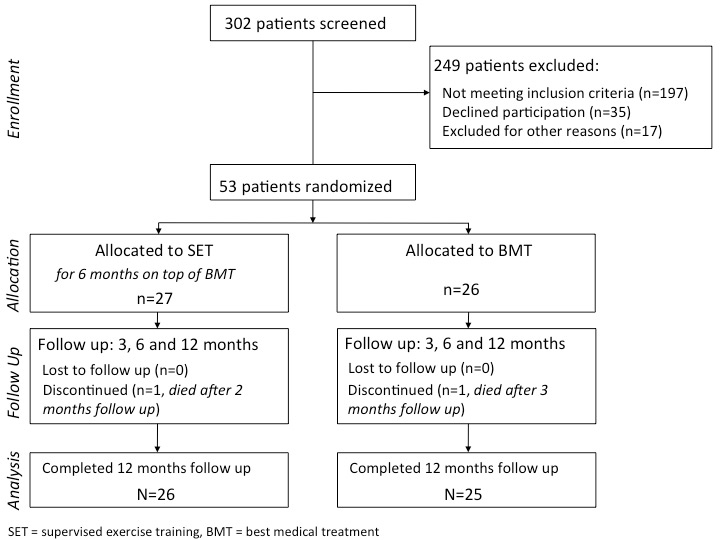
Figure 1
CONSORT flow diagram. * Reasons for exclusion: 15 patients were enrolled in another clinical trial and two patients deteriorated prior to randomization and underwent revascularization.
DOI: https://doi.org/10.4414/smw.2012.13623
Atherosclerosis can be regarded as inflammatory disease of the vascular system and causes thrombotic cardiovascular events [1, 2]. Cardiovascular risk factors promote the atherosclerotic process by altering endothelial properties leading to increased adhesiveness for leukocytes and platelets. These cellular interactions and the subsequent development of atherosclerotic lesions are catalyzed by the release of vasoactive molecules, cytokines and growth factors [3]. Previous studies have demonstrated that inflammatory serum-markers reflect the activity of the atherosclerotic process and might further contribute to platelet aggregation [4, 5]. Thus, in patients with advanced atherosclerosis, such as peripheral arterial disease (PAD), inflammatory serum-markers may predict the occurrence of future cardiovascular adverse events [6–8]. Accordingly, markers of platelet activation were linked to cardiovascular risk in high-risk populations [9].
Patients with PAD face a high risk for cardiovascular adverse events [10]. Thus, a comprehensive therapeutic concept is recommended to reduce cardiovascular morbidity and mortality [11]. Focusing on PAD patients with intermittent claudication (IC) recent data have demonstrated that supervised exercise training (SET) effectively increases walking capacity [12, 13]. However, it remains unclear whether SET could additionally contribute to cardiovascular risk reduction, which is a major therapeutic goal in PAD patients. Regarding patients with coronary artery disease it has been shown that exercise training affects markers of inflammation and platelet activation as surrogates of cardiovascular risk [14, 15]. Comparably, in PAD patients SET might have favorable effects on inflammation and platelet activation and might thereby affect cardiovascular risk. However, data on the impact of SET on markers of inflammation and platelet activation in patients with PAD are scarce [13].
Therefore, we aimed to investigate whether a regular SET program – as it can be provided in a real world setting on top of best medical treatment (BMT) – has favorable effects on markers of inflammation and platelet activation in patients with PAD.
Consecutive PAD patients with IC, who were admitted to our institution were screened for this single center, randomized controlled trial. PAD was diagnosed upon an ankle brachial index (ABI) below 0.9 of the symptomatic lower limb. The clinical inclusion criterion was symptomatic PAD with mild to severe claudication (Rutherford category I–III) lasting longer than two weeks. Asymptomatic patients as well as patients with critical limb ischemia were not included. Further exclusion criteria comprised a reduced exercise tolerance due to other limitations than IC, in particular coronary artery disease, congestive heart failure, dyspnea, uncontrolled blood pressure, any kind of restriction of the musculoskeletal system, which might have had an influence on the efficiency of exercise training.
Moreover, we excluded patients receiving non-steroidal anti-inflammatory drugs, immunosuppressives or cortisone as well as patients with systemic immunological disorders or malignant diseases.
Prior to inclusion, all patients underwent routine laboratory tests and a complete clinical examination to exclude coincident infectious diseases. As previously published [12], patients were randomized using computer generated random digits in sealed envelopes (block-wise randomization by 2) to either SET for six months on top of best medical treatment or to best medical treatment (BMT) only.
The study was performed according to the recommendations of the Declaration of Helsinki and after approval of the institutional review board. Ahead of inclusion written informed consent was obtained from all patients.
The present manuscript was structured according to the CONSORT-checklist ( http://www.consort-statement.org ), which is given in the Appendix.
All patients received BMT according to current guidelines [11, 16]: except for patients who were on oral anticoagulation for other reason than PAD, at least one antiplatelet agent was administered in all patients. Further, BMT comprised administration of statins, blood pressure control as well as an antidiabetic medication to achieve the recommended treatment goals [11, 16]. Finally, all patients were provided with detailed information on PAD and risk factor management and educative brochures on life style modification with respect to exercise and dietary habits.
Patients randomized to SET attended a twice-weekly, physiotherapist-guided training program over a period of six months. According to previously published recommendations patients underwent a warm up period of 5 to 10 minutes [17]. The warm up was followed by intermittent walking training over 35 minutes. This initial duration was increased by 5 minutes each session until 50 minutes of intermittent walking was accomplished. Patients were trained at a walking speed that elicits claudication symptoms within 3 to 5 minutes which was then followed by a brief resting period to allow symptoms to resolve.
