymphocytes that are newly educated from bone marrow derived precursors post-transplant. As with acute GvHD, chronic GvHD is graded according to a clinical system weighing each organ involvement. Almost any organ system can be involved. Manifestations resemble those of autoimmune diseases and present in the skin as a particular form of scleroderma, in the liver as cholestatic hepatitis, but may also induce a Sjögren like syndrome with dry eyes and mouth, and obstructive lung disease termed bronchiolitis obliterans. Chronic GvHD is a major cause of long-term morbidity and mortality in survivors of HSCT and the most significant determinant of post-transplant quality of life. The course of chronic GvHD is variable; some patients gain resolution of the disease and are eventually able to discontinue immunosuppressive treatment, whereas others have severe and treatment-refractory disease [35, 36].
First line treatment of acute and chronic GvHD is i.v. methylprednisolone. Second line treatments for both diseases are not established based on high level evidence studies and remain to be defined through prospective studies.
Common infections
Infections are common in HSCT recipients, and much more so with allogeneic HSCT where immune reconstitution is often slow and incomplete. The peak incidence of infections changes in distinct phases with bacterial infections being most common in the early phase during marrow aplasia, fungal and viral infections may appear early and later in the post-transplant phase. The most common viral infections are with viruses that remain in a latency phase belonging to the herpes family. Of these the cytomegalovirus is most commonly reactivated and there is a relationship between viral infections and GvHD, as viral infections may trigger GvHD events and immunosuppressants used to treat GvHD favour viral replication.
Outcomes in selected diseases
Acute myeloid leukaemia
The most common indication for allogeneic HSCT is acute myeloid leukaemia (AML). Typical long term results are 20%-60% long term leukaemia free survival after allogeneic HSCT, depending on disease stage and donor type. Failures are due in approximately equal parts to relapse of the leukaemia and non-relapse mortality, the majority of which is attributable to GvHD and associated complications. Allogeneic HSCT results in improved disease-free survival in patients with intermediate and high risk disease in first complete remission (CR1) but not in patients with low risk disease, as smaller benefits in relapse reduction are offset by increased treatment related morbidity and mortality. This has been confirmed in a meta-analysis of 24 trials including 6007 patients younger than 60 years [4]. Older patients above 65 years of age with AML have a dismal outcome (<2% 5 year survival) when treated with chemotherapy alone [37]. A study including 274 patients receiving reduced intensity conditioning transplants showed 5‑year overall survival of 33%, with relapse and non-relapse mortalities of 42 and 26% [28] respectively. There are so far no comparative data showing that this strategy is superior to standard conditioning. Autologous HSCT continues to have a place in AML treatment. In a randomised study from the Dutch-Belgian-Swiss group including 500 patients, relapse was reduced with autologous HSCT compared to chemotherapy (58% vs. 70%), and relapse-free survival at 5 years was better (38% vs. 29%) but there were no significant differences in overall survival [8].
Acute lymphoblastic leukaemia
In patients with acute lymphoblastic leukaemia (ALL) indications for allogeneic HSCT is in early disease if the patient presents with high risk features. All other patients undergo allogeneic HSCT only after relapse and reinduction. Due to the slightly higher chemosensitivity of ALL overall, outcomes are similar to AML, although ALL appears to be less sensitive to graft versus tumour effects than myeloid neoplasms. In future trials, treatment decisions will be guided by minimal residual disease as measured quantitatively by immunoglobulin and T-cell receptor gene rearrangements, approaches that have been pioneered by pediatric study groups [38].
Myelodysplastic syndrome
For patients with myelodysplastic syndrome (MDS), allogeneic HSCT is the only treatment with curative potential. MDS is a disease with important heterogeneity, as early disease stage is marked by cytopenia with or without transfusion needs, whereas in more advanced disease accumulating blast cells resemble acute leukaemia. A decision analysis showed that disease stage based on the international prognostic staging system (IPSS) was useful for decision making: delaying HSCT in early disease until disease progression was optimal for patients that did not have an increase of blasts. In contrast, patients with excess of blasts benefited from transplant as soon as feasible [22]. However, this strategy requires regular reassessment of MDS patients to prevent an undue delay of transplant upon progression.
