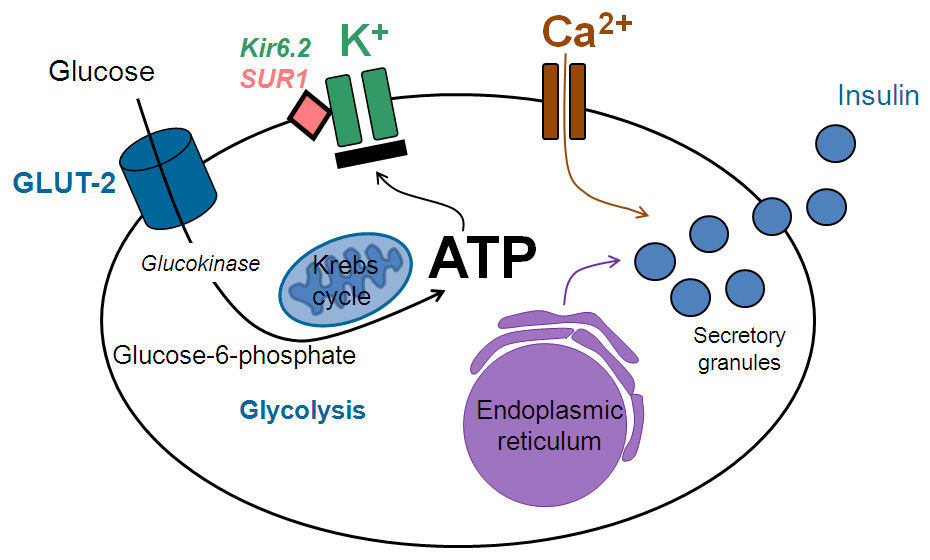
Figure 1
Pancreatic beta cell: the coupling of glucose sensing via GLUT-2 transporters, generation of ATP, membrane depolarization by closing potassium channels, entering of calcium ions, and exocytosis of insulin.
DOI: https://doi.org/10.4414/smw.2012.13690
It is estimated that monogenic forms of diabetes mellitus, i.e. caused by a single gene mutation, account for some 2–5% of all cases of diabetes mellitus [1–3]. Monogenic diabetes mellitus due to defects in insulin secretion comprises a genetically heterogeneous group of diabetes mellitus including MODY (maturity-onset diabetes of the young), mitochondrial diabetes and neonatal diabetes. However, the common pathophysiological pathway in monogenic disorders is impaired insulin secretion of the pancreatic beta cell [1]. The proper diagnosis of monogenic diabetes mellitus and differentiation from type 1 and type 2 diabetes is important in view of the implications for treatment and prognosis as well as for identification of family members at risk of diabetes [4–6]. Although monogenic diabetes mellitus is characterised by typical features such as young age at diagnosis (<25 years), a pronounced family history and non-obesity, low HbA1c and negative pancreatic auto-antibodies, definite diagnosis is possible only by genetic testing, which is expensive and not readily available [7, 8]. Therefore, knowledge of the clinical presentation of monogenic diabetes mellitus is essential to establish high pretest probability and the diagnosis, which on average is delayed by more than 10 years [9]. At present it is estimated that more than 80% of patients with monogenic diabetes are not diagnosed or are misclassified as type 1 or type 2 diabetes [3].
Figure 1
Pancreatic beta cell: the coupling of glucose sensing via GLUT-2 transporters, generation of ATP, membrane depolarization by closing potassium channels, entering of calcium ions, and exocytosis of insulin.
Blood glucose concentration is the main determinant of beta cell insulin secretion: glucose is transported into the beta cell by the GLUT-2 transporter. After glucose phosphorylation by glucokinase, ATP is generated via glycolysis and the Krebs cycle in the mitochondria. ATP then closes sensitive potassium channels leading to membrane depolarisation and opening of calcium channels, which initiates exocytosis of insulin; this is biosynthesised in the endoplasmic reticulum and stored in secretory granules (fig. 1) [10, 11]. A large number of gene mutations have been described to date (e.g. over 800 mutations for the MODY genes) uniformly causing monogenic diabetes mellitus by disturbing the coupling of blood glucose concentration and insulin secretion [1, 12, 13]. Depending on the specific gene mutation, the biosynthesis and secretion of beta cell insulin is altered at various stages (fig. 2): the genes affected in MODY encode the enzyme glucokinase (GCK [MODY 2]), the transcription factors (HNF4A [MODY 1], HNF1A [MODY 3], PDX1 [MODY 4], HNF1B [MODY 5], NeuroD1 [MODY 6]), the tumour suppressor protein KLF-11 (MODY 7), the carboxyl ester lipase CEL (MODY 8), the transcription factor PAX-4 (MODY 9), and the insulin gene (MODY 10) [13–22]. Still more novel gene variants have been described very recently and their possible role in diabetes is under investigation [23]. The mitochondrial DNA mutation A3243G causes mitochondrial dysfunction in mitochondrial diabetes (also designated by the acronym MIDDM = maternally inherited deafness and diabetes mellitus) [24]. Neonatal diabetes mellitus may result from different mutations involving the Kir6.2 genes (KCNJ11 and ABCC8) [25], the transcription factor genes (HNFB1B and IPF-1 gen) [26, 27], or the glucokinase gene [28, 29].
