Reactive oxygen species: from health to disease
DOI: https://doi.org/10.4414/smw.2012.13659
Katharine
Brieger, Stefania
Schiavone, Francis J.
Miller Jr., Karl-Heinz
Krause
Summary
Upon reaction with electrons, oxygen is transformed into reactive oxygen species (ROS). It has long been known that ROS can destroy bacteria and destroy human cells, but research in recent decades has highlighted new roles for ROS in health and disease. Indeed, while prolonged exposure to high ROS concentrations may lead to non-specific damage to proteins, lipids, and nucleic acids, low to intermediate ROS concentrations exert their effects rather through regulation of cell signalling cascades. Biological specificity is achieved through the amount, duration, and localisation of ROS production. ROS have crucial roles in normal physiological processes, such as through redox regulation of protein phosphorylation, ion channels, and transcription factors. ROS are also required for biosynthetic processes, including thyroid hormone production and crosslinking of extracellular matrix. There are multiple sources of ROS, including NADPH oxidase enzymes; similarly, there are a large number of ROS-degrading systems. ROS-related disease can be either due to a lack of ROS (e.g., chronic granulomatous disease, certain autoimmune disorders) or a surplus of ROS (e.g., cardiovascular and neurodegenerative diseases). For diseases caused by a surplus of ROS, antioxidant supplementation has proven largely ineffective in clinical studies, most probably because their action is too late, too little, and too non-specific. Specific inhibition of ROS-producing enzymes is an approach more promising of clinical efficacy.
Introduction
General background
Reactive oxygen species (ROS) have a crucial role in human physiological and pathophysiological processes [1]. All ROS types, including superoxide anions and hydrogen peroxide, have unpaired valence electrons or unstable bonds. At high concentrations, ROS react readily with proteins, lipids, carbohydrates, and nucleic acids, often inducing irreversible functional alterations or even complete destruction. When ROS were initially integrated into biomedical concepts it was thought that they caused exclusively toxic effects and were associated with pathologies.

Figure 1
Variations of ROS generation. (a) Conceptual representation of genetically-determined ROS generation in the human population. Globally, there are significant variations of ROS generation. Note, however, that there is also regulation of ROS generation by epigenetics and external influences. (b) At moderate levels, NOX have crucial roles in health through mechanisms including signaling, biosynthetic processes, and host defence. When ROS levels are too low, the condition may result in decreased antimicrobial defence (e.g. chronic granulomatous disease), hypothyroidosis, low blood pressure or loss of otoconia. When levels are too high, there is increased cardiovascular disease, neurological disorders, cancers, and chronic inflammation.
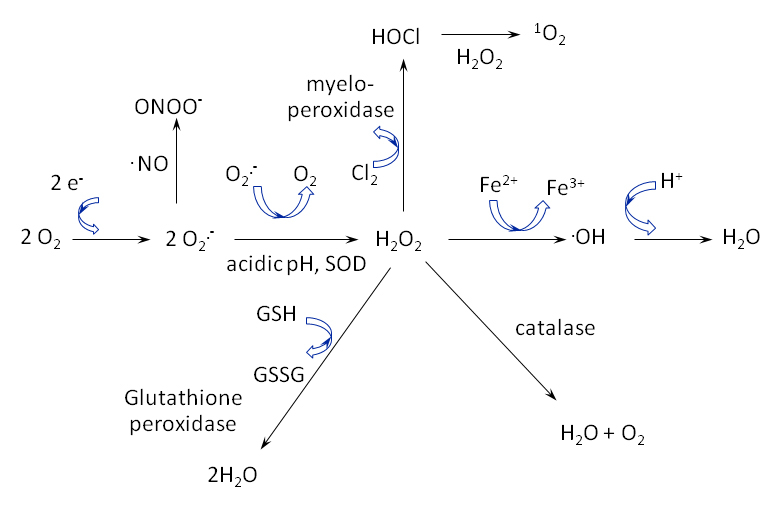
Figure 2
ROS are highly reactive and quickly transition between species. Notably, the superoxide anion (O2
.-) is unstable and quickly dismutates, by action of superoxide dismutase (SOD), into hydrogen peroxide (H2O2). If the anion reacts with nitric oxide
(.NO), then peroxynitrite (ONOO-) is formed. Singlet oxygen (1O2) is formed upon the reaction of hypochlorous acid (HOCl) with hydrogen peroxide.

Figure 3
(a) ROS generation and degradation contributing to the oxidative balance. (b) The NOX2/p22phox complex, consisting of NOX2 (orange) and the subunits p47, p67, and p40 (left) and glutathione defense mechanism (right). The superoxide (O2
.–) generated by the NOX complex is rapidly dismutated by superoxide dismutatase (SOD), where it can enter the glutathione synthesis and recuperation cycle.
ROS were once thought to originate almost entirely from mitochondrial metabolism. Indeed, while with normal mitochondrial function, the final oxygen electron receptor is reduced to water, it is possible, in particular under pathological conditions, that electrons leak out of the system prematurely and create ROS. However, there is mounting evidence that cellular enzymes referred to as NADPH oxidases are an important source of ROS in humans [2]. The seven catalytic isoforms of NADPH oxidase are further explained in subsequent sections, as the isoforms vary substantially in tissue localisation, mechanisms of activation, and roles in pathologies. ROS-generating processes, including mitochondria and NADPH oxidases, have a long and complex evolution and it is possible that ROS took part in early cell-to-cell signalling in unicellular organisms.
It is now clear that organisms have also developed methods of utilising ROS in critical physiological processes [1]. At the cellular level, ROS regulate growth, apoptosis, and other signalling. At the systems level they contribute to complex functions such as blood pressure regulation, cognitive function and immune function. ROS enable the response to growth factor stimulation and the generation of the inflammatory response, as well as having vital roles in the immune system where they directly kill pathogens [3]. Other examples of a biochemical role of ROS are found in primitive organisms, where ROS are involved in the cross-linking of the extracellular matrix [4] and in the hardening of the fertilisation envelope after egg-sperm fusion [5].
