Heart rate, coronary artery disease and plaque rupture – myth, hype, or truth?
DOI: https://doi.org/10.4414/smw.2012.13661
Atherosclerosis – the facts?
Since the end of the 19th century, coronary artery disease (CAD) became and remained the N°1 serial killer in Europe and in most industrialised countries. Because no curative treatment of CAD exists, we strive indefatigably to seek and combat CAD risk factors. This quest began in 1849 when Virchow speculated that atherosclerosis was caused by plasmatic risk factors. Yet, our understanding of the “atherogenesis” has continued to evolve and the current understanding can be summarised as follows: the plaque grows over the years under the influence of risk – as well as protective factors. At a certain time point, the plaque erodes or ruptures. The term plaque “vulnerability” has been coined to reflect the decisive period leading to erosion or rupture [1].
In disciples of Vischow, the majority of the incriminated risk factors (>270 listed by Hopkins and Williams) [2] are biochemical: diabetes mellitus, smoking, dyslipidaemia, chronic renal failure, homocystinuria, C-reactive protein, lipoprotein (a), tissue plasminogen activator, small and dense LDL, fibrinogen, or even age. These factors explain the pathophysiology of plaque growth according to its main components as found at autopsy. Each of these distinct factors has been demonstrated as risk factor in numerous observational studies and epidemiological intervention studies. For example, the persistence of active smoking in patients after myocardial infarction double the 10-year mortality [3], while its cessation restores a normal life expectancy beginning after 36 months [4]. The same holds true for the impact of adequate control of diabetes mellitus, arterial hypertension and dyslipidaemia. These risk factors exist and their inadequate control (as primary and secondary prevention) significantly impacts CAD morbidity and mortality (In the National Health and Nutrition Examination Survey NHANES, patients with >2 insufficiently controlled risk factors have a 5.7-fold increase in mortality at 13-year follow-up) [5].
However and although we rely on many predictive scores integrating several risk factors (i.e., ESC or AGLA scores), the prediction of myocardial infarction or sudden cardiac death remains unsure. Whence this discrepancy? The uncertainty comes from two observations: First, while risk factors for atherosclerotic plaque growth are well known and can be translated into animal models, risk factors for plaque vulnerability and rupture are more difficult to gather. With no question, the Holy Grail remains to identify vulnerable plaque at risk of one particular cardiovascular event. Therefore, the quest continues and the risk factors for rupture are actively investigated.
Risk factors for rupture have been categorised into “intrinsic” or “endogenous” and “extrinsic” or “exogenous”. Intrinsic risk factors are multiple and only scarcely known. These intrinsic factors are mainly associated with hypoxic, inflammatory and thrombogenic stimuli. On the plaque-level, these factors trigger an increased activity of special enzymes excreted by macrophages (matrix metalloproteinases-MMPs), surge neovascularisation with the presence of leaky microvessels that provoke intraplaque haemorrhage and allow subsequent deposition of free cholesterol, macrophage infiltration and enlargement of the necrotic core [6]. On a patient-level, these factors are more often found in patients with diabetes mellitus, smoking habit, hypercoagulable/hypofibrinolytic states and high circulating tissue factor. Based on the most up-to-date researches, some biomarkers of vulnerability have been identified. Such examples are F2-isoprostanes, hs-CRP, urinary microalbumin, myeloperoxidase, or lipoprotein-associated phospholipase A2 . These biomarkers are however difficult to measure at the bedside since gradient up to 1000-fold are found between blood harvested at plaque vicinity and peripheral blood [7]. The second observation is that symptomatic plaque rupture often happens in particular situations. These even more complex triggering elements on cardiovascular events are usually considered as extrinsic risk factors. In most cases, these factors significantly influence the vessel shear stress and the local procoagulable state via multiple biological parameters, such as adrenergic activity, blood pressure, heart rate, blood viscosity, or platelet activity. During the last decade, a high resting heart rate has been postulated to be an extrinsic risk factor for plaque rupture.
Heart rate and atherosclerosis – the myth and the hype?