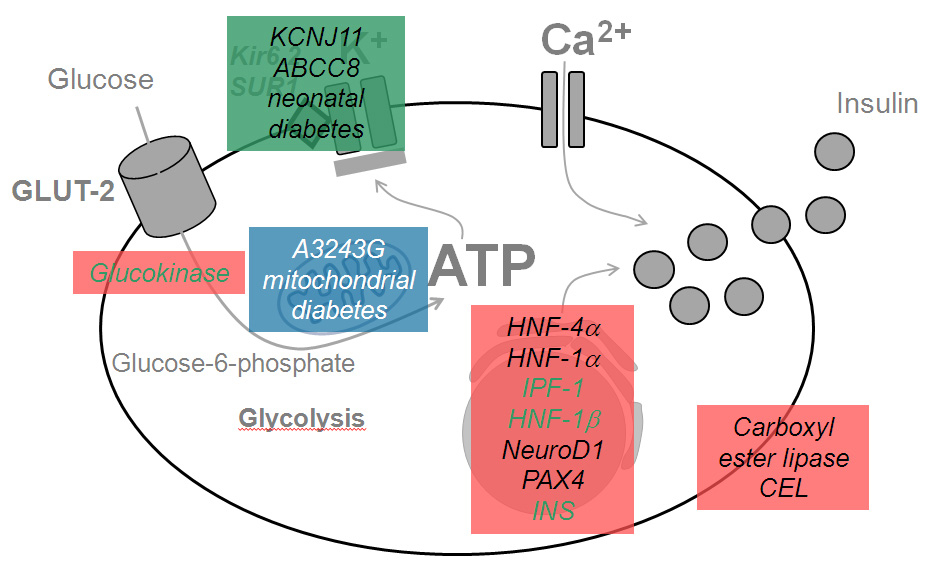
Figure 2
Location of the gene mutations and affected proteins in the pancreatic beta cell in monogenic diabetes mellitus.
MODY (red), mitochondrial diabetes (blue) and neonatal diabetes (green).
The prevalence of the different gene mutations varies widely in patients with MODY: whereas the mutation of HNF1A (encoding hepatic nuclear factor-1α, MODY 3) accounts for 50–70%, the mutation of the GCK (encoding glucokinase, MODY 2) for 20–30%, and the mutation of HNF4A and HNF1B (encoding hepatic nuclear factor-4α and hepatic nuclear factor-1β, MODY 1 and MODY 5 respectively) account for 5% each, the remaining gene mutations are very rare and described only in a few families representing fewer than 1% of all MODY cases [2, 9, 10].
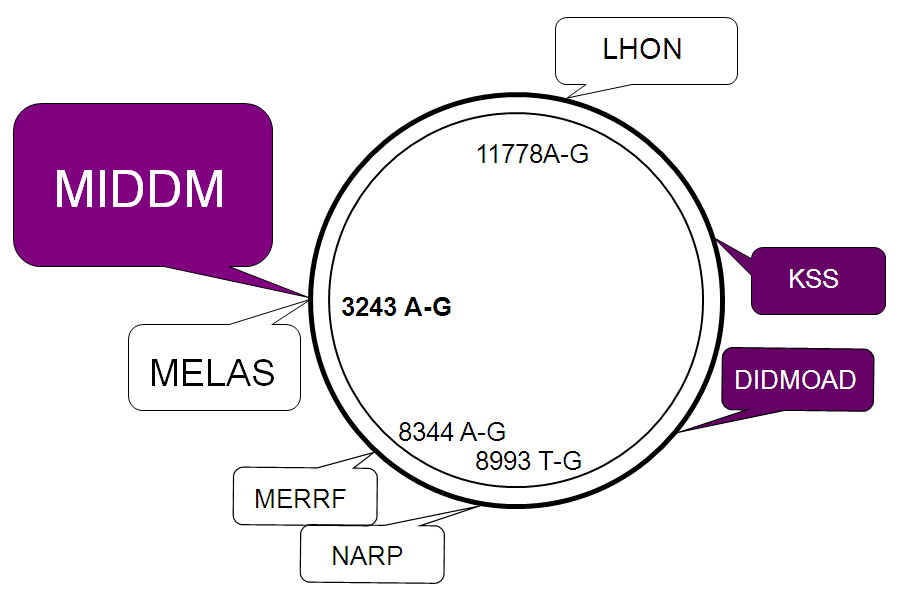
Figure 3
Mitochondrial DNA schematically depicted as circular double-strand DNA with some mutation syndromes.