However, there is a delicate balance between appropriate redox states and oxidative stress, dependent on the relative rates of production and degradation (fig. 1). For specificity in signalling, biological systems recognise the amount and type of ROS, as well as the temporal and spatial distribution. Overproduction of ROS occurs during stimulated disease conditions, but there is also a genetic component to the propensity for ROS generation [6].
Natural human antioxidant defenses are not always sufficient to maintain the proper ROS balance. Also, a normal process can become pathological when it persists long-term. The mechanisms of senescence involve a contribution from free radicals, leading to the proposition more than fifty years ago that the aging process results partly from oxidative damage [7]. Oxidative stress is associated with the general aging process and cell death, affecting all major organ systems [2] and ROS have a part in many age-associated diseases, including Alzheimer's disease [8], Parkinson's disease [9], and virtually all cardiovascular diseases [10]. Large epidemiological studies support the relationship between oxidative state and global health; high consumption of foods rich in antioxidants is associated with lower disease rates and preventive protection [11].
Basic chemistry, biology, and toxicology
ROS can be neutral molecules (eg. hydrogen peroxide), ions (e.g. superoxide anion) or radicals (e.g. hydroxyl radicals). Due to their reactivity, ROS are observed as a cascade of transitions from one species to another (fig. 2). Typically the initial reaction is a one electron transfer to oxygen to form superoxide (O2
.–), which then dismutates (spontaneously or catalysed by superoxide dismutase) to hydrogen peroxide (H2O2). Superoxide does not readily cross membranes and is short-lived and local in its effect, but superoxide dismutase converts superoxide to longer-lasting and membrane-diffusible hydrogen peroxide (H2O2) [12]. When superoxide reacts with nitric oxide (NO), the highly reactive peroxynitrite (ONOO–) is formed. Peroxidases catalyse reactions involving hydrogen peroxide, resulting in the generation of hypochlorous acid (HOCl) and singlet oxygen (1O2), among other species. Finally, the Haber-Weiss reaction uses an iron ion catalyst to generate hydroxyl radicals from superoxide and hydrogen peroxide.
Our present state of knowledge supports the following primary functions of ROS in biology: host defence, cell signalling and biosynthetic processes. In host defence ROS contribute directly and indirectly to the killing of microorganisms. In physiological signalling, ROS can modify redox-sensitive amino acids in a variety of proteins, including phosphatases, ion channels, and transcription factors [13]. The more highly reactive species peroxynitrite and singlet oxygen may be more involved in toxicity, though signalling roles have been described [14]. Certain biosynthetic processes are redox-dependent and ROS are involved in reactions such as iodination of thyroid hormones and crosslinking of matrix proteins.
Because of their high reactivity, ROS readily react with virtually all types of biological molecules. Thus, high and sustained concentrations of ROS can cause damage to many cellular and extracellular constituents, including DNA, proteins and lipids [15]. Of particular concern is DNA oxidation, since this can cause mutations and changes in gene expression [16]. Mitochondrial DNA seems to be more sensitive to oxidative stress-induced mutations because it lacks DNA repair enzymes. Further, oxidation of proteins may lead to the formation of insoluble protein aggregates. Such protein aggregates are the molecular basis of a number of diseases, particularly neurodegenerative pathologies. ROS are also involved in the formation of "advanced glycosylation endproducts", which is an irreversible molecular change typically seen in glycosylated cell surface proteins.
Sources of ROS
Major sources of ROS include cellular respiration and metabolic processes, though ROS may also be generated by radiation. During the process of cellular respiration, oxygen is reduced by the successive transfer of single electrons and the intermediates with odd electrons can escape the chain. Metabolic processes also generate ROS; for instance, the peroxisome catabolises biomolecules using enzymes that remove hydrogen in an oxidative reaction, thereby creating hydrogen peroxide. Indeed, a large number of enzymes (xanthine oxidase, nitric oxide synthetase, p450 cytochromes) and organelles (mitochondria, peroxisomes) produce ROS as a metabolic byproduct. In contrast, the primary function of NADPH oxidases (NOX) is to produce ROS (fig. 3).
Mitochondria
The generation of mitochondrial ROS is a consequence of oxidative phosphorylation. At several sites along the cytochrome chain, electrons derived from NADH or FADH can directly react with oxygen or other electron acceptors and generate free radicals. It has recently been proposed that mitochondrial ROS play a crucial role in several redox-dependent signalling processes as well as in the aging clock. The discovery of superoxide dismutase enzymes (SOD1 and SOD2), was a major step in establishing the generation of ROS or H2O2 in the mitochondria. A number of studies have demonstrated that mitochondrial integrity declines as a function of age. Age-dependent increases in the level of damaged DNA have been commonly assessed through biomarkers such as the formation of 8-oxo-2′-deoxyguanosine (oxo8dG). In postmitotic tissue such as brain, the levels of oxo8dG are significantly higher in mitochondrial compared to nuclear DNA [17].
NOX enzymes
NOX are membrane-bound proteins that function to transfer electrons across membranes, where the final electron receptor is oxygen and superoxide is generated. These enzymes participate in cellular differentiation, growth, proliferation, apoptosis, cytoskeletal regulation, migration, and contraction.
NOX isoforms: structure, distribution, and regulation
All NOX NADPH oxidases are comprised of membrane-bound subunits and catalyse molecular oxygen reduction. However, NOX isoforms are each unique in terms of tissue distribution, domain structure, subunit requirements, and mechanism of activation. The catalytic subunit of all NOX proteins contains a C-terminal dehydrogenase domain with an NADPH binding site and a bound flavin adenine nucleotide (FAD). When NADPH oxidases are activated and their subunits are assembled, NADPH transfers its electrons to FAD, which then passes the electrons to two haem groups bound to the N-terminal domain, which finally pass the two electrons to two molecular oxygens on the opposite side of the membrane, forming superoxide anions [18].
NOX2, also known as the phagocyte NADPH oxidase, is the prototypical NOX and has a distinct role in the host defence against bacteria, where NOX2 in phagosomes generates superoxide to kill bacteria. Besides its abundant presence in phagocytes, NOX2 is expressed at lower levels in many cell types, including cardiomyocytes, endothelial cells, adventitial fibroblasts, neurons, and pancreatic β-cells. NOX2 is activated when the regulatory subunit p47phox is phosphorylated, complexes with the regulatory subunits p67phox and p40phox. At the membrane, the complex binds to NOX2 and p22phox; the binding of Rac protein to these elements completes activation [19].