At baseline, after 3, 6 and 12 months in both groups maximum walking distance (MWD) was determined by standardized treadmill exercise tests using a motorized treadmill with a constant speed of 3.2 km per hour and a 12-degree slope [11, 18]. Two experienced medical technicians, who were blinded to the assigned treatment arm and otherwise not involved in the study, supervised treadmill exercise tests. Patients were instructed to report discomfort occurring during the examination, which was recorded by the attending technician using standardized questionnaires. Treadmill exercise tests were terminated at patients´ request when their symptoms forced them to stop and the MWD was recorded.
Blood pressure was determined from patients’ blood pressure protocols as the average of serial home blood pressure measurements.
After inclusion whole blood cell counts, lipid profile and glycated hemoglobin (HbA1C) were measured. Inflammatory serum markers as well as markers for platelet activation were determined from venous blood samples at baseline, after 3, 6 and 12 months in both groups. Regarding patients undergoing SET blood samples were drawn at least three days after the last exercise training session. All assays were performed by blinded individuals.
High sensitivity C-reactive protein (hs-CRP) was determined using a high sensitivity assay (N Latex CRP Mono®, DADE Behring) with a detection level of 0.03 mg/dl and a coefficient of variation of 4.6%. Interleukin-6 (IL-6) was measured by commercially available immunoassays according to the manufacturers’ instructions (R&D, HS600B). The minimum level of detection of IL-6 was 0.039 pg/ml. Fibrinogen was determined according to Clauss using STA Fibrinogen (Diagnostica Stago) with a detection level of 20 mg/dl and a coefficient of variation of 3.0%.
Further, prothrombin fragment 1+2 (F1.2) and soluble P-Selectin (sP-sel) were determined using Dade Behring, Enzygnost F1.2 (F1.2) and BenderMedSystems BMS219/4 (sP-sel), respectively. The minimum levels of detection were 20 pmol/l for F1.2 and 0.2 ng/ml for sP-sel.
Whole-blood flow cytometry was applied as described previously with little modification [19]. In brief, 100 μL of citrate-anticoagulated whole blood was stained with saturating concentrations of the following fluorochrome-conjugated monoclonal antibodies (mAbs): APC- labeled mAb for the constitutive platelet marker CD42b (glycoprotein Ib of von Willebrand factor receptor complex), PE-Cy5–labeled mAb for monocyte CD14 (endotoxin receptor), and corresponding isotype controls (all antibodies were purchased from Becton Dickinson). After 10 minutes of preincubation with antibodies in the dark at room temperature, samples were fixed and erythrolysed with Optilyse B (instrumentation Laboratories). Flow cytometry was performed on a FACSCalibur (Becton Dickinson). Acquisition was stopped when 5 000 CD14+ events were acquired (Instrumentation Laboratories).
Monocytes were identified by gating CD14+ events, and all additional analyses were performed on this population. The negative and positive delineators were set by gating ~2% background staining on the isotype control fluorescence. The percentage of monocyte platelet aggregates (MPA) characterised by the relative number of monocytes coexpressing the constitutive platelet marker CD42b (CD14+/CD42b+%) was determined.
Categorical data are presented as number (percentage) and were compared by chi-square test. Between group comparisons were performed by Mann-Whitney-U Test. Friedman test for distribution free, repeated-measures analysis of variance was used to test for significant changes within the groups over time. When a significant time effect was identified, the Wilcoxon signed rank test was used for post hoc analysis. All statistical tests were two-sided with a significance level of 5%. SAS 9.2 was used as analytical software.
As we present data from an exploratory analysis of the impact of SET on inflammatory and prothrombotic markers, no sample size calculation was performed for these outcome measures.
Between November 2007 and November 2010 fifty-three patients were randomly assigned either to SET on top of BMT or to BMT only (fig. 1). Patients’ baseline characteristics were similar in both groups and are given in table 1. One patient of the SET-group died from an acute respiratory infection two months after study entry and one patient of the BMT-group died from myocardial infarction three months after study entry. Data on all patients were analyzed on an intention to treat basis.
Figure 1
CONSORT flow diagram. * Reasons for exclusion: 15 patients were enrolled in another clinical trial and two patients deteriorated prior to randomization and underwent revascularization.