High resting heart rate – as cardiovascular risk factor – is motivated by biogerontological considerations in mammals, epidemiological evidence in humans, and indirect and direct observations in animals, and humans. The evidence was extensively summarised in various review articles during last decade’s hype. Briefly, a high resting heart rate is associated with an increased cardiovascular mortality [8]. In line with this concept, the pure heart rate lowering agent ivabradine has been associated with a significant relative risk reduction of myocardial infarction (36%, p = 0.001) in patients with CAD, decreased left ventricular function (<40%) and high resting heart rate (>70 bpm) [9].
Yet, what is new in this issue of the journal? Angela Kohler and colleagues [10] analysed the correlation between resting heart rate and myocardial perfusion single-photon emission computed tomography (MPS) findings in 1465 consecutive naïve patients (= after exclusion of patients with already known CAD, non sinus rhythm, and patients under beta-blockers or calcium channel blockers). The novelty is that the authors demonstrate that resting heart rate was not associated with CAD extent as assess by MPS. The methodology is solid, the results are clear, and are clearly in contradiction with the general feeling. For instance, the results are apparently directly in contraindication with a similar paper from Williams and colleagues [11]. In this latter study, the authors found in 3708 patients that resting heart rate was strongly and independently associated with stress-induced myocardial perfusion defects. So, where is the truth? It is too early to give a definitive answer, but some aspects should be considered: Firstly, the two studies are not mutually exclusive. Indeed, Williams et al. showed that although the correlation was strong in the whole population and in patients under beta-blocker therapy, the association was weaker (and no more significant, p = 0.11) in non beta-blocker users. This is of relevance and in accordance to the findings of Kohler et al. since none of the 1465 patients were under beta-blocker treatment. The corollary of this first remark is however that the risk due to an increased resting heart rate is highest in patients who escape beta-blocker treatment; a point that was already underlined by Barrios and colleagues who suggest that patients with higher heart rate were associated with lessen risk control rates in daily clinical practice [12]. Secondly, the study touches a raw nerve by demonstrating that the analysis of stable patients does not allow identifying vulnerable patients. Undeniably, one would expect to have a good correlation between CAD extent and a putative risk factor when the factor is associated with plaque growth. But, plaque vulnerability is not similar to CAD extent since on one hand plaque rupture may be immediately fatal, whereas on the other hand (and in most cases) it is asymptomatic and does not cause any haemodynamically significant stenosis. In light with this, it is interesting to note that some patients with minimal atherosclerotic changes could suffer sudden cardiac death. This is the case for instance in marathon runners [13]. Therefore, CAD extent as attests by haemodynamic significant stenosis might be similar between patients with and patients without a particular risk factor for plaque instability. Therefore, the question remains: “How to identify the vulnerable patient?”.
Emotion and plaque rupture – the truth?
The present study appropriately diverts our attention from the resting heart rate as single vulnerability risk factor. However, the truth is that intensive physical or emotional stresses are very potent risk factors for plaque rupture, but considering only heart rate is a too simplistic explanation to explain complex pathophysiological mechanisms that constantly interplay. History is full of examples showing the importance of such extrinsic risk factors: For two millennia, the courier Pheidippides collapsed and died at the end of a day-long run in the city of Marathon [14]. For one century, Strazhesko acknowledged emotion as trigger to myocardial infarction in the very first scientific publication on myocardial infarction [15]. Since then, numbers of case reports, observational and epidemiological studies revealed the effect of positive (happiness) or negative (depression, fear, pain) emotions on plaque vulnerability/rupture, acute coronary syndromes and sudden cardiac death [16–21]. Striking examples might be found in (almost) everyday practice [22] and happen during domestic crisis, driving, sport, watching TV, earthquakes, or even coitus [fortunately, exceedingly rare and almost exclusively encountered in extramarital experience!]. These different examples point to the fact that mental stress is a particularly potent extrinsic risk factor for plaque rupture. Mental stress has been shown to be per se a key factor since it has been linked to several components of plaque instability: increased sympathetic tone, blood pressure, heart rate and vascular shear stress; increased viscosity, fibrinogen and von Willebrand factor levels, platelet activity and several inflammatory mediators.