NOX1 is highly expressed in colon epithelium, but is also found in other epithelial cells (e.g. alveolar epithelium), endothelium, smooth muscles, adventitial fibroblasts, and microglia. NOX1 requires subunits to be active. As seen with probably all NOX enzymes, there are spatially distinct mechanisms of receptor-mediated NOX1 activation and generation of ROS, confirming that compartmentalisation is necessary for integration of redox-dependent signaling. Interestingly, endocytosis is required for receptor-mediated signalling in smooth muscle cells [20].
NOX3 is highly expressed in the inner ear, in both the vestibular and auditive components. Like NOX1 and NOX2, activation is subunit-dependent [21].
NOX4 is expressed strongly in the kidney and is present in all vascular cells, fibroblasts, osteoclasts, and neurons. NOX4 is primarily found within intracellular organelles (e.g. ER, nuclear envelope, and possibly also mitochondria). NOX4 stands apart from the rest of the family since it appears to be constitutively active and its activity is therefore primarily regulated by level of expression. The predominant ROS detected from NOX4 is H2O2, but possibly the primary product is O2–, which is rapidly dismutated to H2O2.
NOX5 is highly expressed in the lymphoid tissue and the testis. It has been reported to be found in the cytoskeleton, endoplasmic reticulum, and plasma membrane. The enzyme differs from other NOX enzymes by its additional cytosolic N-terminal regulatory domain. It is activated by elevations of cytosolic free calcium ions (Ca2+) and by phosphorylation [22].
DUOX1 and DUOX2 are especially important in the thyroid, though they are also found in the epithelia of the respiratory and digestive tracts. Like NOX5, DUOX enzymes are activated by Ca2+. They have seven trans-membrane domains and are highly glycosylated. As seen for NOX4, chiefly hydrogen peroxide is detected but this is probably due to rapid dismutation of superoxide [23].
Many cells and virtually all tissues express several NOX isoforms. Expression of NOX enzymes is regulated on a transcriptional level, both under physiological and under pathological conditions.
Natural defence mechanisms
The natural defense against ROS consists of antioxidant enzymes and of antioxidant scavengers (fig. 3). The nuclear transcription factor Nrf2 is the master regulator of gene expression of antioxidant enzymes [24]. Three superoxide dismutases, differing in their subcellular location, catalyse the reaction of superoxide into oxygen and hydrogen peroxide. Catalase decomposes hydrogen peroxide to water. Thioredoxins, which also consist of several isoforms differing in subcellular localisation, enable the reduction of oxidised proteins by cysteine thiol-disulfide exchange. Glutathione peroxidases reduce lipid hydroperoxides to alcohols and reduce hydrogen peroxide to water; glutathione synthetase is responsible for synthesis of the major cellular antioxidant glutathione and therefore also has an important role in ROS detoxification. Peroxiredoxins control cytokine-induced peroxide levels, thereby affecting signal transduction. Antioxidant scavengers are predominantly of dietary origin. These biomolecules include tocopherol (vitamin E), ascorbic acid (vitC), carotenoids, uric acid, and polyphenols.
ROS in health
At low, regulated levels, ROS are involved in many vital physiological processes. They have a role in various signaling cascades, such as response to growth factor stimulation and control of inflammatory responses [25]. They participate in the regulation of many cellular processes, including differentiation, proliferation, growth, apoptosis, cytoskeletal regulation, migration, and contraction [2].
Immune function
Normal immune function requires specific oxidative states. ROS are necessary for microbial killing, for limiting the specific immune response, and for inflammation termination. The evidence for ROS involvement in immune function comes in part from chronic granulomatous disease (CGD) patients. CGD is caused by a lack of the ROS-generating phagocyte NADPH oxidase NOX2. Notably, CGD and the associated lack of ROS leads to immunodeficiency associated with recurrent infections, including pneumonia, abscesses, and osteomyelitis. In response to stimulation, phagocytes of CGD patients do not generate ROS. This is problematic for host defense because macrophages and neutrophils must generate ROS to efficiently kill the bacteria through phagocytosis. Hypochlorous acid is approximately 50 times more potent in microbial killing than hydrogen peroxide; myeloperoxidase catalyses the conversion of hydrogen peroxide and chloride ions into hypochlorous acid [26]. ROS have a vital role in bacterial, fungal, and microbial killing; their role in protection against viruses is not clear.
Recent evidence highlights a central role of the NOD-like receptor (NLR) family in the initiation of the inflammatory responses. Due to its association with numerous inflammatory diseases, the NLRP3 inflammasome has drawn the most attention. A wide variety of danger signals (exogenous, endogenous or damages associated with specific molecula pathways) activate the NLRP3 inflammasome. All these NLRP3 activators lead to ROS generation. In turn, increased levels of ROS are essential for NLRP3 activation. The source of ROS was initially thought to be the NADPH oxidases. However, macrophages deficient in different NOX isoforms (such as NOX1, NOX2 and NOX4) respond normally. This suggests either the presence of a compensatory mechanism by the remaining NADPH oxidase members, or the existence of a different cellular source of inflammasome-activating ROS: the mitochondria [27].
ROS are not only important in host defence, but paradoxically also limit the inflammation and immune response [28]. Thus, there is ample evidence of non-infectious hyperinflammatory conditions in CGD patients, particularly granulomas and colitis [28]. In addition, autoimmune diseases are also associated with lack of ROS generation by the NOX2 complex. Indeed, not only CGD patients but also heterozygous carriers of CGD mutations show an increased frequency of autoimmune diseases. Hyperinflammation and increased immune activation due to NOX2 deficiency might sometimes be beneficial; for instance, CGD mice show that increased inflammation protects against pulmonary infection from influenza [29]. The fact that deficiency of NOX2-dependent ROS generation is associated with enhanced inflammation is surprising and merits further study.