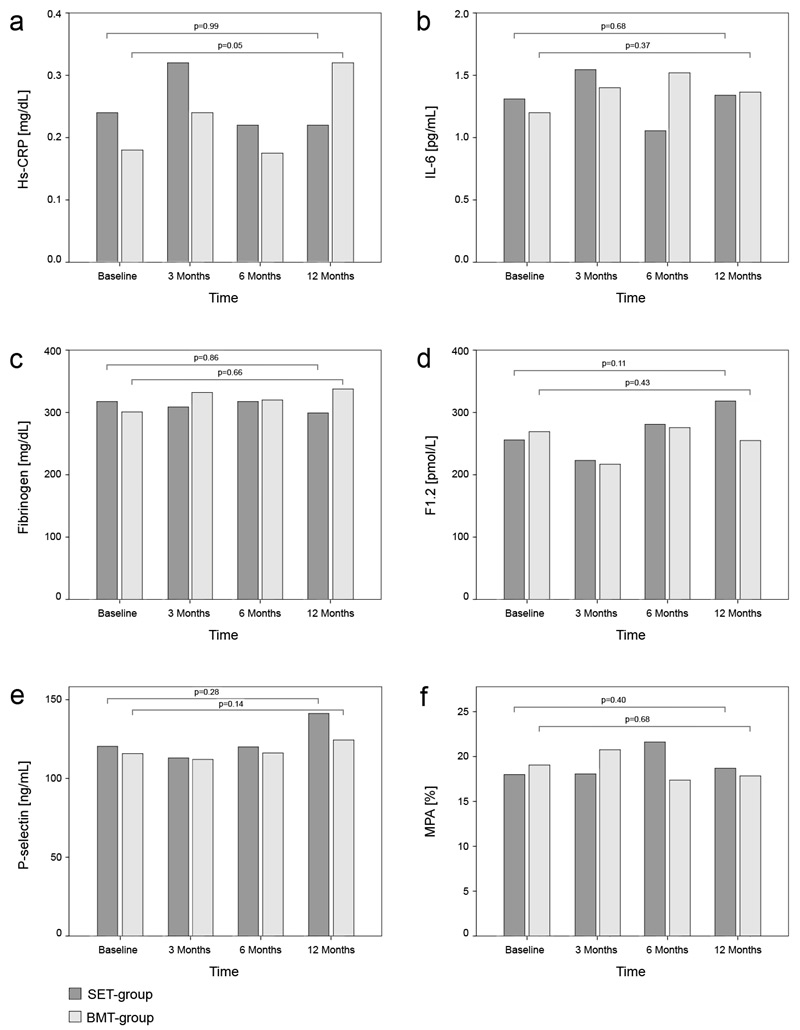
Figure 2
Bar chart indicating median values of markers of high sensitivity C-reactive protein (hs-CRP), interleukin-6 (IL-6), fibrinogen, soluble P-selectin, prothrombin fragment 1+2 (F1.2) and monocyte-platelet aggregates (MPA) at baseline, after three, six and twelve months. Friedman test for distribution free, repeated-measures analysis of variance was used to test for significant changes within the groups over time (p-values).
All included patients received statin treatment through the entire study period – except one patient with statin intolerance in each group. At baseline median (IQR) low density lipoprotein (LDL)-cholesterol was 2.6 (2.1; 2.9) mmol/l in the SET-group and 2.6 (1.8; 3.4) mmol/l in the BMT-group. Regarding the course of LDL-cholesterol during the study we observed no significant change in both groups (SET vs. BMT): median LDL-cholesterol after 3 months 2.2 (1.7; 2.9) mmol/l vs. 2.4 (1.8; 3.1) mmol/l, after 6 months 2.2 (1.6; 3.0) mmol/l vs. 2.3 (1.7; 3.1) mmol/l and after 12 months 2.2 (1.8; 3.4) mmol/l vs. 2.2 (1.7; 2.7) mmol/l.
Considering arterial hypertension we also found no significant change of blood pressure values in both groups over the course of the study (SET vs. BMT): median systolic/diastolic blood pressure (IQR) at baseline 141/80 (126/73,158/84) mm Hg vs. 145/78 (125/70,160/90) mm Hg, after 3 months 139/75 (121/70,153/80) mm Hg vs. 146/80 (130/70,168/88) mm Hg, after 6 months 126/75 (121/70,154/80) mm Hg vs. 153/80 (139/70,168/81) mm Hg and after 12 months 137/80 (123/70,156/84) vs. 143/80 (131/75,159/80) mm Hg.
Among study participants with diabetes the median (IQR) HbA1C was 6.0 (5.5; 6.8) % in the SET-group and 6.0 (5.8; 6.6)% in the BMT-group at baseline. During follow up we found no significant change of HbA1C in both groups (SET vs. BMT): median (IQR) HbA1C after 3 months 6.0 (5.8; 6.6) vs. 6.1 (5.7; 6.8)%, after 6 months 6.1 (5.8; 6.7) vs. 6.3 (5.8; 6.9)% and after 12 months 6.1 (5.7; 6.5) vs. 6.4 (5.7; 6.8)%.
In the SET-group the median MWD increased from 101.5 (IQR 65.0; 168.0) to 115.0 (96.0; 178.0) meters after 3 months (p = 0.015) and to 132.5 (99.0; 175.0) meters after 6 months (p = 0.012). After 12 months we observed a sustained increase of MWD (177.0 meters; IQR 97.0,231.0 meters) when compared with baseline (p = 0.002). In the BMT-group the MWD did not change significantly during follow up: median MWD at baseline 84.5 (IQR 50.0; 150.0), after 3 months 104.0 (IQR 50.0; 170.0) meters, after 6 months 100.0 (72.5; 182.5) meters and after 12 months 100.0 (50.0; 140.0) meters.
Hs-CRP, IL-6 and fibrinogen did not differ significantly between the SET-group and the BMT-group at baseline (table 2). Analyzing the course of hs-CRP, IL-6 and fibrinogen within each group, we observed no significant change of the respective markers in either group (fig. 2a-c). Serum levels of hs-CRP, IL-6 and fibrinogen did not differ significantly between both groups after 3, 6 and 12 months (table 2).