MIDDM = maternally inherited deafness and diabetes mellitus; MELAS = mitochondrial encephalomyopathy, lactic azidosis and stroke-like episodes; DIDMOAD = diabetes insipidus, diabetes mellitus, optic atrophy, and deafness; KSS = Kearns-Sayre syndrome; LHON = Leber‘s hereditary optic neuropathy; MERRF = myoclonic epilepsy, ragged-red fibers; NARP = neuropathy, ataxia, and retinitis pigmentosa.

Figure 4
Pedigree of a 52-year-old woman with MIDDM (arrow), in whom diabetes was diagnosed at the age of 31 years during her first pregnancy with characteristic matrilinear transmission and hearing loss. In her son mitochondrial diabetes mellitus was diagnosed at the age of 16.
The clinical pattern of MODY is characterised by (1.) young age at diagnosis, i.e. usually between age 10 to 45, (2.) a marked family history with diabetes in every generation due to autosomal dominant inheritance, (3.) absence of obesity and insulin resistance, (4.) commonly mild hyperglycaemia without the need for insulin therapy, and (5.) negative testing for auto-antibodies against pancreatic beta cell antigens (GAD 65 and IA2) [1, 2, 4, 9, 30]. A recently published clinical predictive score to calculate the individual risk of contracting MODY [8] considered lower HbA1c, parent with diabetes, female sex and older age to distinguish MODY from type 1 diabetes, and lower BMI, younger age, female sex, lower HbA1c and no treatment with oral antidiabetic drugs or insulin to distinguish from type 2 diabetes. The sensitivity and specificity of the models now available online at http://www.diabetesgenes.org attained over 90% by using optimal cut-offs, and are thus a valuable means of identifying patients with probable MODY and optimising genetic testing. Table 1 summarises the manner of distinguishing clinical features of MODY from type 1 and type 2 diabetes.
MODY 3, the most prevalent MODY form, is denoted by high penetrance and a typical age of manifestation of under 30 years [31]. It presents early glucosuria and accentuated postprandial hyperglycaemia which increases over time due to a decline in beta cell insulin secretion by 1–4% per year [32]. Treatment with antidiabetic drugs is therefore needed, preferably with sulfonylurea and insulin during pregnancy [33]. Strict glycaemic control during pregnancy is mandatory because maternal hyperglycaemia lowers the age of MODY manifestation in the offspring by more than 10 years [34]. The occurrence of microvascular (particularly retinal and renal) and macrovascular complications is comparable to type 1 and type 2 diabetes, likewise depending on the duration and control of diabetes [35, 36].
MODY 2, the other common MODY form, typically causes hyperglycaemia starting at birth [13, 37]. As a consequence of the impaired conversion of glucose to glucose-6-phosphate and hence reduced glucose sensing, insulin secretion starts at a higher threshold (at blood glucose of 6–7 mmol/l instead of 5 mmol/l) resulting in mild and not worsening hyperglycaemia without the need for treatment and without secondary complications [38].
MODY 1 presents in a similar manner to MODY 3 (given the mechanism whereby HNF-4α regulates the expression of HNF-1α) but is relatively uncommon [1, 2].
MODY 5 is a distinct MODY form with additional renal cysts and genitourinary malformation (aplasia of the vasa deferentia and of the vagina) [39]. The spectrum of clinical features varies, in that at one time diabetes dominates and at another time the urinary tract malformations [40–42]. Contrary to the other MODY forms there is a high rate of spontaneous mutations, making family history unreliable and often rendering insulin treatment necessary [43].