Thyroid function
Another example of the importance of ROS in health has been revealed by patients with a rare form of hypothyroidism [30]. Hydrogen peroxide is a necessary cofactor for thyroperoxidase, the enzyme participating in a final step of hormone production. For years, thyroid researchers had been actively looking for an enzyme that produces hydrogen peroxide in an NADPH-dependent manner. Notably, this is another example where the ROS-generating function was described long before the responsible NOX protein and its structure were discovered. It is now clear that DUOX2 (and probably also DUOX1) is the enzyme that generates the hydrogen peroxide required for thyroid peroxidase function; this theory is well supported by the existence of congenital hypothyroid patients with mutations in the DUOX2 gene [31].
Cognitive function
The evidence for ROS involvement in cognitive function comes in large part from CGD patients. While many of these patients are intelligent and work in demanding professions, on average they show decreased cognitive function, with 23% of patients having an IQ of 70 or below [32]. One hypothesis suggests that the deficit is due to chronic illness, which affects growth, development, and education, but no decrease in cognitive function is found in cystic fibrosis patients who have similar rates of infection and hospitalisation [33]. In CGD mouse models, the cognitive defect is reproduced even if the animals are kept under specific pathogen-free conditions [34]. Indeed, there is evidence for a role of ROS in neuronal apoptosis, a key mechanism in brain development [35]. However, no abnormalities in brain structure have yet been observed in CGD patients or in CGD mice. Thus, it is tempting to speculate that ROS have a role in the function of brain cells or in CNS cell-to-cell communication. For example, there is evidence for regulation of neuronal ion channels, kinases, and transcription factors by ROS [36] [28]. NOX2-derived ROS generation also contributes to long-term potentiation and memory function [37]. A key point in understanding of the involvement of ROS in cognitive functions is the molecular and functional link between NOX enzymes and glutamatergic neurotransmission and, in particular, with the ionotropic NMDA receptor (NMDA-R). Indeed, NOX enzymes may regulate the NMDA-R, through regulation of the redox potential. In turn, the NMDA-R may regulate NOX enzymes. Hence, activation of the NMDA-R seems to activate NOX2, leading to ROS production in neurons. Recent evidence also shows that ketamine, an antagonist of the NMDA-R, increases NOX2 activity [35].
Modulation of nutrient sensors and aging
Aging is a physiological process, defined as a series of time-dependent physiological changes that reduce physiological reserve and functional capacity [38]. In eukaryotic cells, this process is regulated by several factors such as the "target of rapamycin" (TOR), a nutrient-sensing protein kinase [39], and the "AMP-activated kinase" (AMPK), a conserved sensor of increased levels of AMP and ADP originating from ATP depletion [40]. Expression and activation of these two factors are finely modulated by ROS, both in physiological and pathological processes [41, 42]. Also, the mitochondrial free radical theory of aging proposes that aging is caused by damage to macromolecules by ROS. However, recent findings suggest that ROS generation is not the primary or initial cause of aging. Thus, it has been proposed that ROS modulate the aging process mediating the stress response to age-dependent damage [43]. Further investigations are required to understand better the mechanisms and the specific targets underlying the positive effects of ROS on the aging process.
ROS in disease
ROS contribute to a wide range of pathologies and many of the implicated diseases are leading causes of death (fig. 4). Cancers, cardiovascular diseases, and neurological diseases all show robust evidence for ROS involvement (table 1).

Figure 4
Overproduction of ROS and its contribution to various disease families.
Cancer
Perhaps most obviously, the DNA damage inflicted by ROS contributes to the initiation and progression of carcinogenesis. ROS-dependent mutations in DNA include base modifications, rearrangement of DNA sequences, miscoding of DNA lesions, gene duplications, and activation of oncogenes [44].
ROS may not only contribute to cancer development through oncogenic mutations, but also through dysregulation, as in renal cell carcinoma. In this disease there is inappropriate upregulation of hypoxia response genes. Elevated levels of the hypoxia-inducible transcription factor (HIF-1α) contribute to tumour growth, angiogenesis, and metastasis [45]. In part, the high levels of HIF-1α in these tumours are explained by loss of the von Hippel-Lindau protein, which normally leads to HIF-1α degradation. Additionally, ROS provide a second signalling pathway to stabilise HIF-1α. Thus, the simultaneous loss of a tumour suppressor and generation of ROS leads to a major alteration of the post-translational processing of the tumour-relevant transcription factor HIF-1α.
|
Table 1: Selected ROS-mediated diseases. |
| Disease family |
Example disease |
Potential mechanisms of ROS involvement |
| Cancer |
Renal cell carcinoma |
HIF-1α expression induces vascular endothelial growth factor (VEGF), a mediator of angiogenesis, tumour growth and metastasis; NOX4 activity is required for HIF-1α expression.
Fumarate hydratase deficiency may induce hypoxia-inducible transcription factor stabilisation by glucose-dependent generation of ROS.
Obligate glycolytic switch is critical to HIF stabilisation via ROS generation.
Decrease in mitochondrial energy metabolism, upregulation of glycolysis.
von Hippel-Lindau tumour suppressor-deficient.
Cells exhibit upregulation of p22phox, NOX4, and NOX-mediated ROS generation.
Tumour cell growth is suppressed by DPI. |
| Cardiovascular |
Hypertension |
Superoxide reacts with .NO, forming peroxynitrite (ONOO–), causing a reduction in .NO bioavailability and endothelium-dependent vasodilation.
NOX4 is strongly expressed in media of small pulmonary arteries and is causally involved in development of pulmonary hypertension.
NOX-derived ROS are a hypertensive signalling element.
Decreased systolic BP response to angiotensin II and to bone morphogenetic protein-4 in p47phox-deficient mice.
Decreases in BP in NOX1-deficient mice. |
| Neurological |
Schizophrenia |
NOX2 involved in neurotransmitter release.
NOX2 contributes to changes in interneurons, including the loss of parvalbumin expression and the capacity to secrete GABA.
Oxidative stress may change the set of active transcription factors within GABAergic interneurons. |
Cardiovascular disease
ROS are involved in a large number of cardiovascular diseases and the causal mechanisms are complex. Vascular cells simultaneously express multiple NOX enzymes and various cardiovascular diseases have been associated with changes in expression of NOX1, 2, 4, and 5. In vascular smooth muscle cells from large arteries, NOX1 is required for migration, hypertrophy, and proliferation, NOX4 mediates differentiation, and NOX1 and 2 are implicated in hypertension. Here we discuss the examples of ischaemia/reperfusion injury and hypertension.
