DOI: https://doi.org/10.4414/smw.2012.13670
Abbreviations
ARVC/D Arrhythmogenic right ventricular cardiomyopathy/dysplasia
DCM Dilated cardiomyopathy
DSC Desmocollin-2 gene
DSG2 Desmoglein-2 gene
DSP Desmoplakin gene
ICD Implantable cardioverter defibrillator
JUP Plakoglobin gene
LV Left ventricular/ventricle
LVEF Left ventricular ejection fraction
MRI Magnetic resonance tomography
PCR Polymerase chain reaction
PKP2 Plakophilin-2 gene
RV Right ventricular/ventricle
RYR2 Cardiac ryanodine receptor 2
SCD Sudden cardiac death
TGFB3 Transforming growth factor-beta 3
TMEM43 Transmembrane protein 43
TTE Transthoracic echocardiography
Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia (ARVC/D) is an autosomal-dominantly inherited disease characterised by fibrofatty replacement predominantly of the right ventricle (RV) and can clinically lead to heart failure, arrhythmias and sudden cardiac death (SCD) [1]. Recently new diagnostic criteria for ARVC/D have been proposed in order to improve diagnosis [2].
In 30–50% of cases ARVC/D is a familial disease with incomplete penetrance. Most genes involved in ARVC/D encode desmosomal proteins, in addition mutations in the genes encoding the transforming growth factor-beta 3 (TGFB3), transmembrane protein 43 (TMEM43) and cardiac ryanodine receptor 2 (RYR2) have been identified recently. RYR2 mutations are still discussed controversially [3]. When genes encoding proteins are systematically screened in ARVC/D patients, most mutations are found in the gene plakophilin-2 (PKP2) (31%), followed by the desmoglein-2 (DSG2) (10%), desmoplakin (DSP) (4.5%) and desmocollin-2 (DSC2) (1.5%) genes [4]. In this recent French study [4] it has been shown that mutations in the DSG2 gene were more frequently responsible for cardiac phenotypes with left ventricular (LV) involvement. In line with the recent French data [4], PKP2 mutations were most frequently identified to be causal for ARVC/D in a study of the Dutch population [5].
Autosomal-recessive forms of ARVC/D often manifest as Naxos and Carvajal syndrome. In Naxos disease caused by homozygous plakoglobin (JUP) mutations, ARVC/D involving predominantly the RV is associated with palmoplantar keratoderma and woolly hair, whereas in Carvajal disease caused by homozygous DSP mutations, patients develop LV involvement combined with palmoplantar keratoderma and woolly hair.
Evidence for wide genetic heterogeneity of ARVC/D and Naxos and Carvajal syndrome was recently summarised in review papers [6, 7] including mutations with pathogenic potential in proteins which are located at or in the composite junction of the intercalated disk in connecting mammalian cardiomyocytes. Besides desmosomal proteins, an increasing number of non-desmosomal proteins seem to be involved in the disease development, e.g. lamin A/C, striatin, titin and desmin [6, 7].
In this study we report on two unrelated patients with Naxos-Carvajal disease caused by heterozygous de novo mutations in the DSP gene. Both patients had severe cardiac phenotypes mainly involving the RV or the LV, resulting in implantation of a cardioverter defibrillator (ICD). The mutations were not identified in the maternal and paternal chromosomes, thus both mutations are considered to be de novo mutations.
The index patient was diagnosed with a bicuspid aortic valve and mild aortic stenosis due to a 2/6 systolic heart murmur at the age of two months. At the age of two years, the patient developed striate palmoplantar keratoderma and progressive erythemato-squamous plaques localised on elbows and knees and woolly hair (fig. 1A–E). The eyebrows and eyelashes were scarce and all nails showed some dystrophies. Dental abnormalities were absent. At the age of seven years, transthoracic echocardiography (TTE) revealed mild LV-involvement with wall motion abnormality of the posterior wall. Three years later the LV was slightly dilated with mild to moderate decreased systolic function and diastolic dysfunction. Holter-ECG revealed no ventricular or atrial arrhythmias. Rapid disease progression was seen in the follow-up TTE with severe dilated cardiomyopathy (DCM) of both ventricles, severely reduced LV systolic function (LVEF 19%) and moderate aortic stenosis (peak gradient 42 mm Hg, underestimated by markedly reduced LVEF). Cardiac magnetic resonance tomography (MRI) confirmed the TTE findings; late-enhancement sequences revealed inhomogeneous areas of probable fibrosis in the LV but no late enhancement of the RV. Balloon valvuloplasty was performed and an ICD was implanted. Last follow up performed at the age of 13 years showed DCM with mild aortic stenosis (peak gradient 21 mm Hg, underestimated by markedly reduced LVEF) and a severely reduced LVEF of 21%. Interrogation of the ICD was uneventful until the age of 14½ years, when ventricular fibrillation occurring during exercise was terminated by adequate ICD shock.