The remaining rare MODY forms are accompanied by disturbances of endocrine (MODY 4, MODY 6) [19, 44] and exocrine (MODY 7, MODY 8, MODY 9) pancreatic development [21, 45], or the mutation in the insulin gene and neonatal diabetes (MODY 10) [46]. Table 2 summarises the features of the various MODY types.
At present sequencing of the most frequent MODY genes is routinely performed, i.e. HNF4A (MODY 1), GCK (MODY 2), HNF1A (or transcription factor-1 TCF-1) (MODY 3), and HNF1B (or transcription factor-2 TCF-2) (MODY 5), confirming the diagnosis of MODY in the majority of cases. However, new gene mutations are regularly detected [23, 47], also expanding the clinical spectrum of the MODY subtypes with sometimes considerable overlap with type 1 and type 2 diabetes. Therefore, other characteristics distinguishing MODY from both common types of diabetes have been investigated, such as serum high-sensitive C-reactive protein (CRP), which has proved to be lower in MODY 3 patients than in healthy individuals (because HNF-1α promotes the expression of CRP [48]). The role of MRI/CT imaging in identifying pancreatic alterations (i.e., pancreatic size, lipomatosis, fibrosis and calcification) in MODY has also been evaluated. Recent studies described a slightly reduced pancreatic volume in MODY 3, pancreatic hypoplasia with agenesis of the dorsal and caudal part in MODY 5, as well as pancreatic atrophy and an increased fat content in MODY 8 [49, 50].
Accurate diagnosis of MODY has important prognostic and therapeutic implications for the individual and the family. Best practice guidelines have been established for molecular genetic testing in patients with suspected MODY [2, 5, 7, 9]. They advocate testing in individuals with a strong family history, presentation at younger age (<25 years) and features atypical for type 1 diabetes (negative antibodies, insulin independence) or type 2 diabetes (absent components of the metabolic syndrome). However, taking further specific clinical features into consideration increases the likelihood of detecting the different MODY subtypes: if the above-mentioned features are present in young-onset diabetes, which remains insulin-independent for more than 3 years and there is marked sensitivity to sulfonylureas, MODY 3 and 1 are likely; additional renal and genital abnormalities indicate MODY 5; if there is persistent mild hyperglycaemia >5.5 mmol/l with only a small increase in <3 mmol/l in the oGTT, typically diagnosed in pregnancy, MODY 2 is suspected and testing for GCK mutations recommended. These MODY subtypes 1, 2, 3 and 5 make up over 95% of all MODY gene mutations. Laboratories performing certified sequencing of the suspected gene(s) are listed on the following website http://www.ncbi.nlm.nih.gov/sites/GeneTests/. The actual cost of one gene sequencing is about CHF 1,200.–, and reimbursement by the insurance companies must be applied for.
| Table 1: Clinical characteristics of MODY vs diabetes type 1 and type 2 (adapted from [1, 2, 9]). | |||
| Characteristic | MODY | Type 1 diabetes | Type 2 diabetes |
| Age at diagnosis | <25 y | 5–20 y | >25 y |
| Parental history | 60–95% | <10% | 10–40% |
| Inheritance | Autosomal dominant autoimmune disease | polygenic | |
| Obesity Insulin resistance Metabolic syndrome | } Uncommon | Uncommon | Common |
| Beta cell antibodies | Absent | Present | Absent |
| C-peptide | Normal | Undetectable | High – low |
| Optimal treatment | Sulfonylurea (MODY 1,3,4) | Insulin | Metformin |
| Table 2: Different MODY subtypes: gene mutations, pathophysiology and clinical manifestations (with prevalence amongst MODY patients). | ||
| MODY | Pathophysiological mechanism | Clinical characteristics |
| 1 | Abnormal beta cell gene transcription leading to deficient insulin secretion | Rare (5%); neonatal hyperinsulinism, low triglycerides, diabetes prone to microvascular complications, sensitive to sulfonylurea |
| 2 | Reduced glucose sensitivity due to deficient phosphorylation, impaired glycogen storage | Common (30-50%); impaired fasting glucose, increment <3 mmol/l in oGTT (<4.6 mmol/l in pregnancy); mild diabetes, usually no antidiabetic treatment needed |
| 3 | Abnormal gene transcription causing deficient insulin secretion, progressive beta cell failure | Common (30-50%) and high penetrance; glucosuria, microvascular complications, treatment with sulfonylurea (and insulin) |
| 4 | Impaired pancreatic development, pancreatic agenesis in homozygous state | Rare (1%); average onset at age 35 years, oral antidiabetic drugs (and insulin) |