At study entry we found no significant differences of sP-sel, MPA and F1.2 between the SET- and the BMT-group (table 3). Regarding markers of platelet activation and procoagulant markers within each group over the study period, we observed no significant changes of sP-sel, MPA and F1.2, neither in the SET-group nor in the BMT group (fig. 2d-f). During the course of the study we observed no differences between both groups with respect to serum levels of F1.2, sP-sel and MPA (table 3).
| Table 1: Demographic and clinical data of 27 Patients randomized to supervised exercise training (SET) on top of best medical treatment (BMT) versus 26 patients receiving BMT only. Part of presented data were published previously [15]. | ||
| SET plus BMT | BMT only | |
| N | 27 | 26 |
| Age (years) | 68.4 ± 7.5 | 70.7 ± 10.6 |
| Male (N) | 18 | 15 |
| Ankle brachial index (mainly affected limb) | 0.63 ± 0.17 | 0.53 ± 0.20 |
| Maximum walking distance (m) | 101.5 (65.0; 168.0) | 84.5 (50.0; 150.0) |
| Smoking history (N) | 19 | 14 |
| Diabetes mellitus (N) | 9 | 12 |
| Coronary artery disease (N) | 11 | 12 |
| Cerebrovascular disease (N) | 7 | 5 |
| BMI (kg/m2) | 25.8 (24.1; 29.2) | 27.8 (23.2; 30.8) |
| HbA1c (patients with and without diabetes) (%) | 6.0 (5.5; 6.8) | 6.0 (5.8; 6.6) |
| LDL-cholesterol (mmol/l) | 2.6 (2.1; 2.9) | 2.6 (1.8; 3.4) |
| HDL-cholesterol (mmol/l) | 1.2 (1.0; 1.4) | 1.3 (1.2; 1.5) |
| Serum amyloid A (mg/dl) | 6.5 (3.8; 8.6) | 6.4 (3.8; 10.0) |
| Serum creatinine (mg/dl) | 1.11 (0.98;1.23) | 1.14 (0.98;1.23) |
| Red blood cell count (*1012/l) | 4.5 (4.3; 4.9) | 4.6 (3.9; 4.8) |
| White blood cell count (*109/l) | 7.7 (7.3; 8.7) | 6.8 (5.8; 7.9) |
| Platelets (*109/l) | 208 (175; 251) | 223 (176; 243) |
| Aspirin (N) | 24 | 21 |
| Clopidogrel (N) | 13 | 8 |
| Statins (N) | 26 | 25 |
| ACE|-Inhibitors (N) | 18 | 16 |
| Angiotensin receptor blockers (N) | 6 | 6 |
| Calcium channel blockers (N) | 11 | 6 |
| Beta Blockers (N) | 10 | 7 |
| BMI = body mass index; HbA1c = glycated hemoglobin A1; LDL = low density lipoprotein; HDL = high density lipoprotein; ACE = angiotensin converting enzyme. Data are shown as number, mean ± standard deviation (SD), or median plus interquartile range (IQR). For all group comparisons p >0.1. | ||
| Table 2: Inflammatory markers of 27 patients receiving supervised exercise training (SET) versus 26 patients receiving BMT only. | ||||
| Time | SET group | BMT group | p-value | |
| Hs-CRP (mg/dl) | Baseline | 0.24 (0.09; 0.64) | 0.18 (0.11; 0.35) | 0.48 |
| 3 Months | 0.32 (0.07; 0.44) | 0.24 (0.14; 0.36) | 0.63 | |
| 6 Months | 0.22 (0.08; 0.41) | 0.18 (0.09; 0.32) | 0.51 | |
| 12 Months | 0.22 (0.09; 0.56) | 0.32 (0.21; 0.55) | 0.40 | |
| IL-6(pg/ml) | Baseline | 1.31 (0.42; 2.91) | 1.20 (0.68; 2.71) | 0.76 |
| 3 Months | 1.55 (0.99; 3.26) | 1.40 (0.66; 2.48) | 0.22 | |
| 6 Months | 1.06 (0.65; 2.18) | 1.52 (0.65; 2.47) | 0.80 | |
| 12 Months | 1.34 (0.75; 2.34) | 1.37 (0.93; 2.56) | 0.52 | |
| Fibrinogen(mg/dl) | Baseline | 397 (349; 435) | 376 (339; 425) | 0.57 |
| 3 Months | 386 (331; 424) | 415 (361; 453) | 0.18 | |
| 6 Months | 397 (319; 442) | 400 (312; 445) | 0.81 | |
| 12 Months | 374 (326; 431) | 422 (354; 440) | 0.31 | |
| Data are shown as median plus interquartile range (IQR). | ||||
| Table 3: Prothrombotic markers of 27 patients receiving supervised exercise training (SET) versus 26 patients receiving BMT only. | ||||
| Time | SET group | BMT group | p-value | |
| F1.2 (pmol/l) | Baseline | 255.91 (155.74; 326.45) | 269.24 (169.98; 434.65) | 0.48 |
| 3 Months | 223.03 (161.18; 414.53) | 216.92 (155.32; 297.95) | 0.67 | |
| 6 Months | 280.91 (181.39; 377.74) | 275.76 (148.34; 356.82) | 0.54 | |
| 12 Months | 274.93 (173.81; 420.90) | 254.96 (171.37; 276.30) | 0.50 | |
| P-selectin (ng/ml) | Baseline | 120.11 (88.66; 185.88) | 115.78 (70.64; 168.59) | 0.43 |
| 3 Months | 112.79 (90.32; 163.75) | 112.08 (70.26; 193.25) | 0.80 | |
| 6 Months | 119.76 (90.78; 156.21) | 116.20 (71.01; 218.84) | 0.76 | |
| 12 Months | 140.96 (88.62; 175.26) | 120.90 (83.48; 190.25) | 0.64 | |
| MPA (%) | Baseline | 18.0 (15.2; 21.3) | 19.1 (13.0; 21.9) | 0.74 |
| 3 Months | 17.9 (14.4; 23.2) | 20.8 (14.0; 25.8) | 0.42 | |
| 6 Months | 21.1 (17.8; 24.3) | 17.4 (13.2; 22.6) | 0.15 | |
| 12 Months | 18.7 (13.5; 22.1) | 17.8 (15.1; 22.3) | 0.95 | |
| Data are shown as median plus interquartile range (IQR). | ||||
In the present study we found that SET on top of BMT had no additional effect on markers of inflammation and platelet activation in patients with PAD. Recently, a randomized controlled trial has demonstrated that SET effectively improves walking performance in patients with IC and thereby might even be superior over stent revascularization [13]. Similarly, we observed a significant increase of MWD in patients with PAD undergoing regular SET. Apart from symptom improvement, however, reduction of cardiovascular risk is a major therapeutic goal in patients with PAD. In PAD cardiovascular risk can be predicted by serum markers, which are sensitive for low-grade inflammation and thereby mirror the activity of the atherosclerotic process [6, 7, 20]. Notably, the inflammatory process resulting from atherosclerosis has to be differentiated from primarily immunological inflammatory vascular diseases [21]. Regarding the atherosclerotic process the release of inflammatory cytokines promotes the recruitment of monocytes and T-cells to the vessel wall [1]. Subsequently, immigrated leukocytes secrete further cytokines, which provoke proliferation of smooth muscle cells of the arterial vessel wall [2]. Within the spectrum of cytokines IL-6 stimulates hepatic production of CRP and fibrinogen [2, 22]. Thus, both, CRP and fibrinogen, potentially predict disease progression in patients with advanced atherosclerosis [6, 7].