Ischaemia/reperfusion injury
Ischaemia/reperfusion refers to a situation where a tissue is deprived of oxygen for a long period of time, followed by an abrupt increase in oxygen concentrations – for instance, after thrombolysis in stroke and myocardial infarction or after surgical intervention. In all of these situations, a dysregulation of oxygen-using enzyme systems may occur during hypoxia, followed by transient overactivity during reperfusion. Recent research, both in animals and in humans, has shown that NOX enzymes are particularly relevant in this context.
In an in vivo human model of flow-mediated dilation to assess endothelial function of endothelial ischaemia/reperfusion (IR) injury, the response of healthy volunteers was compared with that of CGD patients. In controls, IR caused significant reduction in flow-mediated dilation, while in CGD patients IR had no effect on endothelial function. ROS produced by NOX are therefore determinants of endothelial function after IR injury [46].
Hypertension
There is abundant evidence that ROS contribute to the pathophysiology of hypertension and many mechanisms have been implicated. Notably, increased superoxide will lead to a reduction in bioavailability of the vasodilator nitric oxide. Further, ROS have been implicated in proliferation and hypertrophy of vascular smooth muscle cells, which leads to increased vascular resistance. There is genetic evidence from mice, rats, and humans to suggest a role of NOX enzymes in hypertension:
– NOX1-deficient mice have decreased basal and angiotensin II-stimulated blood pressure [47].
– Spontaneously hypertensive rats have a polymorphism in the promoter of the p22phox gene, which leads to an overexpression of this NOX subunit and a subsequent increase in ROS.
– Humans homozygous for a polymorphism in the gene encoding p22phox have reduced oxidative stress in the vascular system and probably also reduced blood pressure [48].
Neurological disease
ROS have a role in neurological disease progression, primarily through the expression of NOX enzymes in microglia cells. Other brain cells, such as astrocytes and neurons, also generate ROS by mechanisms only partially understood. While low ROS concentrations are required for brain function (see above), high ROS concentrations contribute to disease due to neurotoxicity [35, 49]. Alzheimer's disease is a good example of how a neuroinflammatory loop can contribute to neurodegeneration and dementia. Amyloid, particularly soluble forms, can lead to microglia activation and long-lasting ROS generation, contributing to neuronal damage and ultimately dementia. ROS also appears to have an important pathological role in the pathogenesis of another neurodegenerative disorder: Parkinson's disease [35]. Thus, NOX-derived oxidative stress is thought to be a key factor for the degeneration of dopaminergic neurons, both in patient studies, in animal and in vitro models of this disorder, such as the MPTP model. Interestingly, degeneration of dopaminergic neurons after administration of MPTP was attenuated in NOX2-deficient mice as compared with wild-type controls, suggesting the contribution of NOX2 in this process. Oxidative stress also plays a central role in the development of amyotrophic lateral sclerosis (ALS), a progressive degenerative disease affecting motor neurons. Although the primary cause of this pathology is largely unknown, point mutations in the SOD1 gene are associated with familial forms of ALS. Increased presence of markers of oxidative stress has been found in the spinal cord and other biological fluids (such as plasma and cerebrospinal fluid) of ALS patients, together with increased expression of NOX2 enzyme. A crucial role of NOX2 has been also reported in studies using the SOD1 animal models and the in vitro models (such as organotypic spinal cord slices) of this disease [35].
Sensory impairment
Many age-associated diseases of the eye, such as cataract and retinal degeneration, are thought to involve oxidative stress. Similarly, age-associated hearing loss is thought to be ROS-mediated disease. It is thus interesting to note that NOX3 is highly expressed in the inner ear, where it plays a physiological role in the biogenesis of otoliths, structures involved in gravity perception [50]. However, there is also evidence that increased NOX3 activity is associated with hearing loss; in particular cisplatin ototoxicity has been associated with overactivation of NOX3 donc [51].
Psychiatric disease
ROS have been implicated in several psychiatric diseases, including depression and autism. The most thoroughly studied example is schizophrenia, which illustrates yet more complicated and interesting roles for ROS.
For more than fifty years it has been known that there are signs of increased oxidative stress in the brain of schizophrenic patients. To understand the possible causative role of oxidative stress in the development of psychosis, animal models have been utilised, particularly that of social isolation animals who are deprived of stimuli that are critical to behavioral and neurobiological development [52]. Isolated animals have alterations in striatal dopamine function, abnormalities in the hippocampus and frontal cortex, and abnormal firing of pyramidal cells in the prefrontal cortex [53]. NOX2 is below detection level in the brains of control animals but highly expressed in isolated rats [52] [54]. Strikingly, when rats with a loss of function mutation in a NOX2 subunit are subjected to social isolation, the behavioral and histopathological alterations are prevented [55]. This indicates that NOX2-derived ROS are involved in the alterations after social isolation.
ROS and ROS-generating systems as drug targets
Antioxidants
In epidemiological studies, people consuming high levels of antioxidant-rich fruits and vegetables are in better health. Hence several antioxidant supplementation strategies have been tested in humans, based on the assumption that they will increase degradation of ROS and thereby reduce ROS-associated diseases. Notable treatments have included superoxide dismutase and mimetics, peroxidase and mimetics, Vitamins A, C, and E, coenzymeQ10, beta carotene, and bioflavoids. These clinical studies using antioxidant food supplements were largely disappointing [56]. In fact, the long-term health consequences of many supplements are dubious. As part of the Iowa Women's Health Study, vitamin and mineral supplements were assessed for their relationship to total mortality in more than 30 000 elderly women. The use of food supplements, including those with antioxidant activity (multivitamins, vitamin B6), was associated with increased risk of total mortality [57]. The only supplements found to decrease mortality risk were calcium and vitamin D, certainly through mechanisms unrelated to oxidative stress.