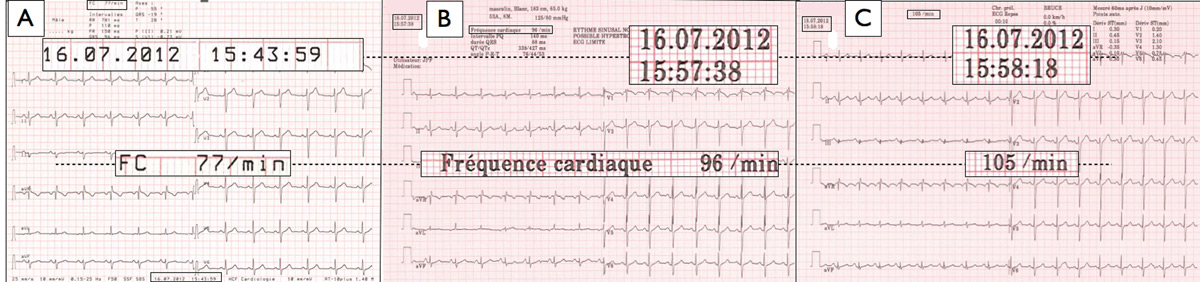
Figure 1
Example of the effect of mental stress due to exercise testing on “resting” heart rate. A 53 year old male with normal physical status was considered for exercise testing. His resting heart rate – as measured by a 30 seconds palpation the right radial pulse – was 72 beats per minute (bpm). Panel A depicts his “resting” 12-lead ECG recording made 15 minutes before initiation of exercise testing on a treadmill ergometer. Heart rate was 77 bpm. Panels B and C show his “baseline” ECGs as acquired on the treadmill ergometer 2 minutes (panel B), and 1 minute (panel C) before the test start. Heart rate was 96 bpm, and 105 bpm respectively.
In light with this consideration, the study of Kohler and colleagues once again turns our attention to the somewhat neglected issue of what is a “resting” or “baseline” heart rate? The authors considered that the heart rate prior to the stress test was similar to the “resting” heart rate. This is of course a very pragmatic postulation and the majority of us will report this value in ambulatory patients. However, the cumulative impact of “white coats", performance anxiety and fear of the subsequent findings induces a mental stress that reaches a climax by the recording of this baseline value. An illustration is depicted in (fig. 1). This “white coat” mental stress has been demonstrated in athletes, medical students and common ambulatory patients. As the response to mental stress is individual and is associated with plaque vulnerability, the variation of the heart rate from the resting to the baseline might be possibly used as forecast to predict plaque rupture. This area is speculative and should be further investigated. Nevertheless, the clinician must continue to pay attention to heart rate in clinical practice since it is cheap, easily performed and very useful. Is it really a warning about an increased risk of coronary event? Signals indicate this direction. So, why not continue using it since emerging modalities like biochemical test cocktails (“CVD inflammatory profile tests”), functional imaging (MRI or infrared) or structural intravascular imaging (OCT, OFDI) are very expensive, hardly available, and still clearly immature.
Finally and as suggested by Kohler and colleagues, heart rate cannot be used to wager CAD extent, especially in low-risk patients. To determine CAD extent, only coronary angiography, SPECT, stress echocardiography or MRI should be considered. Inversely, the study from Kohler and colleagues can be used in order to relax patients in stress due to high resting heart rate. Additionally and based on the current evidence, it seems that risk factors for plaque rupture and acute cardiac events are short-term. Conventional studies are not appropriate for isolating triggers for cardiac events, mainly because acute coronary syndrome is a storm that warrants timely revascularisation, and such instant exposures are likely to be hidden by established (plaque-growth) factors, as well as cognitive barriers (in case of anger, fear or sexual activities). In light of this ephemeral nature, research in the field remains challenging. Who knows, perhaps, only the integration of advanced neuropsychological research will enable us to render acute cardiac events into an orphan disease.
References
1 Cook S, Hess OM. Resting heart rate and cardiovascular events: time for a new crusade? Eur Heart J. 2010;31(5):517–9.
2 Hopkins PN, Williams RR. Identification and relative weight of cardiovascular risk factors. Cardiol Clin. 1986;4(1):3–31.
3 Aberg A, Bergstrand R, Johansson S, Ulvenstam G, Vedin A, Wedel H, et al. Cessation of smoking after myocardial infarction. Effects on mortality after 10 years. British heart journal. 1983;49(5):416-22.
4 Rea TD, Heckbert SR, Kaplan RC, Smith NL, Lemaitre RN, Psaty BM. Smoking status and risk for recurrent coronary events after myocardial infarction. Ann Intern Med. 2002;137(6):494–500.