Aplastic anaemia
HSCT in severe aplastic anaemia (SAA) is standard of care for young patients below the age of 40 to 50 years. Reported outcomes vary between 75–80% long term survival if transplanted from a sibling donor with successful reconstitution of the hematopoietic system. Results with transplants from unrelated donors have been historically inferior, but recently improvement with this type of transplant has resulted in outcomes similar to sibling donor transplants [39]. Older patients are usually transplanted after having failed immunosuppressive treatment, which may also result in hematopoietic reconstitution; patients without sibling donors will receive transplants from alternative donors if failing immunosuppression. In contrast to other indications, in SAA bone marrow is still the stem cell source of choice. As in SAA no GvL effect is needed, the increased risk of GvHD mediated by PBSCT is not justified [40].
Lymphoma
Autologous HSCT is standard of care in many instances in the treatment of lymphoma. Standard indications include Hodgkin lymphoma with early relapse, and diffuse large B cell lymphoma in 2nd or later remission. These indications have been shown in randomised clinical trials to result in superior disease free survival [14]. Issues include age limits of these treatments and the extrapolation to other situations. There is much less agreement on first line treatment in some types of Non-Hodgkin lymphoma (NHL), where conflicting results have been obtained in studies of consolidation treatment by autologous HSCT. Allogeneic HSCT is used as salvage therapy in both NHL and Hodgkin lymphoma. Most of these transplants are done with reduced intensity conditioning, and myeloablative conditioning is occasionally used in selected young patients. Reported outcomes vary. The most promising results have been reported with indolent NHL, where progression-free survival ranges from 45 to 85% depending on patient selection, timing and other factors. Several studies show that allogeneic HSCT is effective for poor-risk or refractory chronic lymphocytic leukaemia (CLL).
Plasma-cell Myeloma
Plasma-cell Myeloma is the most frequent indication for autologous HSCT, where survival benefit has been demonstrated in large clinical trials. With the advent of more potent new drugs this is challenged, and trials are repeated where autologous HSCT is used as part of the upfront treatment or delayed for the relapsed patient. The role of allogeneic HSCT in multiple myeloma is less clear. Data from several clinical trials was inconclusive regarding overall and relapse free survival [9]. Currently allogeneic HSCT is reserved for very high risk patients in first remission, or for patients relapsing early after first line treatment.
Long term care, quality of life and follow-up by the practitioner
The survey by the European Group for Blood and Marrow Transplantation (EBMT) 1990–2010 included 376,000 patients transplanted over this period. Overall survival at 5 years is approximately 53%, and at 10 years 44% based on pooled data obtained from the EBMT. Extrapolating from this information, physicians outside of transplant centres have an increasing likelihood of encountering long-term survivors of HSCT. Approximately, 200,000 patients may be alive in Europe after having had an HSCT in the past.
Caring for these patients will often occur in collaboration with the transplant centres, but given increasing numbers and given that HSCT is no longer considered an experimental procedure with a lifelong narrow attachment of these patients to the transplant centres, practitioners will be increasingly involved in their care. Many patients enjoy an excellent quality of life but others may suffer from important complications requiring prompt attention. Both, recipients of autologous and allogeneic HSCT are at risk of late toxicity of the treatment, which is quantitatively but not qualitatively different from other patients requiring specific follow-up after exposure to genotoxic agents. Furthermore, recipients of allogeneic HSCT require much added attention because of the complications related to chronic GvHD.
Recommendations have been published as to the follow-up as well as re-vaccination schedules [41]. There needs to be an awareness of the issues involved related to chronic GvHD, immune reconstitution and long term toxicity of intensive treatments with consequences on all organ systems. It is beyond the scope of this paper to discuss them in great detail.
Some principles may be listed here [42–45]:
Patients after HSCT need to recover immunity. Full T-cell immunity may require 1 to 2 years to be established and longer if chronic GvHD persists. Patients may be at risk of infection with viruses, encapsulated bacteria, pneumocystis and fungal infections.