Thus, there is an apparent contradiction: on the one hand there is ample evidence for a role of ROS in various diseases, and antioxidant-rich food is generally associated with health, but antioxidant supplements do not prevent disease and are even associated with a poor health outcome. What are possible reasons for this contradiction? We hypothesise that antioxidant supplementation is too late, too little, and too non-specific:
1 Tissue concentrations that can be achieved with antioxidants might be far below levels required to counteract a ROS-generating system. The best example for this is thyroid function, where attaching an iodide to thyroglobulin requires hydrogen peroxide generated by the NOX enzyme DUOX2 (see above). An antioxidant strategy capable of efficiently counteracting the activity of DUOX2 would therefore lead to hypothyroidism, yet hypothyroidism in response to antioxidants has never been reported.
2 Antioxidants preferentially localise to subcellular compartments based on solubility. This is obviously not a problem with antioxidant of dietary origin (1 glass of red wine provides more than 1000 different compounds with antioxidant capacity). However, this is a limiting factor for single molecule supplements.
3 Antioxidant food supplements (e.g., vitamin C) may also have pro-oxidant activity under certain circumstances, typically upon interaction with ROS.
4 Antioxidants scavenge ROS after their production. They are incapable of preventing oxidation of molecules that have a very high affinity for ROS, such as nitric oxide which has an extremely rapid rate of reaction with superoxide.
5 Antioxidants are most effective in combating low levels of ROS generation, but have a limited capacity to reduce high levels.
On a more affirmative note, the antioxidant supplement N-acetyl cysteine has been successful in some clinical studies, e.g. with schizophrenia [58] and in pulmonary fibrosis [59]. However, N-acetyl cysteine is not a simple antioxidant since it replenishes cellular glutathione levels and feeds into an enzymatic detoxification system. This might be the reason why among all therapeutic "antioxidant strategies," the only one showing consistent, albeit modest, protective results is supplementation with high doses of N-acetyl cysteine.
Inhibitors of ROS generation
While antioxidants increase rates of ROS degradation, enzyme inhibitors decrease rates of production. Inhibitors of ROS production have the potential of specificity, as drugs can be designed to inhibit specific ROS-generating systems. This is of importance, since compounds that would completely inhibit ROS production could have fatal consequences (e.g., inhibition of thyroid function; inhibition of the mitochondrial respiratory chain).
NOX inhibitors
Although multiple sources of ROS contribute to disease development, the family of NADPH oxidases appears particularly important and thus NOX enzymes could be good drug targets. NOX isoforms are cell- and tissue-specific, have different modes of activation and serve distinct roles. When the concept of NOX inhibitors was developed, there was much hesitation because of possible "on-target" side effects (i.e. major problems linked to the inhibition of NOX enzymes in vivo). For example, inhibition of NOX2 was thought to be undesirable because it might lead to immunodeficiency. In the light of our present knowledge, however, these on-target effects are unlikely to be a major concern. Drugs never inhibit 100% of an enzyme's activity for 100% of the time and, taking the example of NOX2, recent results show that very small amounts of ROS generation (~0.1% of normal) already provide substantial protection against severe infection [60]. Also, NOX1 and NOX4 knock-out mice are doing well and have no obvious signs of disease, suggesting that pharmacological inhibition of these NOX isoforms should be well tolerated. However, DUOX2 inhibition would pose problems for thyroid function and therefore should be avoided.
First generation NOX inhibitors have been chiefly non-specific and too toxic for medical use. For example, the widely-used compound diphenyliodonium (DPI), not only inhibits all NOX enzymes, but also other electron transporters, such as xanthine oxidase, as well as the mitochondrial respiratory chain.
Currently a new generation of NOX inhibitors is being developed. These compounds achieve increasing specificity, not only discriminating from other electron transport systems, but also discriminating between isoforms within the NOX family. Also, based on animal experimentation, new NOX inhibitors have less "off-target" side effects (i.e. problems not related to inhibition of the targeted NOX isoform). Thus, NOX inhibition is a promising pharmacological approach.
Inhibitors of other ROS-metabolising systems
In addition to NOX enzymes, other ROS-metabolising systems can be targeted. For instance, clinical conditions such as gout have been managed with xanthine oxidase inhibitors for decades. Notably, the xanthine oxidase inhibitor allopurinol has been used in gout and other conditions presenting with hyperuricaemia; in recent trials, allopurinol has benefited patients suffering from vascular injuries, inflammatory diseases, and chronic heart failure [61]. Further, inhibitors of superoxide dismutase (SOD) have applications in the treatment of cancer [62] since malignant cells are highly dependent on SOD for antioxidant protection, though this approach merits careful study because SOD is also necessary in normal cells.
Conclusion
Recent decades have seen a surge of interest in the role of ROS in health and disease. From basic science research to clinical trials, the biomedical community has rapidly advanced toward a better understanding of ROS-metabolising systems and their contribution to specific conditions. In particular, identification of the NOX family of enzymes has opened an exciting avenue for research and targeted therapy [63].
Loss of function of NOX enzymes is rare, but causes clinically severe disease, such a chronic granulomatous disease (NOX2 deficiency) or certain forms of hypothyroidism (DUOX2 deficiency). Disease due to increased NOX function appears very common, notably cardiovascular and neurodegenerative diseases. In many instances the increased NOX activity is due to NOX activation by specific stimuli, as in the activation of microglial NOX2 by amyloid in Alzheimer's disease. However, it is likely that genetic variation within NOX genes may contribute to the extent of NOX activation in response to a stimulus. For example, this might explain why some individuals develop severe neurodegeneration despite a relatively low amyloid load.
Independent of whether the enhanced NOX activity is due to enzymatic activation, genetic predisposition or a combination, NOX inhibitors could be useful pharmaceutical tools. First-generation NOX inhibitors lack selectivity and potency, but new compounds are more specific and therefore of greater potential for clinical use.
NOX enzymes are only one of the relevant systems involved in regulation of redox processes. It is possible that several sources of ROS contribute to the same disease. Also, decreased antioxidant defense may mimic the effect of increased ROS generation and the predominant mechanism could vary depending on the genetic predisposition of an individual. Thus, individualised medicine may become an important approach in the treatment of oxidative stress-related diseases.
References
1 D'Autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8(10):813–24.
2 Krause KH. Aging: a revisited theory based on free radicals generated by NOX family NADPH oxidases. Exp Gerontol, 2007;42(4):256–62.