In patients with coronary manifestation of atherosclerosis physical exercise has shown beneficial effects on surrogate markers of inflammation [23]. Importantly, the exercise induced effect on inflammation seems to depend on intensity of exercise training: strenuous exercise initially causes oxidative stress as well as muscle injury and interferes with intramuscular signaling which increases the release of chemokines, most notably IL-6 [24]. In the long term, however, it has been shown that endurance training effectively contributes to a decline of inflammatory activity [25]. Regarding our findings, the lack of response of inflammatory markers to SET might partly be attributed to the overall low intensity of exercise-induced strain in the respective study cohort. As previously published, SET was not accompanied by a substantial increase of heart rate [12], which might indicate a low level of overall physical strain during SET. According to current recommendations for exercise training in PAD the duration of training sessions was set to 50 minutes comprising sequences of intermittent walking that elicited claudication symptoms within 3 to 5 minutes [11]. Notably, recommendations of SET for PAD focus on a gain in walking capacity by exercising the muscles of the lower limbs that are affected by PAD. Such a training concept succeeded in gaining MWD, but the overall amount of physical strain might have been insufficient to impact inflammatory activity. Consequently, to affect inflammation and platelet activation a more comprehensive training concept involving the entire muscular system might be required.
Apart from muscular strain other mechanisms have been linked to the potential impact of exercise on inflammatory activity: especially multidisciplinary rehabilitation programs, which additionally aim at weight reduction have shown to yield anti-inflammatory effects [23]. It has been demonstrated that adipocytes, especially from omental and subcutaneous adipose tissue, are mainly involved in IL-6 production [26, 27]. Therefore, the potential influence of exercise on inflammatory serum markers might at least partly be attributed to a reduction of adipose tissue upon training. However, in the present study training sessions primarily aimed at a gain of pain-free walking distance rather than weight reduction, which might further explain the missing change of inflammatory markers.
Regarding clinical measures blood pressure could have been controlled more strictly. Focusing on serum lipids and glucose control, however, a considerable number of study participants met recommended targets of cardiovascular risk factor control before study entry. Further, all patients – apart from one subject with statin intolerance in each group – were on statin therapy. Hence, the potential anti-inflammatory effect of statins might explain the low baseline levels of inflammatory serum markers in both groups (SET and BMT). Although it previously has been shown that exercise potentially yields an additional anti-inflammatory effect in hypercholesterolemic subjects when provided on top of statin treatment, the low baseline levels of both, LDL-cholesterol and inflammatory markers, might have made it difficult to further decrease these markers by training [28]. Further, PAD can be regarded as systemic disease or, better still, as clinical manifestation of advanced atherosclerosis, where it might be difficult to achieve a significant reduction of low-grade inflammation by a twice-weekly training program.
As inflammation has been linked to activation of platelet activation in the atherosclerotic process we further investigated the impact of SET on sP-sel, MPA and F1.2 in PAD patients [5]. P-selectin is an adhesion molecule mainly stemming from platelet a-granules upon platelet activation [29, 30]. Platelet surface expression of P-selectin facilitates heterogenic aggregate formation with leukocytes and monocytes [31], which leads to monocyte activation. Subsequent monocytic tissue factor expression contributes to a prothrombotic state and might trigger generation of F1.2 by cleavage of thrombin [32]. Increased F1.2 levels might not only reflect a prothrombotic state, but seem to be associated with carotid intima media thickness as a surrogate marker of atherosclerosis and cardiovascular risk [33]. In addition, both sP-sel and MPA have also been reported to predict future cardiovascular events [29, 31]. However, similar to the course of inflammatory markers, we found no significant change of these parameters in response to training.
According to the association of inflammatory activity and platelet activation the lacking effect of SET on platelet activation in the present study might mainly be attributed to the absence of an inflammatory response upon SET. Furthermore, the comprehensive medication including an anti-thrombotic therapy might have attenuated the interference of SET and platelet activity. Additionally, the predefined period of three days between the last training session and blood withdrawal has to be kept in mind for interpretation of our results. Even though a shorter time period between training sessions and taking blood might have revealed short-term changes of respective serum markers, we aimed to investigate long-term effects of SET on inflammation and platelet activation, which might better reflect a sustained impact of SET on cardiovascular risk of PAD patients.