Chronic GvHD may present with many different manifestations. Onset is often slow, and signs and symptoms might be misinterpreted or initially considered non-significant. The skin is a prime target of chronic GvHD which can manifest with altered skin pigmentation, superficial or deep sclerosis – called sclerodermatous GvHD – which may lead to decreased joint mobility or impaired wound healing. Involvement of the lacrimal or salivary glands leads to a Sjögren like sicca-syndrome with dry eyes and dry mouth, often combined with vaginal dryness in women. The skin in HSCT recipients is particularly sensitive to UV damage and UV toxicity may induce GvHD. Therefore sun protection is of great importance. Eye involvement requires particular attention and care by a specialist with experience in ocular GvHD, as erosions or ulceration of the cornea may occur which can ultimately lead to an irreversible loss of vision. Dental assessment is of importance as low and altered salivary flow may promote rapidly progressive tooth decay. Lung involvement leading to bronchiolitis obliterans is often triggered by respiratory viral infection and starting with a chronic dry cough. A high level of attention and repeated lung function tests are required since loss of lung function is often irreversible if diagnosis is delayed.
The liver is another prime target of chronic GvHD, and other patients may suffer from rapidly progressive chronic hepatitis if infected prior to HSCT. Iron overload parameters need to be watched as patients often have an extensive transfusion history. After transplantation excess iron may be removed by venesection.
Cardiovascular risk profile needs to be assessed as patients with chronic inflammation may suffer from progressive atherosclerosis with increased risks of cardiac complications. Arterial hypertension and hyperlipidemia are frequent findings and at least in part due to ongoing immunosuppressive treatment, further increasing the cardiovascular risk profile.
Long term kidney toxicity is often precipitated by use of calcineurin inhibitors, such as cyclosporine and tacrolimus with their specific type of renal toxicity. Obviously high blood pressure needs to be treated if detected.
Bone density needs to be assessed as patients may be in a state of hypogonadism and may lose bone mass through steroid treatment for chronic GvHD. Aseptic osteonecrosis is a common complication of chronic GvHD.
Endocrine function needs to be watched, alteration in thyroid function, diabetes, and gonadal dysfunction are common. In children, growth velocity needs to be monitored.
Fertility is an issue in young patients and needs to be addressed. In male patients sperm cryopreservation prior to treatment is mandatory, whereas in female patients preservation of ovarian tissue and oocytes is still highly experimental.
Second cancers may occur, as many types of cancers are increased in frequency in long term survivors. Screening for second cancers is strongly recommended.
Transplant centres usually use checklists to follow patients and these may be distributed to practitioners who are following patients (table 2). Treatment strategies need to be defined in collaboration with the transplant centre. They are essentially the same as for individuals without transplantation for findings like arterial hypertension, diabetes mellitus or hyperlipidemia but may differ for more unique post-transplant complications like manifestations of cGvHD underlining the need for a successful partnership between practitioners and transplant centres.
|
Table 2: Checklist to follow HSCT patients at 1 year post-transplant and annually thereafter.
Additional testing may be required depending on the individual status of the patients. A helpful summary of recommendations for post-transplant follow up can be accessed at: http://marrow.org/Physicians/Medical_Education/Clinical_Guidelines/Clinical_Guidelines.aspx |
| |
1 year
|
Annually
|
Remark
|
| Patient history and complete clinical status, including psychosocial, neurologic and cognitive evaluation, assessment of sexual function, muscle strengths and physical activity |
X |
X |
Including risk factors for cardiovascular risk or malignancies (personal and family history), fatigue, depression and anxiety disorders |
| Screening for second cancers (solid tumours and hematologic malignancies) and second cancer vigilance counselling |
X |
X |
Includes the currently recommended cancer screening practice for the general population, counselling for sun-exposure, skin self-examination, stop of smoking etc. Additional screening depends on individual risk factors (history, type of transplantation, cGvHD) and should be defined in collaboration with the transplant centre team. |