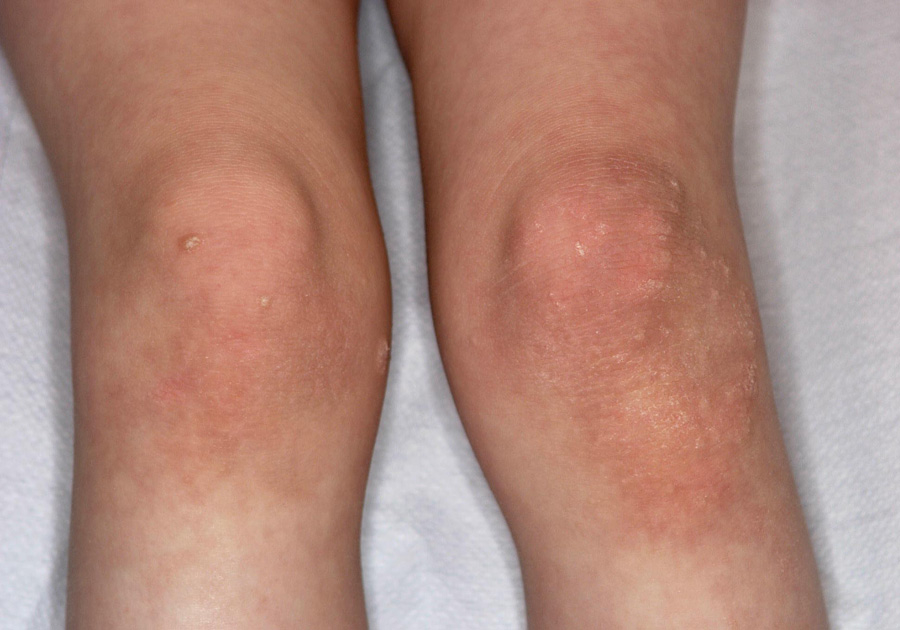
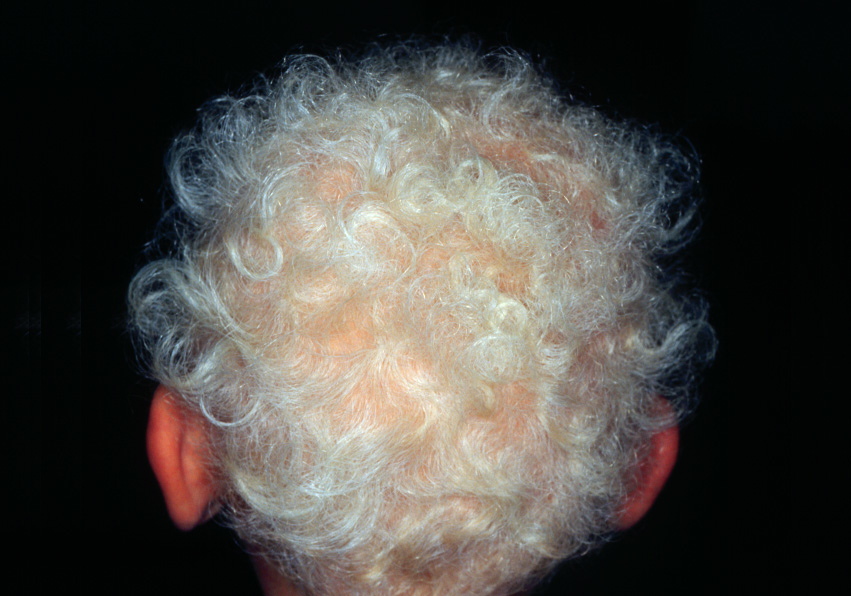
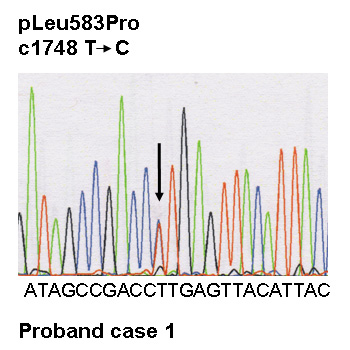

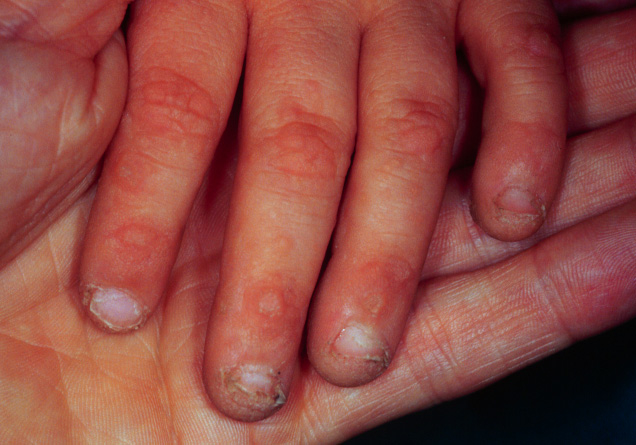
Figure 1
Dermatological phenotype,and genetic screening of case 1.
A All nails show dystrophic nail plates.
B Striate keratoderma of the palms.
C Plantar keratoderma.
D Erythemato-squamous skin lesions on the knee.
E Curly woolly hair.
F Electropherograms of the desmoplakin mutation and of the wild type: a heterozygous variant in DSP, c.1748 T>C, was identified, resulting in the missense mutation p.Leu583Pro at a heterozygous state.
Family history was completely normal. Both parents (Caucasians) and one brother had no cardiac, dental or dermatological diseases. History for SCD was negative.
The index patient is a female born from Caucasian non consanguineous parents at term with normal birth and weight. Her first tooth appeared at 11 months of age and, both cognition and growth were normal during the following years of life.
At the age of thirteen, the clinical examination was consistent with curly and woolly blond hair with high frontal hairline. Although shape and dental enamel of teeth were normal, she had dental agenesis with missing teeth numbers 14, 15, 17, 25, 27, 35, 37, 45 and 47. Furthermore she exhibited slight plantar keratosis, toe intertrigo and minimal palmar keratosis. Nail examination revealed bilateral leuconychia of third and fourth toe whereas finger nails were normal. While she had no cardiac symptoms, her resting electrocardiogram showed regular sinus rhythm, left hemi bundle-branch block and an epsilon wave. Her Holter ECG recordings showed frequent ventricular premature beats. Echocardiographic findings were consistent with severe RV dilatation and slight LV enlargement from the apical-4 chamber view (fig. 2A, B). At 22 years, an ICD was implanted following three episodes of syncope.
The study complies with the Declaration of Helsinki. After obtaining the written informed consent of each patient and family member, DNA extraction was performed from peripheral leucocytes using a classical phenol chloroform protocol. All coding exons of the genes PKP2, DSG2, DSC2, DSP and JUP were amplified by polymerase chain reaction (PCR) and directly sequenced in both strands. Analysis of the entire coding sequence was performed by direct sequencing with the use of the Big Dye dideoxy-terminator chemistry (Perkin Elmer) on ABI 3830 DNA sequencer (PE Applied Biosystems) [4].