| 5 | Abnormal gene transcription causing deficient insulin secretion | Rare (5%); diabetes with extrapancreatic manifestations (renal cysts or dysplasia, genital abnormalities in female, azoospermia in male), large variability in phenotype, insulin treatment needed |
| 6 | Abnormal development of beta cell function | Very rare (<1%); adult onset of diabetes |
| 7 | Reduced glucose sensitivity of the beta cell, tumour suppressor gene in pancreas (KLF-11) | Very rare (<1%); phenotype similar to type 2 diabetes |
| 8 | Impaired control of endocrine and exocrine pancreatic function (pathophysiology?) | Very rare (<1%); diabetes with typical autosomal dominant heredity |
| 9 | Impaired gene transcription in pancreatic beta cells on apoptosis and proliferation | Very rare (<1%); ketoacidosis possible |
| 10 | Heterozygous mutation of the insulin gen | Very rare (<1%); diabetes diagnosed <20 years, sulfonylurea and insulin late in follow-up |
| 11 | Heterozygous mutation of the BLK gene (tyrosin kinase expressed in pancreatic islets) affecting insulin secretion | Very rare (<1%); higher penetrance with increasing body mass index |
Mitochondrial DNA (mtDNA), which is almost exclusively of maternal origin (fig. 3), harbours 37 genes many of which encode enzymes involved in oxidative phosphorylation [51]. Hence mutations in the mtDNA predominantly affect organs with a high energy demand such as skeletal muscle, brain, sensory organs and pancreatic beta cells causing a wide spectrum of syndromes (fig. 4): the point mutation A→G at position 3243 generates mitochondrial diabetes with deafness (MIDDM) at an estimated prevalence of 0.4 to 1.5% [52], and the MELAS syndrome (mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes). The clinical manifestations of MIDDM, including age of onset and requirement of insulin treatment depend on the heteroplasmy, i.e. the level of mutated mtDNA in the individual patient [53, 54]. Thus the age when diabetes is diagnosed ranges from 8–70 years with a mean of 35 years, and ketoacidosis occurs in almost one third of patients. Hearing loss affects 90% and myopathy 30% of patients with MIDDM. Because diabetes develops due to failure of insulin secretion, most patients will eventually need insulin treatment, whereas metformin should be not be given because of possible myopathy [55]. Diabetes is also associated with other mtDNA mutation syndromes such as the Wolfram syndrome (the acronym DIDMOAD stands for diabetes insipidus, diabetes mellitus, optic atrophy and deafness) [57] or Kearns-Sayre syndrome (KSS, external ophthalmoplegia, cardiac conduction block, retinal degeneration, deafness, and ragged red muscle fibers) [57].
Neonatal diabetes mellitus describes a heterogeneous group of diabetes forms occurring until the age of six months in about 1:200,000 live births [58], caused by mutations of different genes involved in pancreatic organogenesis, formation of beta cells and insulin synthesis. Depending on the underlying gene mutations, neonatal diabetes is classified into transient or permanent. Due to the intrauterine insulin deficiency children are small for gestational age, with diminished subcutaneous fat.
1 Fajans SS, Graeme IB, Polonksy KS. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N Engl J Med. 2001;345:971–80
2 Gardner D, Shyong Tai E. Clinical features and treatment of maturity onset diabetes of the young (MODY). Diabet Metab Syndr Obes. 2012;5:101–8.
3 Shields BM, Hicks S, Shepherd MH, Colclough K, Hattersley AT, Ellard S. Maturity-onset diabetes of the young (MODY): how many cases are we missing? Diabetologia. 2010;53:2504–8.
4 Schober E, Rami B, Grabert M, Thon A, Kapellen T, Reinehr T, et al. Phenotypical aspects of maturity-onset diabetes of the young (MODY diabetes) in comparison with type 2 diabetes mellitus (T2DM) in children and adolescents: experience from a large multicentre database. Diabet Med. 2009;26:466–73.
5 Shepherd M, Shields B, Ellard S, Rubio-Cabezas O, Hattersley AT. A genetic diagnosis of HNF1A diabetes alters treatment and improves glycaemic control in the majority of insulin-treated patients. Diabet Med. 2009;26:437–41.
6 Pearson ER, Starkey BJ, Powell RJ, Gribble FM, Clark PM, Hattersley AT, et al. Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet. 2003;362:1275–81.
7 Ellard S, Bellanné-Chantelot C, Hattersley AT. Best practice guidelines for the molecular genetic diagnosis of maturity-onset diabetes of the young. Diabetologia. 2008;51:546–53.