| Complete blood count
Renal and liver function tests
Lipid status
Ferritin
Urine analysis |
X
X
X
X
X |
X
X
X
X
X |
Frequency of testing during annual follow up depends on previous results and new signs or symptoms, e.g. if lipids and ferritin are within the normal range testing does not need to be repeated annually but in larger intervals. In contrast, at least annual blood count is recommended independent of previous results during the first 5 years to detect treatment-related myelodysplastic syndrome/acute myeloid leukaemia after autologous or relapse after allogeneic HSCT. |
|
Endocrine testing
|
|
|
Focus on hypothyroidism, hypogonadism, growth retardation and hypoadrenalism. |
| Thyroid function test (TSH) |
X |
X |
| Children: assessment of growth velocity and pubertal development |
X |
X |
| Assessment of gonadal function in post-pubertal patients |
X |
X |
Basically hormonal replacement therapy (HRT) is indicated in the majority of women below the age of 50 years. Decision has to be made based on the individual risk assessment (history of breast cancer, thromboembolism, liver disease etc. vs. sequela of premature ovarian failure). HRT in post-pubertal male is rarely indicated. |
| adrenal function test |
(X) |
(X) |
Only after prolonged treatment with steroids. |
|
Assessement for immunity and vaccination for
|
|
|
|
| Encapsulated bacteria prophylaxis |
X |
X |
Recommended for any patient with ongoing immunosuppression and/or cGvHD or signs of functional hyposplenism. |
| PcP prophylaxis |
X |
X |
|
| Vaccination |
X |
X |
Cf. (40) |
| Ocular evaluation |
X |
X |
Including assessment for development of sicca syndrome, premature cataract and retinopathy. |
| Dental assessment |
X |
X |
Focus on sicca syndrome, caries and –after allogeneic HSCT and cGvHD- intraoral malignancies. |
| Lung function testing (LFT) |
X |
(X) |
More frequent LFT (every 3 months) is recommended in patients with active cGvHD.
In patients without smoking, immunosuppression or active cGvHD longer intervals in follow up may be justified. |
| Bone density |
X |
(X) |
Follow up examination every two years if previous osteopenia/-porosis under treatment, occurrence of new fractures or steroid treatment, |
| Gynaecologic examination and mammography |
X |
(X) |
PAP smear
Mammography is recommended:
– Before start of hormonal replacement therapy;
– After the age of 25 years or 8 years after HSCT, whichever is later but starting at the age of 40 years at the latest in women after TBI or chest irradiation;
– Follow up mammography is recommended every 2 years. |
| Assessment of minimal residual disease markers |
X |
X |
Type of marker and frequency of assessment depends on the initial disease and time after transplantation. Needs to be defined in collaboration with the transplant centre. |
Future developments
In the next years, progress will be made in donor selection. More knowledge will be available to include information on polymorphisms in the HLA and non HLA regions to define lower risk donors for particular patients [46]. Supportive care will benefit as it has in the past from better antiviral and antifungal drugs. Progress will come from the knowledge gained in the pathophysiology of GvHD and the importance of cytokine networks to design better drugs to prevent and treat this complication. There are high hopes for cellular therapy, be it exploiting the immunomodulatory properties of mesenchymal progenitor cells [47] cultured ex vivo; the use of in vitro expanded cell populations of the donor such as NK cells, or infusing cells with engineered T-cell receptors to exert anti-tumour effects through target recognition with these modified T-cell receptors [48, 49]; or ultimately using a suicide gene approach to exert “safe” anti-tumour effects with T-lymphocytes that can be stopped by activating the suicide gene if these cells induce GvHD [50].
References
1 Copelan EA. Hematopoietic stem-cell transplantation. N Engl J Med. 2006;354(17):1813–26.
2 Gyurkocza B, Rezvani A, Storb RF. Allogeneic hematopoietic cell transplantation: the state of the art. Expert Rev Hematol. 2010;3(3):285–99.
3 Passweg JR, Baldomero H, Gratwohl A, Bregni M, Cesaro S, Dreger P, et al.; for the European Group for Blood and Marrow Transplantation (EBMT). The EBMT activity survey: 1990–2010. Bone Marrow Transplant. 2012;47(7):906-23
4 Koreth J, Schlenk R, Kopecky KJ, et al. Allogeneic stem cell transplantation for acute myeloid leukemia in first complete remission: a systematic review and meta-analysis of prospective clinical trials. JAMA. 2009;301(22):2349–60.
5 Wynn R. Stem cell transplantation in inherited metabolic disorders. Hematology Am Soc Hematol Educ Program. 2011;2011:285–91.