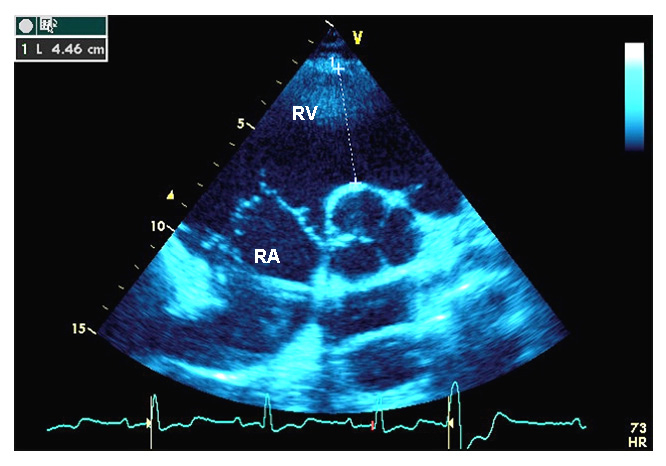
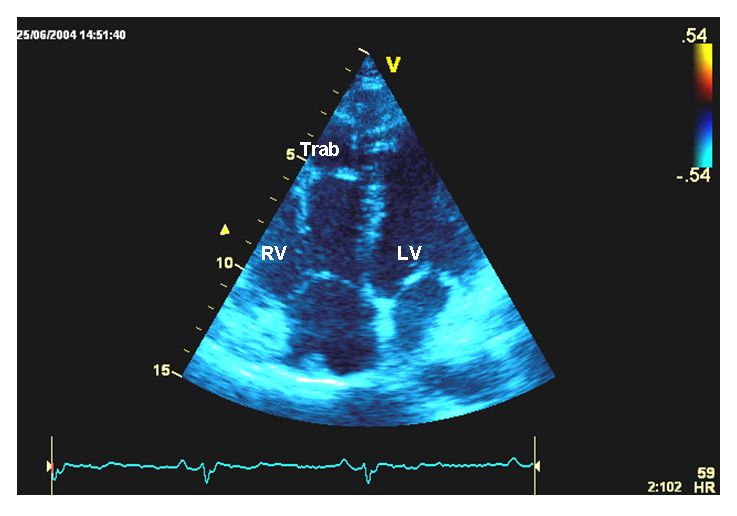
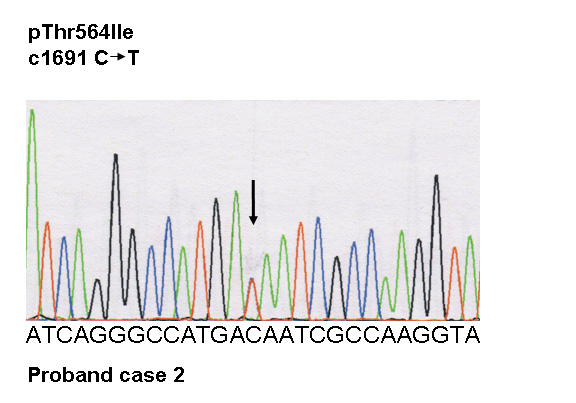
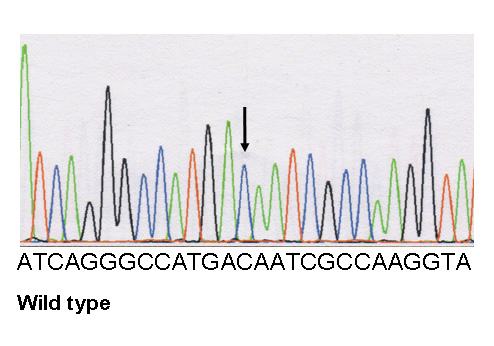
Figure 2
Transthoracic echocardiography, and genetic screening of case 2.
A Parasternal short axis view. Right atrium and right ventricular infundibulum are indicated. The right ventricular infundibulum diameter is measured at the level of the aortic valve.
RA = right atrium; RV = right ventricular infundibulum.
B Apical four chamber view. Right ventricular trabeculations are visible at the free wall level.
RV = right ventricle; Trab = trabeculations; LV = left ventricle.
C Electrophoregrams of the desmoplakin mutation and of wild type: a heterozygous variant in DSP, c.1691 C>T, was identified, resulting in the missense mutation p.Thr564Ile at a heterozygous state.
In case 1 a heterozygous variant in DSP, c.1748 T>C (NM004415.2) was identified, resulting in a miss-sense mutation p.Leu583Pro at a heterozygous state (fig. 1F).
In case 2, a heterozygous variant in DSP, c.1691 C>T (NM004415.2) was identified, resulting in a miss-sense mutation p.Thr564Ile at a heterozygous state (fig. 2C).
Both mutations were absent in 600 control chromosomes from a Caucasian population and were not reported in the exome variant server as well as the 1000 genome server databases. No additional mutation was found in the other four genes encoding desmosomal proteins.