We are aware of the moderate sample size of the present study. However, previous studies on exercise training in patients with coronary artery disease and congestive heart failure were able to demonstrate statistically relevant effects on inflammatory biomarkers utilizing similar sample sizes [34, 35]. Within the spectrum of randomized controlled trials on SET in PAD the majority of studies comprised sample sizes between 20 and 49 patients [36, 37]. Further, in a recently published multi-center study, which was conducted in 22 study centers, 119 patients were randomized after screening of 999 patients over a time period of four-years [13]. This mirrors the required endeavors to implement a SET-program in both clinical trials and routine. Infrastructural feasibility of regular SET as well as acceptance by patients and doctors might limit the implementation of training programs as a comprehensive therapeutic strategy for PAD patients. In addition, several patients had to be excluded due to co-morbidities, such as cardiac, respiratory or musculoskeletal restrictions.
A limitation of the present study might be the chosen frequency of training sessions. Although three sessions per week are recommended by current guidelines [11, 16], previous studies have demonstrated a substantial increase of walking distance upon a twice-weekly regimen [37, 38]. Since our aim was to ensure patients´ adherence we set up a twice-weekly training regimen. To further enhance compliance of included patients training groups were limited to a maximum size of six patients per group over the entire SET period. Therefore, the feasibility of this SET-program was limited by local resources in terms of time and personnel of physiotherapists at our institution. Both, the twice-weekly training regimen as well as the high-quality physiotherapeutic supervision in small groups might have conjointly contributed to the excellent adherence of patients to the SET program. However, we cannot exclude that a higher training frequency might have had an impact on inflammation and platelet activation.
Further, we have to acknowledge that we did not track home-based activities of included patients. However, all study participants – irrespective of the allocated treatment group – received the same information on cardiovascular risk factor modification as well as the same educative brochures on life style modification including exercise training and dietary habits.
SET resulted in a gain of walking capacity in PAD, however, further studies are required to assess whether a more comprehensive training concept might additionally affect markers of inflammation and platelet activation as surrogates of cardiovascular risk.
In PAD patients SET – when provided on top of best medical treatment – effectively improved claudication symptoms but failed to reduce markers of inflammation and platelet activity as surrogates for cardiovascular risk.
| CONSORT 2010 checklist of information to include when reporting a randomised trial*. | ||||
| Section/topic | Item No | Checklist item | Reported in the article | |
| Title and abstract | ||||
| 1a | Identification as a randomised trial in the title. | Not applicable | ||
| 1b | Structured summary of trial design, methods, results, and conclusions (for specific guidance see CONSORT for abstracts). | See 'Summary' | ||
| Introduction | ||||
| Background and objectives | 2a | Scientific background and explanation of rationale. | See 'Introduction' | |
| 2b | Specific objectives or hypotheses. | See 'Introduction' | ||
| Methods | ||||
| Trial design | 3a | Description of trial design (such as parallel, factorial) including allocation ratio. | See 'Study design' | |
| 3b | Important changes to methods after trial commencement (such as eligibility criteria), with reasons. | Not applicable | ||
| Participants | 4a | Eligibility criteria for participants. | See 'Study design' | |
| 4b | Settings and locations where the data were collected. | See 'Study design' | ||
| Interventions | 5 | The interventions for each group with sufficient details to allow replication, including how and when they were actually administered. | See 'Best medical treatment (BMT)' and 'Supervised exercise training' | |
| Outcomes | 6a | Completely defined pre-specified primary and secondary outcome measures, including how and when they were assessed. | See 'Clinical measurements' and 'Laboratory measurements' | |
| 6b | Any changes to trial outcomes after the trial commenced, with reasons. | Not applicable | ||
| Sample size | 7a | How sample size was determined. | See 'Statistical analysis' | |
| 7b | When applicable, explanation of any interim analyses and stopping guidelines. | Not applicable | ||
| Randomisation: | ||||
| Sequence generation | 8a | Method used to generate the random allocation sequence. | See 'Study design' | |
| 8b | Type of randomisation; details of any restriction (such as blocking and block size). | See 'Study design' | ||
| Allocation concealment mechanism | 9 | Mechanism used to implement the random allocation sequence (such as sequentially numbered containers), describing any steps taken to conceal the sequence until interventions were assigned. | See 'Study design' | |
| Implementation | 10 | Who generated the random allocation sequence, who enrolled participants, and who assigned participants to interventions. | See 'Study design' | |
| Blinding | 11a | If done, who was blinded after assignment to interventions (for example, participants, care providers, those assessing outcomes) and how. | Not applicable | |
| 11b | If relevant, description of the similarity of interventions. | Not applicable | ||
| Statistical methods | 12a | Statistical methods used to compare groups for primary and secondary outcomes. | Not applicable | |
| 12b | Methods for additional analyses, such as subgroup analyses and adjusted analyses. | See 'Statistical analysis' | ||
| Results | ||||
| Participant flow (a diagram is strongly recommended) | 13a | For each group, the numbers of participants who were randomly assigned, received intended treatment, and were analysed for the primary outcome. | See fig. 1 | |