8 Shields BM, McDonald TJ, Ellard S, Campbell MJ, Hyde C, Hattersley AT. The development and validation of a clinical prediction model to determine the probability of MODY in patients with young-onset diabetes. Diabetologia. 2012;55:1265–72.
9 Thanabalasingham G, Owen KR. Diagnosis and management of maturity onset diabetes of the young (MODY). BMJ. 2011;343:d6044
10 Pickup J, Williams G. Textbook of diabetes. 2nd ed. 1997; Blackwell Science Ltd. Oxford UK.
11 Stride A, Vaxillaire M, Tuomi T, Barbetti F, Njølstad PR, Hansen T, et al. The genetic abnormality in the beta cell determines the response to an oral glucose load. Diabetologia. 2002;45:427–35.
12 Nyunt O, Wu JY, McGown IN, Harris M, Huynh T, Leong GM, et al. Investigating maturity onset diabetes of the young. Clin Biochem Rev. 2009;30:67–74.
13 Pearson ER, Velho G, Clark P, Stride A, Shepherd M, Frayling TM, et al. β-Cell genes and diabetes: quantitative and qualitative differences in the pathophysiology of hepatic nuclear factor-1α and glucokinase mutations. Diabetes 2001;50:101–7.
14 Schnyder S, Mullis PE, Ellard S, Hattersley AT, Flück CE. Genetic testing for glucokinase mutations in clinically selected patients with MODY: a worthwhile investment. Swiss Med Wkly. 2005;135:352–6.
15 Vionnet N, Stoffel M, Takeda J, Yasuda K, Bell GI, Zouali H, et al. Nonsense mutation in the glucokinase gene causes early-onset non-insulin-dependent diabetes mellitus. Nature. 1992;356:721–2.
16 Bellanné-Chantelot C, Clauin S, Chauveau D, Chauveau D, Collin P, Daumont M, et al. Large genomic rearrangements in the hepatocyte nuclear factor-1beta (TCF2) gene are the most frequent cause of maturity-onset diabetes of the young type 5. Diabetes. 2005;54:3126–32.
17 Stoffers DA, Ferrer J, Clarke WL, Habener JF. Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nat Genet. 1997;17:138–9.
18 Horikawa Y, Iwasaki N, Hara M, Furuta H, Hinokio Y, Cockburn BN, et al. Mutation in hepatocyte nuclear factor-1β gene (TCF2) associated with MODY. Nat Genet. 1997;17:384–5.
19 Malecki MT, Jhala US, Antonellis A, Fields L, Doria A, Orban T, et al. Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat Genet. 1999;23:323–8.
20 Fernandez-Zapico ME, van Velkinburgh JC, Gutiérrez-Aguilar R, Neve B, Froguel P, Urrutia R, Stein R. MODY7 gene, KLF11, is a novel p300-dependent regulator of Pdx-1 (MODY4) transcription in pancreatic islet beta cells. J Biol Chem. 2009;284:36482–90.
21 Johansson BB, Torsvik J, Bjørkhaug L, Vesterhus M, Ragvin A, Tjora E, et al. Diabetes and pancreatic exocrine dysfunction due to mutations in the carboxyl ester lipase gene-maturity onset diabetes of the young (CEL-MODY): a protein misfolding disease. J Biol Chem. 2011;286:34593–605.
22 Biason-Lauber A, Boehm B, Lang-Muritano M, Gauthier BR, Brun T, Wollheim CB, Schoenle EJ. Association of childhood type 1 diabetes mellitus with a variant of PAX4: possible link to beta cell regenerative capacity. Diabetologia. 2005;48:900–6.
23 Johansson S, Irgens H, Chudasama KK, Molnes J, Aerts J, Roque FS, et al. Exome sequencing and genetic testing for MODY. PLoS One. 2012;7:e38050. Epub 2012 May 25
24 Zeviani M, Tiranti V, Piantadosi C. Mitochondrial disorders. Medicine. (Baltimore) 1998;77:59–72.
25 Oçal G, Flanagan SE, Hacihamdioğlu B, Berberoğlu M, Siklar Z, Ellard S, et al. Clinical characteristics of recessive and dominant congenital hyperinsulinism due to mutation(s) in the ABCC8/KCNJ11 genes encoding the ATP-sensitive potasium channel in the pancreatic beta cell. J Pediatr Endocrinol Metab. 2011;24:1019–23.