3 Krause KH, Bedard K. NOX enzymes in immuno-inflammatory pathologies. Semin Immunopathol. 2008;30(3):193–4.
4 Edens WA, Sharling L, Cheng G, Shapira R, Kinkade JM, Lee T, et al. Tyrosine cross-linking of extracellular matrix is catalyzed by Duox, a multidomain oxidase/peroxidase with homology to the phagocyte oxidase subunit gp91phox. J Cell Biol. 2001;154(4):879–91.
5 Warburg O. Beobachtungen über die Oxydationsprozesse im Seeigelei. Zeitschrift fur Physiologische Chemie. 1908;57:1–16.
6 Bedard K, Attar H, Bonnefont J, Jaquet V, Borel C, Plastre O, et al. Three common polymorphisms in the CYBA gene form a haplotype associated with decreased ROS generation. Hum Mutat. 2009;30(7):1123–33.
7 Harma D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11(3):298–300.
8 Qin B, Cartier L, Dubois-Dauphin M, Li B, Serrander L, Krause KH. A key role for the microglial NADPH oxidase in APP-dependent killing of neurons. Neurobiol Aging. 2006;27(11):1577–87.
9 Zhang Y, Dawson VL, Dawson TM. Oxidative stress and genetics in the pathogenesis of Parkinson's disease. Neurobiology of Disease. 2000;7(4):240–50.
10 Dhalla NS, Temsah RM, Netticadan T. Role of oxidative stress in cardiovascular diseases. Journal of Hypertension. 2000;18(6):655–73.
11 Wannamethee SG, Lowe GD, Rumley A, Bruckdorfer KR, Whincup PH. Associations of vitamin C status, fruit and vegetable intakes, and markers of inflammation and hemostasis. Am J Clin Nutr. 2006;83(3):567–74; quiz 726–7.
12 Guzik TJ, West NE, Black E, McDonald D, Ratnatunga C, Pillai R, et al. Vascular superoxide production by NAD(P)H oxidase: association with endothelial dysfunction and clinical risk factors. Circ Res. 2000;86(9):E85–90.
13 Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med. 2008;45(5):549–61.
14 Schieke SM, von Montfort C, Buchczyk DP, Timmer A, Grether-Beck S, Krutmann J, et al. Singlet oxygen-induced attenuation of growth factor signaling: possible role of ceramides. Free Radic Res. 2004;38(7):729–37.
15 Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82(1):47–95.
16 Wiseman H, Halliwell B. Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem J. 1996;313(Pt 1):17–29.
17 Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120(4):483–95.
18 Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: Physiology and Pathophysiology. Physiological Reviews. 2007;87(1):245–313.
19 Babior BM, Lambeth JD, Nauseef W. The neutrophil NADPH oxidase. Arch Biochem Biophys. 2002;397(2):342–4.
20 Miller FJ Jr, Chu X, Stanic B, Tian X, Sharma RV, Davisson RL, et al. A differential role for endocytosis in receptor-mediated activation of Nox1. Antioxid Redox Signal. 12(5):583–93.
21 Bánfi B, Malgrange B, Knisz J, Steger K, Dubois-Dauphin M, Krause KH. NOX3, a superoxide-generating NADPH oxidase of the inner ear. J Biol Chem. 2004:279(44):46065–72.
22 Bedard K, Jaquet V, Krause KH. NOX5: from basic biology to signaling and disease. Free Radic Biol Med. 2012;52(4):725–34.
23 De Deken X, Wang D, Many MC, Costagliola S, Libert F, Vassart G, Dumont JE, et al. Cloning of two human thyroid cDNAs encoding new members of the NADPH oxidase family. Journal of Biological Chemistry. 2000;275(30):23227–33.
24 Hybertson BM, Gao B, Bose SK, McCord JM. Oxidative stress in health and disease: the therapeutic potential of Nrf2 activation. Molecular Aspects of Medicine. 2011;32(4–6):234–46.
25 Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 194(1):7–15.
26 Mauch L, Lun A, O'Gorman MR, Harris JS, Schulze I, Zychlinsky A, et al. Chronic granulomatous disease (CGD) and complete myeloperoxidase deficiency both yield strongly reduced dihydrorhodamine 123 test signals but can be easily discerned in routine testing for CGD. Clin Chem. 2007;53(5):890–6.
27 Tschopp J. Mitochondria: Sovereign of inflammation? European Journal of Immunology. 2011;41(5):1196–202.
28 Schäppi MG, Jaquet V, Belli DC, Krause KH. Hyperinflammation in chronic granulomatous disease and anti-inflammatory role of the phagocyte NADPH oxidase. Semin Immunopathol. 2008;30(3):255–71.
29 Snelgrove RJ, Edwards L, Rae AJ, Hussell T. An absence of reactive oxygen species improves the resolution of lung influenza infection. European Journal of Immunology. 2006;36(6):1364–73.
30 Erdamar H, Demirci H, Yaman H, Erbil MK, Yakar T, Sancak B, et al. The effect of hypothyroidism, hyperthyroidism, and their treatment on parameters of oxidative stress and antioxidant status. Clin Chem Lab Med. 2008;46(7):1004–10.
31 Moreno JC, Bikker H, Kempers MJ, van Trotsenburg AS, Baas F, de Vijlder JJ, et al. Inactivating mutations in the gene for thyroid oxidase 2 (THOX2) and congenital hypothyroidism. N Engl J Med. 2002;347(2):95–102.
32 Pao M, Wiggs EA, Anastacio MM, Hyun J, DeCarlo ES, Miller JT, et al. Cognitive function in patients with chronic granulomatous disease: a preliminary report. Psychosomatics. 2004;45(3):230-4.
33 Thompson RJ Jr, Gustafson KE, Meghdadpour S, Harrell ES, Johndrow DA, Spock A. The role of biomedical and psychosocial processes in the intellectual and academic functioning of children and adolescents with cystic fibrosis. J Clin Psychol. 1992;48(1):3–10.
34 Kishida KT, Hoeffer CA, Hu D, Pao M, Holland SM, Klann E. Synaptic plasticity deficits and mild memory impairments in mouse models of chronic granulomatous disease. Mol Cell Biol. 2006;26(15):5908–20.