| 13b | For each group, losses and exclusions after randomisation, together with reasons. | See fig. 1 | ||
| Recruitment | 14a | Dates defining the periods of recruitment and follow-up. | See 'Results' | |
| 14b | Why the trial ended or was stopped. | Not applicable | ||
| Baseline data | 15 | A table showing baseline demographic and clinical characteristics for each group. | See table 1 | |
| Numbers analysed | 16 | For each group, number of participants (denominator) included in each analysis and whether the analysis was by original assigned groups. | See fig. 1 | |
| Outcomes and estimation | 17a | For each primary and secondary outcome, results for each group, and the estimated effect size and its precision (such as 95% confidence interval). | See table 2 and table 3 | |
| 17b | For binary outcomes, presentation of both absolute and relative effect sizes is recommended. | Not applicable | ||
| Ancillary analyses | 18 | Results of any other analyses performed, including subgroup analyses and adjusted analyses, distinguishing pre-specified from exploratory. | See 'Results' | |
| Harms | 19 | All important harms or unintended effects in each group (for specific guidance see CONSORT for harms). | See 'Results' | |
| Discussion | ||||
| Limitations | 20 | Trial limitations, addressing sources of potential bias, imprecision, and, if relevant, multiplicity of analyses. | See 'Discussion' | |
| Generalisability | 21 | Generalisability (external validity, applicability) of the trial findings. | See 'Discussion' | |
| Interpretation | 22 | Interpretation consistent with results, balancing benefits and harms, and considering other relevant evidence. | See 'Discussion' | |
| Other information | ||||
| Registration | 23 | Registration number and name of trial registry. | NCT00926081 | |
| Protocol | 24 | Where the full trial protocol can be accessed, if available. | Not applicable | |
| Funding | 25 | Sources of funding and other support (such as supply of drugs), role of funders. | No funding | |
| * We strongly recommend reading this statement in conjunction with the CONSORT 2010 Explanation and Elaboration for important clarifications on all the items. If relevant, we also recommend reading CONSORT extensions for cluster randomised trials, non-inferiority and equivalence trials, non-pharmacological treatments, herbal interventions, and pragmatic trials. Additional extensions are forthcoming: for those and for up to date references relevant to this checklist, see http://www.consort-statement.org . | ||||
Acknowledgements: The authors acknowledge the excellent support of the physiotherapists Nadine Graszler, Barbara Mick and Cornelia Hein who instructed the patients through the entire SET program.
1 Lusis AJ. Atherosclerosis. Nature. 2000;407(6801):233–41.
2 Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105(9):1135–43.
3 Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med. 1999;340(2):115–26.
4 Blake GJ, Ridker PM. Novel clinical markers of vascular wall inflammation. Circ Res. 2001;89(9):763–71.
5 Danenberg HD, Kantak N, Grad E, Swaminathan RV, Lotan C, Edelman ER. C- reactive protein promotes monocyte-platelet aggregation: an additional link to the inflammatory-thrombotic intricacy. Eur J Haematol. 2007;78(3):246–52.
6 Schillinger M, Exner M, Amighi J, Mlekusch W, Sabeti S, Rumpold H, et al. Joint effects of C-reactive protein and glycated hemoglobin in predicting future cardiovascular events of patients with advanced atherosclerosis. Circulation. 2003;108(19):2323–8.
7 Sabeti S, Exner M, Mlekusch W, Amighi J, Quehenberger P, Rumpold H, et al. Prognostic impact of fibrinogen in carotid atherosclerosis: nonspecific indicator of inflammation or independent predictor of disease progression? Stroke. 2005;36(7):1400–4.
8 Luc G, Bard JM, Juhan-Vague I, Ferrieres J, Evans A, Amouyel P, et al. C-reactive protein, interleukin-6, and fibrinogen as predictors of coronary heart disease: the PRIME Study. Arterioscler Thromb Vasc Biol. 2003;23(7):1255–61.
9 Varughese GI, Patel JV, Tomson J, Blann AD, Hughes EA, Lip GY. Prognostic value of plasma soluble P-selectin and von Willebrand factor as indices of platelet activation and endothelial damage/dysfunction in high-risk patients with hypertension: a sub-study of the Anglo-Scandinavian Cardiac Outcomes Trial. J Intern Med. 2007;261(4):384–91.
10 Caro J, Migliaccio-Walle K, Ishak KJ, Proskorovsky I. The morbidity and mortality following a diagnosis of peripheral arterial disease: long-term follow-up of a large database. BMC Cardiovasc Disord. 2005;5:14.
11 Hirsch AT, Haskal ZJ, Hertzer NR, Bakal CW, Creager MA, Halperin JL, et al. ACC/AHA 2005 guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): executive summary a collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease) endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; and Vascular Disease Foundation. J Am Coll Cardiol. 2006;47(6):1239–312.
12 Schlager O, Giurgea A, Schuhfried O, Seidinger D, Hammer A, Groger M, et al. Exercise training increases endothelial progenitor cells and decreases asymmetric dimethylarginine in peripheral arterial disease: A randomized controlled trial. Atherosclerosis. 2011;217(1):240–8.
13 Murphy TP, Cutlip DE, Regensteiner JG, Mohler ER, Cohen DJ, Reynolds MR, et al. Supervised exercise versus primary stenting for claudication resulting from aortoiliac peripheral artery disease: six-month outcomes from the claudication: exercise versus endoluminal revascularization (CLEVER) study. Circulation. 2012;125(1):130–9.
14 Pamukcu B, Oflaz H, Acar RD, Umman S, Koylan N, Umman B, et al. The role of exercise on platelet aggregation in patients with stable coronary artery disease: exercise induces aspirin resistant platelet activation. J Thromb Thrombolysis. 2005;20(1):17–22.
15 Lara Fernandes J, Serrano CV Jr., Toledo F, Hunziker MF, Zamperini A, Teo FH, et al. Acute and chronic effects of exercise on inflammatory markers and B-type natriuretic peptide in patients with coronary artery disease. Clin Res Cardiol. 2011;100(1):77–84.