The two mutations are located in the highly conserved domain. The effect of mutated residues was predicted as damaging by two among the three appropriate softwares (Polyphen, SIFT, Align AGVGD) used in our laboratory. Thus, we considered these miss-sense mutations as causal for the phenotypes. Mutation screening of the first degree relatives was secondarily performed but did not identify the family mutation in either of the parents of both patients, or in the sister of case 2. A paternity test was carried out in each of the families using the AmpF/STR SGM Plus® Pcr Amplification Kit (Applied Biosystems) that includes the study of ten different short tandem repeats, confirming that the alleged father was the biological one in each case. We therefore conclude that the probands both carried a de novo mutation in DSP.
In this study we identified a single de novo heterozygous DSPmutation in each of the patients leading to combined cardiac/dermatological and cardiac/dermatological/dental phenotypes predominantly involving the LV or the RV. The phenotypes mostly resemble Naxos-Carvajal syndrome, which belong to the large spectrum of ectodermal dysplasias [8].
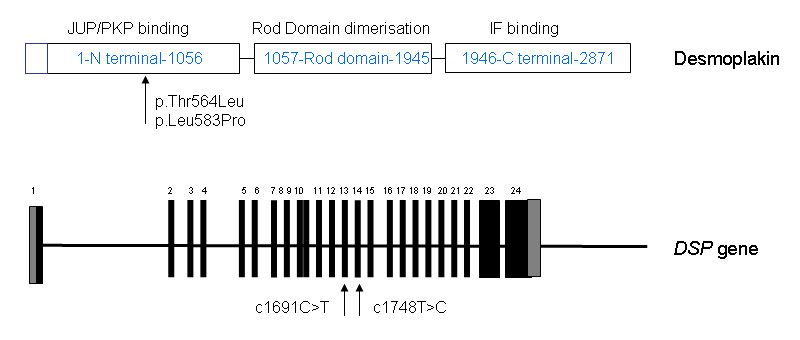
Figure 3
Structural organisation of desmoplakin and the desmoplakin gene (DSP). Desmoplakin, a big molecule (2871 AA) composed of three domains, is a critical protein of the desmosomal junction that serves to anchor the cytoskeleton to the plasma membrane. Desmoplakin exists in two isoforms: the longer desmoplakin I and the shorter desmoplakin II which is not expressed in the heart.
The phenotype of patient 1 was characterised by biventricular predominant LV involvement with palmoplantar keratoderma and woolly hair. In patient 2 the phenotype was predominant RV involvement with mainly RV dilation, associated with palmoplantar keratoderma and dental involvement.
Today numerous allelic variants of DSP mutations are known. Striate keratosis palmoplantares [9] and skin fragility-woolly hair syndrome [10] are phenotypes affecting the skin and are mostly caused by compound heterozygous DSP mutations.
In contrast, the genetic basis of acantholytic lethal epidermiolysis bullosa is compound heterozygous or homozygous DSP mutations [11, 12].
Heterozygous DSP mutations can either lead to a pure cardiac or a pure dermatological phenotype: ARVC/D type 8 is caused by heterozygous DSP mutations [13]. Only a few heterozygous DSP mutations have been identified leading to pure dermatological phenotypes. In a family with a striate subtype of palmoplantar keratoderma a nonsense DSP mutation in the amino terminal domain of desmoplakin was identified [9].
The composed cardiac/dermatological phenotype caused by homozygous DSP mutations is known as Carvajal disease, characterised by epidermolytic palmoplantar keratoderma with woolly hair and dilated cardiomyopathy [14]. The cardiac phenotype of the patients in this study [14] was exclusively DCM.
The first recessive mutation, a deletion in DSP, was associated with a phenotype consistent for generalised striate keratoderma, woolly hair and DCM of the LV [15]. In a study of Alcalai et al. [16], a homozygous DSP miss-sense mutation was identified in a large family with a high rate of consanguinity. The clinical phenotype caused by DSP homozygosity was characterised by foliaceous pemphigous, woolly hair and ARVC/D without LV involvement [15]. The heterozygous mutation carriers did not reveal any cardiac or dermatological abnormalities [15].
In our study both patients showed mixed cardiac/dermatological or cardiac/dermatological/dental phenotypes and each carried a single heterozygous mutation in DSP. Compound heterozygosity was at least excluded for the PKP2, DSG2, DSC2and JUPgenes.