26 D’Amato E, Lorini R. Neonatal diabetes mellitus and mutation in the HNF-1beta gene. J Clin Endocrinol Metab. 2005;90:5906–7.
27 Thomas IH, Saini NK, Adhikari A, Lee JM, Kasa-Vubu JZ, Vazquez DM, et al. Neonatal diabetes mellitus with pancreatic agenesis in an infant with homozygous IPF-1 Pro63fsX60 mutation. Pediatr Diabetes. 2009;10:492–6.
28 Njolstad PR, Sovik O, Cuesta-Munoz A, Bjørkhaug L, Massa O, Barbetti F, et al. Neonatal diabetes mellitus due to complete glucokinase deficiency. N Engl J Med. 2001;344:1588–92.
29 Russo L, Iafusco D, Brescianini S, Nocerino V, Bizzarri C, Toni S, et al. (ISPED Early Diabetes Study Group). Permanent diabetes during the first year of life: multiple gene screening in 54 patients. Diabetologia. 2011;54:1693–701.
30 McDonald TJ, Colclough K, Brown R, Shields B, Shepherd M, Bingley P, et al. Islet autoantibodies can discriminate maturity-onset diabetes of the young (MODY) from type 1 diabetes. Diabet Med. 2011;28:1028–33.
31 Frayling TM, Bulman MP, Appleton M, Hattersley AT, Ellard S. A rapid screening method for hepatocyte nuclear factor 1 alpha frameshift mutations; prevalence in maturity-onset diabetes of the young and late-onset non-insulin dependent diabetes. Hum Genet. 1997;101:351–4.
32 Stride A, Ellard S, Clark P, Shakespeare L, Salzmann M, Shepherd M, et al. Beta-cell dysfunction, insulin sensitivity, and glycosuria precede diabetes in hepatocyte nuclear factor-1alpha mutation carriers. Diabetes Care. 2005;28:1751–6.
33 Fajans SS, Brown MB. Administration of sulfonylureas can increase glucose-induced insulin secretion for decades in patients with maturity-onset diabetes of the young. Diabetes Care. 1993;16:1254–61.
34 Stride A, Shepherd M, Frayling TM, Bulman MP, Ellard S, Hattersley AT. Intrauterine hyperglycemia is associated with an earlier diagnosis of diabetes in HNF-1alpha gene mutation carriers. Diabetes Care. 2002;25:2287–91.
35 Isomaa B, Henricsson M, Lehto M, Forsblom C, Karanko S, Sarelin L, et al. Chronic diabetic complications in patients with MODY3 diabetes. Diabetologia. 1998;41:467–73.
36 Steele AM, Shields BM, Shepherd M, Ellard S, Hattersley AT, Pearson ER. Increased all-cause and cardiovascular mortality in monogenic diabetes as a result of mutations in the HNF1A gene. Diabet Med. 2010;27:157–61.
37 Prisco F, Iafusco D, Franzese A, Sulli N, Barbetti F. MODY2 presenting as neonatal hyperglycaemia: a need to reshape the definition of “neonatal diabetes”? Diabetologia. 2000;43:1331–2.
38 Martin D, Bellanné-Chantelot C, Deschamps I, Froguel P, Robert JJ, Velho G. Long-term follow-up of oral glucose tolerance test-derived glucose tolerance and insulin secretion and insulin sensitivity indexes in subjects with glucokinase mutations (MODY2) Diabetes Care. 2008;31:1321–3.
39 Lindner TH, Njolstad PR, Horikawa Y, Bostad L, Bell GI, Sovik O. A novel syndrome of diabetes mellitus, renal dysfunction and genital malformation associated with a partial deletion of the pseudo-POU domain of hepatocyte nuclear factor-1beta. Hum Mol Genet. 1999;8:2001–8.
40 Bellanné-Chantelot C, Chauveau D, Gautier JF, Dubois-Laforgue D, Clauin S, Beaufils S, et al. Clinical spectrum associated with hepatocyte nuclear factor-1beta mutations. Ann Intern Med. 2004;6140:510–7.
41 Ulinski T, Lescure S, Beaufils S, Guigonis V, Decramer S, Morin D, et al. Renal phenotypes related to hepatocyte nuclear factor-1beta (TCF2) mutations in a pediatric cohort. J Am Soc Nephrol. 2006;17:497–503.
42 Raile K, Klopocki E, Holder M, Wessel T, Galler A, Deiss D, et al. Expanded clinical spectrum in hepatocyte nuclear factor 1b-maturity-onset diabetes of the young. J Clin Endocrinol Metab. 2009;94:2658–64.