6 Ljungman P, Urbano-Ispizua A, Cavazzana-Calvo M, Demirer T, Dini G, Einsele H, et al.; European Group for Blood and Marrow. Allogeneic and autologous transplantation for haematological diseases, solid tumours and immune disorders: definitions and current practice in Europe. Bone Marrow Transplant. 2006;37(5):439–49.
7 Cornelissen JJ, van der Holt B, Verhoef GE, et al. Myeloablative allogeneic versus autologous stem cell transplantation in adult patients with acute lymphoblastic leukemia in first remission: a prospective sibling donor versus no-donor comparison. Blood. 2009;113(6):1375–82.
8 Vellenga E, van Putten W, Ossenkoppele GJ, Verdonck LF, Theobald M, Cornelissen JJ, et al.; Dutch-Belgian Hemato-Oncology Cooperative Group (HOVON); Swiss Group for Clinical Cancer Research Collaborative Group (SAKK). Autologous peripheral blood stem cell transplantation for acute myeloid leukemia. Blood. 2011;118(23):6037–42.
9 Bruno B, Rotta M, Patriarca F, et al. A comparison of allografting with autografting for newly diagnosed myeloma. N Engl J Med. 2007;356(11):1110–20.
10 Peggs KS, Hunter A, Chopra R, Parker A, Mahendra P, Milligan D, et al. Clinical evidence of a graft-versus-Hodgkin’s-lymphoma effect after reduced-intensity allogeneic transplantation. Lancet. 2005;365(9475):1934–41.
11 Harousseau JL, Avet-Loiseau H, Attal M, Charbonnel C, Garban F, Hulin C, et al. Achievement of at least very good partial response is a simple and robust prognostic factor in patients with multiple myeloma treated with high-dose therapy: long-term analysis of the IFM 99-02 and 99-04 Trials. J Clin Oncol. 2009;27(34):5720–6. Epub 2009 Oct 13.
12 Moreau P, Garban F, Attal M, Michallet M, Marit G, Hulin C, et al.; IFM Group. Long-term follow-up results of IFM99-03 and IFM99-04 trials comparing nonmyeloablative allotransplantation with autologous transplantation in high-risk de novo multiple myeloma. Blood. 2008;112(9):3914–5.
13 Attal M, Harousseau JL. Randomized trial experience of the Intergroupe Francophone du Myélome. Semin Hematol. 2001;38(3):226–30.
14 Gisselbrecht C. Autologous stem cell transplantation in aggressive non-Hodgkin’s lymphoma. Recent Results Cancer Res. 1998;144:15–26.
15 Mounier N, Canals C, Gisselbrecht C, Cornelissen J, Foa R, Conde E, et al.; for the Lymphoma Working Party of the European Blood and Marrow Transplantation Registry (EBMT). High-dose therapy and autologous stem cell transplantation in first relapse for diffuse large B cell lymphoma in the rituximab era: an analysis based on data from the European Blood and Marrow Transplantation Registry. Biol Blood Marrow Transplant. 2012;18(5):788–93. Epub 2011 Oct 17.
16 Schmitz N, Pfistner B, Sextro M, Sieber M, Carella AM, Haenel M, et al. German Hodgkin’s Lymphoma Study Group; Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. Aggressive conventional chemotherapy compared with high-dose chemotherapy with autologous haemopoietic stem-cell transplantation for relapsed chemosensitive Hodgkin's disease: a randomised trial. Lancet. 2002;359(9323):2065–71.
17 Bensinger WI, Martin PJ, Storer B, Clift R, Forman SJ, Negrin R, et al. Transplantation of bone marrow as compared with peripheral-blood cells from HLA-identical relatives in patients with hematologic cancers. N Engl J Med. 2001;344(3):175–81.
18 Claudio Anasetti, Brent R. Logan, Stephanie J. Lee, Edmund K Waller, Daniel J. Weisdorf, John R. Wingard, et al. Increased incidence of chronic graft-versus-host disease (GVHD) and no survival advantage with filgrastim-mobilized peripheral blood stem cells (PBSC) compared to bone marrow (BM) transplants from unrelated donors: results of Blood and Marrow Transplant Clinical Trials Network (BMT CTN) protocol 0201, a phase III, prospective, randomized trial blood (ASH Annual Meeting Abstracts). 2011:118: Abstract 1.