It is still under investigation why heterozygous DSP mutation can lead to wide phenotype heterogeneity. Genotype heterogeneity with unknown variants in the major genes or polymorphisms and unknown factors might be modulating factors. Evidence for genetic heterogeneity specifically in Carjaval syndrome with dental involvement was discussed in a paper of Nehme recently [17]. In this paper [17] DSP mutations were excluded in a patient with Carjaval syndrome. Due to the wide genotype and phenotype heterogeneity in ARVC/D and Naxos and Carvajal syndrome it can be speculated that the complex phenotypes in our patients might be a “common” desmoplakin mutation condition. Such conditions could be based on an autosomal recessive or autosomal dominantly inheritance background.
The desmosomal plaque consists of two distinct electron dense regions: one close to the membrane of the low density region and one to the high density region further from the membrane, separated by a clear region [18]. The desmosomal proteins involved in desmosomal structure include plakoglobin, plakophilins, desmocollins, desmogleins and the plakins with desmoplakin. Desmoplakin is a critical protein of the desmosomal junction that serves to anchor the cytoskeleton to the plasma membrane. Historically the desmoplakin localisation was first demonstrated in 1981 (for review, see [19]). Desmoplakin exists in two isoforms: the longer desmoplakin I and the shorter desmoplakin II which is not expressed in the heart. Desmoplakin is a big molecule (2871 AA) composed of three domains (fig. 3). The desmoplakin N-terminal of 1056 AA contains a plakin domain (580AA) that is thought to be responsible for targeting desmoplakin to junction by mediating interactions with plakophilin and plakoglobin [20].
The C-terminal domain (1946-2871 AA) is predicted to interact with intermediate filaments. The structure of the C-terminal domains features three plakin repeat domains called A, B, and C. The core of the molecule (1057-1945 AA) is composed of a helical domain. Two molecules of desmoplakin are predicted to form dimers based on a coiled formation between their two central cores [21, 22].
It is likely that various mechanisms are involved in explaining the different phenotypes, depending on the type of mutation and on their positions.
The c7901Del homozygous mutation located in c-terminal domain of DSP causes a premature stop and results in a truncated protein. The loss of the desmoplakin tail impairs interaction with intermediate filaments in the heart, the skin and the hair resulting in a combination of woolly hair, palmoplantar keratoderma and LV DCM [14]. C-terminal compound mutations (nonsense and miss-sense mutations) have been reported by Mahoney et al. [23]. The nonsense mutation is thought to lead to a truncated protein, and the associated missense mutation (c.7964 C>A, p.Ala2655Asn) is thought to alter intermolecular interactions with intermediate filaments. The resulting phenotype includes cutaneous blisters, epidermolytic palmoplantar keratoderma, nail dystrophy, enamel dysplasia, woolly hair and cardiomyopathy [23]. Compound heterozygosity with amino terminal missense mutations (p.Arg2366Cys) and carboxy terminal nonsense mutations (p.Gln664X) are involved in severe keratoderma of hands and feet and woolly hair without cardiomyopathy [10].
For the heterozygous nonsense mutation, pGln331X, haploinsufficiency was the proposed physiopathological mechanism [9]. The relevance of haploinsufficiency for desmoplakin causing inherited dermatological phenotypes was further proposed by Whittock et al. [24].
Almost 50 mutations have been described in the first 15 exons coding N terminal domain. Among these mutations, 10 to 15 are variants of unknown significance. Four of these mutations are in the region of interest of the protein (table 1). The missense mutation p.Ala566Thr reported by Basso et al [25] was associated with a second missense mutation in DSP, p.Lys470Glu. All the patients in this study [25] were diagnosed with ARVD.