35 Sorce S, Krause KH. NOX enzymes in the central nervous system: from signaling to disease. Antioxid Redox Signal. 2009;11(10):2481–504.
36 Thomas MP, Chartrand K, Reynolds A, Vitvitsky V, Banerjee R, Gendelman HE. Ion channel blockade attenuates aggregated alpha synuclein induction of microglial reactive oxygen species: relevance for the pathogenesis of Parkinson's disease. J Neurochem. 2007;100(2):503–19.
37 Massaad CA Klann E. Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxid Redox Signal. 2011;14(10):2013–54.
38 Ahmed A, Tollefsbol T. Telomeres and telomerase: basic science implications for aging. Journal of the American Geriatrics Society. 2001;49(8):1105–9.
39 Kapahi P, Chen D, Rogers AN, Katewa SD, Li PW, Thomas EL, et al. With TOR, Less is more: a key role for the conserved nutrient-sensing TOR pathway in aging. Cell Metabolism. 2010;11(6):453–65.
40 Salminen A, Kaarniranta K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Research Reviews. 2012;11(2):230–41.
41 Park IJ, Hwang JT, Kim YM, Ha J, Park OJ. Differential modulation of AMPK signaling pathways by low or high levels of exogenous reactive oxygen species in colon cancer cells. Annals of the New York Academy of Sciences. 2006;1091(1):102–9.
42 Sandström ME, Zhang SJ, Bruton J, Silva JP, Reid MB, Westerblad H, et al. Role of reactive oxygen species in contraction-mediated glucose transport in mouse skeletal muscle. The Journal of Physiology. 2006;575(1):251–62.
43 Hekimi S, Lapointe J, Wen Y. Taking a "good" look at free radicals in the aging process. Trends in Cell Biology. 2011;21(10):569–76.
44 Waris G, Ahsan H. Reactive oxygen species: role in the development of cancer and various chronic conditions. J Carcinog. 2006;5:14.
45 Liao D, Corle C, Seagroves TN, Johnson RS. Hypoxia-inducible factor-1α is a key regulator of metastasis in a transgenic model of cancer initiation and progression. Cancer Res. 2007;67(2):563–72.
46 Loukogeorgakis SP, van den Berg MJ, Sofat R, Nitsch D, Charakida M, Haiyee B, et al. Role of NADPH oxidase in endothelial ischemia/reperfusion injury in humans. Circulation. 121(21):2310–6.
47 Gavazzi G, Banfi B, Deffert C, Fiette L, Schappi M, Herrmann F, et al. Decreased blood pressure in NOX1-deficient mice. FEBS Lett. 2006;580(2):497–504.
48 Wyche KE, Wang SS, Griendling KK, Dikalov SI, Austin H, Rao S, et al. C242T CYBA polymorphism of the NADPH oxidase is associated with reduced respiratory burst in human neutrophils. Hypertension. 2004;43(6):1246–51.
49 Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8(1):57–69.
50 Ueno N, Takeya R, Miyano K, Kikuchi H, Sumimoto H. The NADPH oxidase Nox3 constitutively produces superoxide in a p22phox-dependent manner: its regulation by oxidase organizers and activators. J Biol Chem. 2005;280(24):23328–39.
51 Bánfi B, Malgrange B, Knisz J, Steger K, Dubois-Dauphin M, Krause KH. NOX3, a superoxide-generating NADPH oxidase of the inner ear. J Biol Chem. 2004;279(44):46065–72.
52 Schiavone S, Sorce S, Dubois-Dauphin M, Jaquet V, Colaianna M, Zotti M, et al. Involvement of NOX2 in the development of behavioral and pathologic alterations in isolated rats. Biol Psychiatry. 2009;66(4):384–92.
53 Pascual R, Zamora-Leon SP, Valero-Cabre A. Effects of postweaning social isolation and re-socialization on the expression of vasoactive intestinal peptide (VIP) and dendritic development in the medial prefrontal cortex of the rat. Acta Neurobiol Exp (Wars). 2006;66(1):7–14.
54 Tucci P, Morgese MG, Colaianna M, Zotti M, Schiavone S, Cuomo V, et al. Neurochemical consequence of steroid abuse: stanozolol-induced monoaminergic changes. Steroids. 2012;77(3):269–75.
55 Schiavone S, Jaquet V, Sorce S, Dubois-Dauphin M, Hultqvist M, Bäckdahl L, et al. NADPH oxidase elevations in pyramidal neurons drive psychosocial stress-induced neuropathology. Transl Psychiatry. 2012;2(5):e111.
56 Stanner SA, Hughes J, Kelly CN, Buttriss J. A review of the epidemiological evidence for the 'antioxidant hypothesis'. Public Health Nutr. 2004;7(3):407–22.
57 Mursu J, Robien K, Harnack LJ, Park K, Jacobs DR Jr. Dietary supplements and mortality rate in older women: the Iowa Women's Health Study. Arch Intern Med. 2011;171(18):1625–33.
58 Berk M, Copolov D, Dean O, Lu K, Jeavons S, Schapkaitz I, et al. N-acetyl cysteine as a glutathione precursor for schizophrenia – a double-blind, randomized, placebo-controlled trial. Biol Psychiatry. 2008;64(5):361–8.
59 Lassegue B. Griendling KK. NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol. 2012;30(4):653–61.
60 Kuhns DB, Alvord WG, Heller T, Feld JJ, Pike KM, Marciano BE, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. 2010;363(27):2600–10.
61 Pacher P, Nivorozhkin A, Szabo C. Therapeutic effects of xanthine oxidase inhibitors: renaissance half a century after the discovery of allopurinol. Pharmacol Rev. 2006;58(1):87–114.
62 Huang P, Feng L, Oldham EA, Keating MJ, Plunkett W. Superoxide dismutase as a target for the selective killing of cancer cells. Nature. 2000;407(6802):390–5.
63 Streeter J, Thiel W, Brieger K, Miller Jr FJ . Opportunity nox: the future of NADPH oxidases as therapeutic targets in cardiovascular disease. Cardiovasc Ther. 2012. DOI 10.1111.