19 Keating GM. Plerixafor: a review of its use in stem-cell mobilization in patients with lymphoma or multiple myeloma. Drugs. 2011;71(12):1623–47.
20 To LB, Levesque JP, Herbert KE. How I treat patients who mobilize hematopoietic stem cells poorly. Blood. 2011;118(17):4530–40. Epub 2011 Aug 10.
21 Raffoux C, Gluckman E. Suggested strategies for establishing an HLA-typed cord blood bank. J Hematother. 1993;2(2):263–4.
22 Eapen M, Rubinstein P, Zhang MJ, et al. Outcomes of transplantation of unrelated donor umbilical cord blood and bone marrow in children with acute leukaemia: a comparison study. Lancet. 2007;369(9577):1947–54.
23 Cutler CS, Lee SJ, Greenberg P, Deeg HJ, et al. A decision analysis of allogeneic bone marrow transplantation for the myelodysplastic syndromes: delayed transplantation for low-risk myelodysplasia is associated with improved outcome. Blood. 2004;104(2):579–85. Epub 2004 Mar 23.
24 Saussele S, Lauseker M, Gratwohl A, Beelen DW, Bunjes D, Schwerdtfeger R, et al. German CML Study Group. Allogeneic hematopoietic stem cell transplantation (allo SCT) for chronic myeloid leukemia in the imatinib era: evaluation of its impact within a subgroup of the randomized German CML Study IV. Blood. 2010;115(10):1880–5. Epub 2009 Nov 18.
25 Gratwohl A, Stern M, Brand R, Apperley J, Baldomero H, de Witte T, et al. European Group for Blood and Marrow Transplantation and the European Leukemia Net. Risk score for outcome after allogeneic hematopoietic stem cell transplantation: a retrospective analysis. Cancer. 2009;115(20):4715–26.
26 Sorror ML, Maris MB, Storb R, Baron F, Sandmaier BM, Maloney DG, Storer B. Hematopoietic cell transplantation (HCT)-specific comorbidity index: a new tool for risk assessment before allogeneic HCT. Blood. 2005;106(8):2912–9.
27 Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366(12):1079–89. Epub 2012 Mar 14.
28 Reisner Y, Hagin D, Martelli MF. Haploidentical hematopoietic transplantation: current status and future perspectives. Blood. 2011;118(23):6006–17. Epub 2011 Sep 14
29 Lee SJ, Klein J, Haagenson M, et al. High-resolution donor-recipient HLA matching contributes to the success of unrelated donor marrow transplantation. Blood. 2007;110(13):4576–83.
30 Bettens F, Passweg J, Schanz U, Chalandon Y, Heim D, Güngör T, et al. Impact of HLA-DPB1 haplotypes on outcome of 10/10 matched unrelated hematopoietic stem cell donor transplants depends on MHC-linked microsatellite polymorphisms. Biol Blood Marrow Transplant. 2012;18(4):608–16. Epub 2011 Oct 1.
31 Cooley S, Weisdorf DJ, Guethlein LA, Klein JP, Wang T, Le CT, et al. Donor selection for natural killer cell receptor genes leads to superior survival after unrelated transplantation for acute myelogenous leukemia. Blood. 2010;116(14):2411–9. Epub 2010 Jun 25.
32 Passweg JR, Koehl U, Uharek L, Meyer-Monard S, Tichelli A. Natural-killer-cell-based treatment in haematopoietic stem-cell transplantation. Best Pract Res Clin Haematol. 2006;19(4):811–24.
33 Gyurkocza B, Storb R, Storer BE, et al. Nonmyeloablative allogeneic hematopoietic cell transplantation in patients with acute myeloid leukemia. J Clin Oncol. DOI:10.1200/JCO.2009.27.1460 (2010).
34 Horwitz ME. Reduced intensity versus myeloablative allogeneic stem cell transplantation for the treatment of acute myeloid leukemia, myelodysplastic syndrome and acute lymphoid leukemia. Curr Opin Oncol. 2011;23(2):197–202.