| Table 1: Desmoplakin gene (DSP) missense mutations in exons 13–15. | |||||
| Clinical phenotype | Inheritance | Exons | Sequence change | Amino acid change | Reference |
| Naxos-Carvajal phenotype | De Novo (heterozygous) | 13 | c.1691 C>T | p.Thr564Ile | This article |
| Patient referred as Naxos-Carvajal phenotype | Unknown heterozygous | 13 | c.1691 C>T | p.Thr564Ile | Ref. [4] |
| Patient referred as ARVD | Unknown Compound heterozygous | 13 | c.1696 G>A | p.Ala566Thr associated with p.Lys470Glu | Ref. [25] |
| Naxos-Carvajal phenotype | De Novo (heterozygous) | 14 | c.1748 T>C | pLeu583Pro | This article |
| Oligodontia and Carvajal-Naxos syndrome | Heterozygous | 14 or 15 | c.1790 C>T | pSer597Leu | Ref. [26] |
Recently, a heterozygous mutation has been described by Chalabreysse et al. [26] in a family with oligodontia associated with Carvajal/Naxos disease. The missense mutation p.Ser597Leu [26] is in the same region of the desmoplakin protein as the present mutations and the patients’ phenotypes resemble each other except for the presence of hypo-/oligodontia. This observation suggests a key role for the desmoplakin region (564-597), which may be involved in the interactions with plakophilin or plakoglobin. Additional studies are needed in order to explain the role of this key region.
Besides the direct effects of the desmoplakin mutations on the myocardial composite, indirect mechanisms may play a key role in early phenotype development [27, 28].
In a murine model, conditional genetic deletion of one desmoplakin allele caused connexin mislocation which precedes any overt histological abnormalities in ARVD/C [27]. The importance of the connexin-desmosomal protein interdependence resulting in ventricular conduction abnormalities preceding the histopathological ARVC/D phenotype has also been shown in an in vitro study of an ARVC/D patient’s myocardial tissue [28].
In the present study, two single heterozygous DSP missense mutations in two unrelated patients were found in the N terminal domain of the desmoplakin protein. These variants involve high conserved residues. Moreover, these mutations are de novo and localised in critical protein domains that appear to be mutation hot spots in residues 250 to 604 [29]. We assume that these variants are the causal mutations because of their absence in 300 controls.
1 Basso C, Corrado D, Marcus FI, Nava A, Thiene G. Arrhythmogenic right ventricular cardiomyopathy. Lancet. 2009;373:1289–300.
2 Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010;121:1533–41.
3 Ellinor PT, MacRae CA, Thierfelder L. Arrhythmogenic right ventricular cardiomyopathy. Heart Fail Clin. 2010;6:161–77.
4 Fressart V, Duthoit G, Donal E, Probst V, Deharo JC, Chevalier P, et al. Desmosomal gene analysis in arrhythmogenic right ventricular dysplasia/cardiomyopathy: spectrum of mutations and clinical impact in practice. Europace. 2010;12:861–8.
5 Cox MG, van der Zwaag PA, van der Werf C, van der Smagt JJ, Noorman M, Bhuiyan ZA, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: pathogenic desmosome mutations in index-patients predict outcome of family screening: Dutch arrhythmogenic right ventricular dysplasia/cardiomyopathy genotype-phenotype follow-up study. Circulation. 2011;123:2690–700.
6 Rickelt S, Piephoff S. Mutations with pathogenic potential in proteins located in or at the composite junctions of the intercalated disk connecting mammalian cardiomyocytes: a reference thesaurus for arrhythmogenic cardiomyopathies and for Naxos and Carvajal diseases. Cell Tissue Res. 2012;348:325–33.
7 Murray B. Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy (ARVD/C): a review of molecular and clinical literature. J Genet Couns. 2012;21:494–504.
8 Itin PH. Rationale and background as basis for a new classification of the ectodermal dysplasias. Am J Med Genet. 2009;149A:1973–6.
9 Armstrong DK, McKenna KE, Purkis PE, Green KJ, Eady RA, Leigh IM, et al. Haploinsufficiency of desmoplakin causes a striate subtype of palmoplantar keratoderma. Hum Mol Genet. 1999;8:143–8.
10 Whittock NV, Wan H, Morley SM, Garzon MC, Kristal L, Hyde P, et al. Compound heterozygosity for non-sense and mis-sense mutations in desmoplakin underlies skin fragility/woolly hair syndrome. J Invest Derm. 2002;118:232–8.
11 Jonkman MF, Pasmooij AM, Pasmans SG, van den Berg MP, Ter Horst HJ, Timmer A, et al. Loss of desmoplakin tail causes lethal acantholytic epidermolysis bullosa. Am J Hum Genet. 2005;77:653–60.