5 Qureshi AI, Suri MF, Kirmani JF, Divani AA. The relative impact of inadequate primary and secondary prevention on cardiovascular mortality in the United States. Stroke; a journal of cerebral circulation. 2004;35(10):2346–50.
6 Finn AV, Nakano M, Narula J, Kolodgie FD, Virmani R. Concept of vulnerable/unstable plaque. Arterioscler. Thromb. Vasc. Biol. 2010;30(7):1282–92.
7 Cook S, Ladich E, Nakazawa G, Eshtehardi P, Neidhart M, Vogel R, et al. Correlation of intravascular ultrasound findings with histopathological analysis of thrombus aspirates in patients with very late drug-eluting stent thrombosis. Circulation. 2009;120:i (5):391–9.
8 Cook S, Togni M, Schaub MC, Wenaweser P, Hess OM. High heart rate: a cardiovascular risk factor? Eur Heart J. 2006;27(20):2387–93.
9 Fox K, Ford I, Steg PG, Tendera M, Ferrari R. Ivabradine for patients with stable coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): a randomised, double-blind, placebo-controlled trial. Lancet. 2008;372(9641):807–16.
10 Kohler A, Muzzarelli S, Leibundgut G, Brand J-M, Brinkert M, Zellweger MJ. Relationship between the resting heart rate and the extent of coronary artery disease as assessed by myocardial perfusion. Swiss Med Wkly. 2012;142:w13660.
11 Williams BA, Merhige ME. The prognostic association between resting heart rate and cardiac death-myocardial perfusion defects as a potential mechanism. Atherosclerosis. 2012;221(2):445–50.
12 Barrios V, Escobar C, Bertomeu V, Murga N, de Pablo C, Asin E. High heart rate: more than a risk factor. Lessons from a clinical practice survey. Int J Cardiol. 2009;137(3):292–4.
13 Mittleman MA, Maclure M, Tofler GH, Sherwood JB, Goldberg RJ, Muller JE. Triggering of acute myocardial infarction by heavy physical exertion. Protection against triggering by regular exertion. Determinants of Myocardial Infarction Onset Study Investigators. N Engl J Med. 1993;329(23):1677–83.
14 Frost FJ. The dubious srigins of the marathon. American Journal of Ancient History. 1979;4:159–63.
15 Obraztsov VP, Strazhesko ND. The symptomatology and diagnosis of coronary thrombosis. In: Vorobeva VA, Konchalovski MP (eds). Works of the first Congress of Russian Therapists Comradeship Typography of AE Moscow: Mamontov; 1910. p. 26–43.
16 Moller J, Hallqvist J, Diderichsen F, Theorell T, Reuterwall C, Ahlbom A. Do episodes of anger trigger myocardial infarction? A case-crossover analysis in the Stockholm Heart Epidemiology Program (SHEEP). Psychosomatic medicine. 1999;61(6):842–9.
17 Masoomi M, Zare J, Kahnooj M, Mirzazadeh A, Sheikhvatan M. Sex differences in potential daily triggers of the onset of acute myocardial infarction: a case-crossover analysis among an Iranian population. J Cardiovasc Med. 2010;11(10):723–6.
18 Muller JE, Mittleman MA, Maclure M, Sherwood JB, Tofler GH. Triggering myocardial infarction by sexual activity. Low absolute risk and prevention by regular physical exertion. Determinants of myocardial infarction onset study investigators. JAMA. 1996;275(18):1405–9.
19 Baylin A, Hernandez-Diaz S, Siles X, Kabagambe EK, Campos H. Triggers of nonfatal myocardial infarction in Costa Rica: heavy physical exertion, sexual activity, and infection. Ann Epidemiol. 2007;17(2):112–8.
20 Zheng ZJ, Croft JB, Giles WH, Mensah GA. Sudden cardiac death in the United States, 1989 to 1998. Circulation. 2001;104(18):2158–63.
21 von Kanel R. Psychosocial stress and cardiovascular risk: current opinion. Swiss Med Wkly. 2012;142:0.
22 Kalberer L, Wahl P, Cook S. Dislocated artificial hip as trigger for ST-elevation myocardial infarction. Cardiovasc Med. 2012;15(10):in press.