35 Martin PJ, Rizzo JD, Wingard JR, Ballen K, Curtin PT, Cutler C, et al. First and second-line systemic treatment of acute graft-versus-host disease: recommendations of the American Society of Blood and Marrow Transplantation. Biol Blood Marrow Transplant. 2012 Apr 14. [Epub ahead of print]
36 Pavletic SZ, Lee SJ, Socie G, Vogelsang G. Chronic graft-versus-host disease: implications of the National Institutes of Health consensus development project on criteria for clinical trials. Bone Marrow Transplant. 2006;38(10):645–51.
37 Yee KW, Keating A. Older patients with acute myeloid leukemia. Expert Rev Hematol. 2010;3(6):755–74.
38 Brandwein JM. Treatment of acute lymphoblastic leukemia in adolescents and young adults. Curr Oncol Rep. 2011;13(5):371–8. doi: 10.1007/s11912-011-0185-9.
39 Viollier R, Socié G, Tichelli A, Bacigalupo A, Korthof ET, Marsh J, et al. Recent improvement in outcome of unrelated donor transplantation for aplastic anemia. Bone Marrow Transplant. 2008;41(1):45–50. Epub 2007 Nov 5.
40 Schrezenmeier H, Passweg JR, Marsh JC, Bacigalupo A, Bredeson CN, Bullorsky E, et al. Worse outcome and more chronic GVHD with peripheral blood progenitor cells than bone marrow in HLA-matched sibling donor transplants for young patients with severe acquired aplastic anemia. Blood. 2007;110(4):1397–400. Epub 2007 May 2.
41 Empfehlungen zur Impfung von Empfängerinnen und Empfängern von Blut-Stammzellen Bull BAG 2012;21:363–70.
42 Majhail NS, Rizzo JD, Lee SJ, Aljurf M, Atsuta Y, Bonfim C, et al. Center for International Blood and Marrow Transplant Research; American Society for Blood and Marrow Transplantation; European Group for Blood and Marrow Transplantation; Asia-Pacific Blood and Marrow Transplantation Group; Bone Marrow Transplant Society of Australia and New Zealand; East Mediterranean Blood and Marrow Transplantation Group; Sociedade Brasileira de Transplante de Medula Ossea. Recommended screening and preventive practices for long-term survivors after hematopoietic cell transplantation. Bone Marrow Transplant. 2012;47(3):337–41.
43 Tichelli A, Rovó A, Passweg J, Schwarze CP, Van Lint MT, Arat M, et al. Late Effects Working Party of the European Group for Blood and Marrow Transplantation. Late complications after hematopoietic stem cell transplantation. Expert Rev Hematol. 2009;2(5):583–601.
44 Bieri S, Roosnek E, Helg C, Verholen F, Robert D, Chapuis B, et al. Quality of life and social integration after allogeneic hematopoietic SCT. Bone Marrow Transplant. 2008;42(12):819–27.
45 Ljungman P, Small TN; Vaccination Recommendations Writing Group. Vaccination of SCT recipients. Bone Marrow Transplant. 2011;46(4):621.
46 Fleischhauer K, Shaw BE, Gooley T, Malkki M, Bardy P, Bignon JD, et al.; on behalf of the International Histocompatibility Working Group in Hematopoietic Cell Transplantation. Effect of T-cell-epitope matching at HLA-DPB1 in recipients of unrelated-donor haemopoietic-cell transplantation: a retrospective study. Lancet Oncol. 2012;13(4):366–74. Epub 2012 Feb 15.
47 Le Blanc K, Frassoni F, Ball L, Locatelli F, Roelofs H, Lewis I, et al. Developmental Committee of the European Group for Blood and Marrow Transplantation Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: a phase II study. Lancet. 2008;371(9624):1579–86.
48 Provasi E, Genovese P, Lombardo A, Magnani Z, Liu PQ, Reik A, et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat Med. 2012 Apr 1. doi: 10.1038/nm.2700. Epub ahead of print.
49 Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–33.
50 Ciceri F, Bonini C, Stanghellini MT, Bondanza A, Traversari C, Salomoni M, et al. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): a non-randomised phase I-II study. Lancet Oncol. 2009;10(5):489–500.