12 Hobbs RP, Han SY, van der Zwaag PA, Bolling MC, Jongbloed JD, Jonkman MF, et al. Insights from a desmoplakin mutation identified in lethal acantholytic epidermolysis bullosa. J Invest Derm. 2010;130:2680–3.
13 Rampazzo A, Nava A, Malacrida S, Beffagna G, Bauce B, Rossi V, et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2002;71:1200–6.
14 Carvajal-Huerta L. Epidermolytic palmoplantar keratoderma with woolly hair and dilated cardiomyopathy. J Am Acad Dermatol. 1998;39:418–21.
15 Norgett EE, Hatsell SJ, Carvajal-Huerta L, Cabezas JC, Common J, Purkis PE, et al. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and cause dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet. 2000;9:2761–6.
16 Alcalai R, Metzger S, Rosenheck S, Meiner V, Chajek-Shaul T. A recessive mutation in desmoplakin causes arrhythmogenic right ventricular dysplasia, skin disorder, and woolly hair. J Am Coll Cardiol. 2003;42:319–27.
17 Nehme N, El Malti R, Roux-Buisson N, Caignault JR, Bouvagnet P. Evidence for genetic heterogeneity in Carvajal syndrome. Cell Tissue Res. 2012;348:261–4.
18 Al-Amoudi A, Castano-Diez D, Devos DP, Russell RB, Johnson GT, Frangakis AS. The three-dimensional molecular structure of desmosomal plaque. Proc Natl Acad Sci U S A 2011;108:6480–5.
19 Franke WW. Discovering the molecular components of intercellular junctions – a historical view. Cold Spring Harb Perspect Biol. 2009;1:a003061.
20 Getsios S, Huen AC, Green KJ. Working out the strength and flexibility of desmosomes. Nat Rev Mol Cell Biol. 2004;5:271–81.
21 Stokes DL. Desmosomes from a structural perspective. Curr Opin Cell Biol. 2007;19:565–71.
22 Yang Z, Bowles NE, Scherer SE, Taylor MD, Kearney DL, Ge S, et al. Desmosomal dysfunction due to mutations in desmoplakin causes arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Res. 2006;99:646–55.
23 Mahoney MG, Sadowski S, Brennan D, Pikander P, Saukko P, Wahl J, et al. Compound heterozygous desmoplakin mutations result in a phenotype with a combination of myocardial, skin, hair, and enamel abnormalities. J. Invest Dermat. 2010;130:968–78.
24 Whittock NV, Ashton GH, Dopping-Hepenstal PJ, Gratian MJ, Keane FM, Eady RA, et al. Striate palmoplantar keratoderma resulting from desmoplakin haploinsufficiency. J Invest Dermatology. 1999;113:940–6.
25 Basso C, Czarnowska E, Della Barbera M, Bauce B, Beffagna G, Wlodarska EK, et al. Ultrastructural evidence of intercalated disc remodelling in arrhythmogenic right ventricular cardiomyopathy: an electron microscopy investigation on endomyocardial biopsies. Eur Heart J. 2006;27:1847–54.
26 Chalabreysse L, Senni F, Bruyère P, Aime B, Ollagnier C, Bozio A, et al. A new hypo/oligodontia syndrome: Carvajal/Naxos syndrome secondary to desmoplakin-dominant mutations. J Dent Res. 2011;90:58–64.
27 Gomes J, Finlay M, Ahmed AK, Ciaccio EJ, Asimaki A, Saffitz JE, et al. Electrophysiological abnormalities precede overt structural changes in arrhythmogenic right ventricular cardiomyopathy due to mutations in desmoplakin-A combined murine and human study. Eur Heart J. 2012 Jan 11. [Epub ahead of print].
28 Gehmlich K, Lambiase PD, Asimaki A, Ciaccio EJ, Ehler E, Syrris P, et al. A novel desmocollin-2 mutation reveals insights into the molecular link between desmosomes and gap junctions. Heart Rhythm. 2011;8:711–8.
29 Kapplinger JD, Landstrom AP, Salisbury BA, Callis TE, Pollevick GD, Tester DJ, et al. Distinguishing arrhythmogenic right ventricular cardiomyopathy/dysplasia-associated mutations from background genetic noise. J Am Coll Cardiol. 2011;57:2317–27.
Funding / potential competing interests: No financial support and no other potential conflict of interest relevant to this article were reported.