16 Tendera M, Aboyans V, Bartelink ML, Baumgartner I, Clement D, Collet JP, et al. ESC Guidelines on the diagnosis and treatment of peripheral artery diseases: Document covering atherosclerotic disease of extracranial carotid and vertebral, mesenteric, renal, upper and lower extremity arteries: The Task Force on the Diagnosis and Treatment of Peripheral Artery Diseases of the European Society of Cardiology (ESC). Eur Heart J. 2011;32(22):2851–906.
17 Stewart KJ, Hiatt WR, Regensteiner JG, Hirsch AT. Exercise training for claudication. N Engl J Med. 2002;347(24):1941–51.
18 Cachovan M, Rogatti W, Creutzig A, Diehm C, Heidrich H, Scheffler P, et al. Treadmill testing for evaluation of claudication: comparison of constant-load and graded-exercise tests. Eur J Vasc Endovasc Surg. 1997;14(4):238–43.
19 Steiner S, Speidl WS, Pleiner J, Seidinger D, Zorn G, Kaun C, et al. Simvastatin blunts endotoxin-induced tissue factor in vivo. Circulation. 2005;111(14):1841–6.
20 Wilhelmsen L, Svardsudd K, Korsan-Bengtsen K, Larsson B, Welin L, Tibblin G. Fibrinogen as a risk factor for stroke and myocardial infarction. N Engl J Med. 1984;311(8):501–5.
21 Kesten F, Aschwanden M, Gubser P, Glatz K, Daikeler T, Hess C. Giant cell arteritis – a changing entity. Swiss Med Wkly. 2011;141:w13272.doi: 10.4414/smw.2011.13272.
22 Tzoulaki I, Murray GD, Lee AJ, Rumley A, Lowe GD, Fowkes FG. C-reactive protein, interleukin-6, and soluble adhesion molecules as predictors of progressive peripheral atherosclerosis in the general population: Edinburgh Artery Study. Circulation. 2005;112(7):976–83.
23 Milani RV, Lavie CJ, Mehra MR. Reduction in C-reactive protein through cardiac rehabilitation and exercise training. J Am Coll Cardiol. 2004;43(6):1056–61.
24 Kasapis C, Thompson PD. The effects of physical activity on serum C-reactive protein and inflammatory markers: a systematic review. J Am Coll Cardiol. 2005;45(10):1563–9.
25 Yakeu G, Butcher L, Isa S, Webb R, Roberts AW, Thomas AW, et al. Low-intensity exercise enhances expression of markers of alternative activation in circulating leukocytes: roles of PPARgamma and Th2 cytokines. Atherosclerosis. 2010;212(2):668–73.
26 Fried SK, Bunkin DA, Greenberg AS. Omental and subcutaneous adipose tissues of obese subjects release interleukin-6: depot difference and regulation by glucocorticoid. J Clin Endocrinol Metab. 1998;83(3):847–50.
27 Yudkin JS, Stehouwer CD, Emeis JJ, Coppack SW. C-reactive protein in healthy subjects: associations with obesity, insulin resistance, and endothelial dysfunction: a potential role for cytokines originating from adipose tissue? Arterioscler Thromb Vasc Biol. 1999;19(4):972–8.
28 Coen PM, Flynn MG, Markofski MM, Pence BD, Hannemann RE. Adding exercise to rosuvastatin treatment: influence on C-reactive protein, monocyte toll-like receptor 4 expression, and inflammatory monocyte (CD14+CD16+) population. Metabolism. 2010;59(12):1775–83.
29 Ridker PM, Buring JE, Rifai N. Soluble P-selectin and the risk of future cardiovascular events. Circulation. 2001;103:491–5.
30 McEver RP. Selectins. Curr Opin Immunol. 1994;6(1):75–84.
31 Michelson AD, Barnard MR, Krueger LA, Valeri CR, Furman MI. Circulating monocyte-platelet aggregates are a more sensitive marker of in vivo platelet activation than platelet surface P-selectin: studies in baboons, human coronary intervention, and human acute myocardial infarction. Circulation. 2001;104(13):1533–7.
32 Aronson DL, Stevan L, Ball AP, Franza BR Jr., Finlayson JS. Generation of the combined prothrombin activation peptide (F1-2) during the clotting of blood and plasma. J Clin Invest. 1977;60(6):1410–8.
33 Paramo JA, Orbe J, Beloqui O, Benito A, Colina I, Martinez-Vila E, et al. Prothrombin fragment 1+2 is associated with carotid intima-media thickness in subjects free of clinical cardiovascular disease. Stroke. 2004;35(5):1085–9.
34 Niessner A, Richter B, Penka M, Steiner S, Strasser B, Ziegler S, et al. Endurance training reduces circulating inflammatory markers in persons at risk of coronary events: Impact on plaque stabilization? Atherosclerosis. 2006;186(1):160–5.
35 Adamopoulos S, Parissis J, Karatzas D, Kroupis C, Georgiadis M, Karavolias G, et al. Physical training modulates proinflammatory cytokines and the soluble fas/soluble fas ligand system in patients with chronic heart failure. J Am Coll Cardiol. 2002;39(4):653–63.
36 Watson L, Ellis B, Leng GC. Exercise for intermittent claudication. Cochrane Database Syst Rev. 2008;(4):CD000990.
37 Leng GC, Fowler B, Ernst E. Exercise for intermittent claudication. Cochrane Database Syst Rev. 2000;(2):CD000990.
38 Nicolai SP, Hendriks EJ, Prins MH, Teijink JA. Optimizing supervised exercise therapy for patients with intermittent claudication. J Vasc Surg. 2010;52(5):1226–33.
Trial registration number: NCT00926081.
Funding / potential competing interests: The authors declare that there was no financial support and that there is no duality of interest associated with this manuscript.