43 Bellanné-Chantelot C, Lévy DJ, Carette C, Saint-Martin C, Riveline JP, Larger E, et al. (French Monogenic Diabetes Study Group). Clinical characteristics and diagnostic criteria of maturity-onset diabetes of the young (MODY) due to molecular anomalies of the HNF1A gene. J Clin Endocrinol Metab. 2011;96:1346–51.
44 Rubio-Cabezas O, Minton JA, Kantor I, Williams D, Ellard S, Hattersley AT. Homozygous mutations in NEUROD1 are responsible for a novel syndrome of permanent neonatal diabetes and neurological abnormalities. Diabetes. 2010;59:2326–31.
45 Chavali S, Mahajan A, Tabassum R, Dwivedi OP, Chauhan G, Ghosh S, et al. Association of variants in genes involved in pancreatic β-cell development and function with type 2 diabetes in North Indians. J Hum Genet. 2011;56:695–700.
46 Raile K, O’Connell M, Galler A, Werther G, Kühnen P, Krude H, et al. Diabetes caused by insulin gene (INS) deletion: clinical characteristics of homozygous and heterozygous individuals. Eur J Endocrinol. 2011;165:255–60.
47 Bowman P, Flanagan SE, Edghill EL, Damhuis A, Shepherd MH, Paisey R, et al. Heterozygous ABCC8 mutations are a cause of MODY. Diabetologia. 2012;55:123–7.
48 Thanabalasingham G, Shah N, Vaxillaire M, Hansen T, Tuomi T, Gašperíková D, et al. A large multicentre European study validates high-sensitivity C-reactive protein (hsCRP) as a clinical biomarker for the diagnosis of diabetes subtypes. Diabetologia. 2011;54:2801–10.
49 Haldorsen IS, Raeder H, Vesterhus M, Molven A, Njolstad PR. The role of pancreatic imaging in monogenic diabetes mellitus. Nat Rev Endocrinol. 2012;8:148–59.
50 Vesterhus M, Haldorsen IS, Raeder H, Molven A, Njølstad PR. Reduced pancreatic volume in hepatocyte nuclear factor 1A-maturity-onset diabetes of the young. J Clin Endocrinol Metab. 2008;93:3505–9.
51 de Andrade PB, Rubi B, Frigerio F, van den Ouweland JM, Maassen JA, Maechler P. Diabetes-associated mitochondrial DNA mutation A3243G impairs cellular metabolic pathways necessary for beta cell function. Diabetologia. 2006;49:1816–26.
52 Nagata H, Kumahara K, Tomemori T, Arimoto Y, Isoyama K, Yoshida K, et al. Frequency and clinical features of patients with sensorineural hearing loss associated with the A3243G mutation of the mitochondrial DNA in otorhinolaryngic clinics. J Hum Genet. 2001;46:595–9.
53 Laloi-Michelin M, Meas T, Ambonville C, Bellanné-Chantelot C, Beaufils S, Massin P, et al. Mitochondrial Diabetes French Study Group. The clinical variability of maternally inherited diabetes and deafness is associated with the degree of heteroplasmy in blood leukocytes. J Clin Endocrinol Metab. 2009;94:3025–30.
54 Lynn S, Borthwick GM, Charnley RM, Walker M, Turnbull DM. Heteroplasmic ratio of the A3243G mitochondrial DNA mutation in single pancreatic beta cells. Diabetologia. 2003;46:296–9.
55 Maassen JA, ’T Hart LM, Van Essen E, Heine RJ, Nijpels G, Jahangir Tafrechi RS, et al. Mitochondrial diabetes: molecular mechanisms and clinical presentation. Diabetes. 2004;53:103–9.
56 Domenech E, Gomez-Zaera M, Nunes V. Wolfram/DIDMOAD syndrome, a heterogenic and molecularly complex neurodegenerative disease. Pediatr Endocrinol Rev. 2006;3:249–57.
57 Laloi-Michelin M, Virally M, Jardel C, Meas T, Ingster-Moati I, Lombès A, et al. Kearns Sayre syndrome: an unusual form of mitochondrial diabetes. Diabetes Metab. 2006;32:182–6.
58 Rubio-Cabezas O, Klupa T, Malecki MT. CEED3 Consortium. Permanent neonatal diabetes mellitus – the importance of diabetes differential diagnosis in neonates and infants. Eur J Clin Invest. 2011;41:323–33.
Funding / potential competing interests: No financial support and no other potential conflict of interest relevant to this article was reported.