New insights in acute kidney failure in the critically ill
DOI: https://doi.org/10.4414/smw.2012.13662
Zaccaria
Ricci, Claudio
Ronco
Summary
The term acute kidney injury (AKI) has been recently coined by a large panel of international experts in place of the former expression “acute renal failure”. This change has been motivated by a double intention: first it served to definitely find a conventional definition for acute changes of renal function, previously lacking in the medical community. In fact, any attempt to compare scientific papers and different centres experiences on AKI was essentially impossible. The second aim was to remark that this syndrome is characterised by a spectrum of progressive damage, from mild creatinine increase to renal injury to a more severe form, failure: this important concept should increase clinicians awareness to every form of renal dysfunction, even milder ones, in order to improve epidemiologic analyses, potentially preventing eventual AKI progression and finally helping standardisation of medical and supportive therapy. This review will describe such “new era” of critical care nephrology by presenting current literature (and its many controversies) about AKI diagnosis, physiopathology and management.
Epidemiology of AKI
Acute kidney injury (AKI) is common: its incidence, according to most recent reports, ranges from 0.25% in general population, 18% in hospitalised patients, 30 to 60% in critically ill patients [1]. AKI incidence is similar to that of myocardial infarction [2]. In a recent survey [3] conducted in 10 Italian intensive care units (ICU) through a web-based data collection, showed that AKI had a high crude ICU mortality (28.8% vs non-AKI 8.1%) and long ICU length of stay (median 7 days vs non-AKI 3 days). According to this survey, almost two thirds of AKI cases were diagnosed within 24 hours of ICU admission. About 12% of AKI patients were treated with renal replacement therapy (RRT) in the ICU. Patients were started on RRT a median of 2 (IQR 0–6) days after ICU admission. 60% of AKI patients had complete recovery of renal function, 13.5% had partial renal recovery, while about 30% did not recover renal function at the time of death or ICU discharge. Septic patients had more severe AKI, and were more likely to receive RRT with less frequency of renal function recovery. Patients with sepsis had higher ICU mortality and longer ICU stay.
Physiopathology
Physiopathology of AKI is multifactorial: several variables and extremely different clinical conditions are involved in reduction of renal function and make it difficult to exactly fit all types of AKI into a single pathophysiologic pathway. The traditional classification of AKI physiopathology into “pre-renal” (in case of mild to moderate renal hypoperfusion), “intra-renal” (mostly characterised by acute tubular necrosis) and “post-renal” (obstructive) has recently been argued [4]. As a matter of fact, a histological diagnosis is rarely performed: hence a clear distinction between most severe forms of pre-renal AKI and tubular damage is never possible and it can only be retrospectively hypothesised. Furthermore it is possible that tubular necrosis occurrence is limited to few AKI cases and that structure of renal parenchyma is not frequently involved. A recent 10-year retrospective multicentre French study on 77 critically ill patients with AKI who underwent renal biopsy showed that, in spite of a relatively high number of complications, only 18% were diagnosed with ATN [5]. In this light, even if the actual role of parenchymal modifications during AKI occurrence remains to be definitely clarified, from a conceptual point of view AKI should be looked at as a continuum of injury and not classified by presence/absence of histological damage [4].
Critical reappraisal of current literature on experimental studies, generally conducted on models of ischaemia reperfusion, revealed multiple pathways and mechanisms of organ injury: local activation of the coagulation system [6] infiltration of the kidney by leukocytes [7], endothelial injury [8], expression of adhesion molecules [9], release of cytokines [10] induction of toll-like receptors [11] activation of intra-renal vasoconstrictor pathways [12] and induction of apoptosis [13] There are also associated changes in tubular cells with loss or inversion of polarity [14] and loss of adhesion to the basement membrane [15]. Unfortunately, these models are quite “extreme” and likely far from any clinical manifestation of human AKI, where renal arteries are not clamped and then unclamped but less severe forms of low flow state followed by reperfusion generally occur.
On the other side, pathogenetic investigations in humans are frequently focused on renal perfusion either due to the empirical observation that severely septic shock patients and haemodynamically unstable patients are most frequently affected by AKI. Furthermore normotensive AKI has been described as a consequence of intra-renal haemodynamic changes [15]. However, a recent editorial remarked that since renal blood flow (RBF) is not routinely assessed in critically ill patients, we currently do not know if it is really decreased during septic shock [16]. Notably, a clinical observation on invasive RBF measurement described complex and unpredictable association with changes (either increase and reduction) in intra-renal microcirculation and glomerular haemodynamics [17]. A small interesting study on established septic AKI from the same group, evaluated by cine phase-contrast magnetic resonance, confirmed that RBF fraction of cardiac output appears significantly reduced compared to normal healthy individuals (7 vs 20%) [18]. However, measurement of RBF in patients with established AKI must rely on organ oedema, tubular injury, back-leak, increased tubular luminal pressure [4]: hence, RBF reduction may be the consequence of, rather than the mechanism responsible for AKI. Causes of RBF reduction may be identified in sympathetic system activation [19], kidney-specific neuro-hormonal activation of the renin-angiotensin system (RAAS) [20] and activation of the tubulo-glomerular feedback (TGF) system [21]. In particular, during septic AKI, infection might lead to induction of nitric oxide synthase and nitric oxide mediated arterial vasodilatation and secondary activation of the sympathetic system, RAAS activity and renal vasoconstriction [20]. Arginine vasopressin seems to have a role too, contributing to water retention [20]. Complex neurohormonal feedbacks may also cause intra-renal shunting with decreased glomerular filtration rate (GFR) and ischaemia of the renal medulla. Hence, microcirculation is finally affected, suggesting that, even if one could measure global RBF, unless the microcirculation is also assessed, estimation of renal perfusion is significantly limited [22].
Organ “cross-talk” is also becoming an important object of recent pathogenetic investigations: after many years from the description of hepato-renal syndrome (and the description of many neurohormonal pathways and links between the liver and the kidneys [4]) it has been observed that both heart failure and kidney disease are frequently combined: heart insufficiency can be superimposed on acute or chronic renal dysfunction; otherwise kidney disease may be linked to an acute or chronic decompensation of heart failure (cardiorenal and renocardiac syndromes) [23]. Interestingly, it has been observed that a congestive state (right heart failure), due to renal oedema causing a form of kidney tamponade, may be as important to the pathogenesis of AKI as low blood pressure or low cardiac output (left heart failure and other shock states) [24]. Similarly, it is currently evident that pathogenesis of severe lung injury and acute respiratory distress syndrome, especially after aggressive ventilation have been instituted, are strictly linked to AKI occurrence [25]: inflammation mediators produced by the two organs are probably involved, together with the difficulty of achieving fluid restriction in patients with damaged lungs when kidney function is hampered.
Definition and diagnosis
Apart from any pathophysiologic consideration, during AKI sudden reduction in renal function occurs: consequently, creatinine and urea accumulate. Commonly, but not necessarily, urine production is also reduced. The term acute kidney injury (AKI) has been recently coined [26] in place of the former expression “acute renal failure”. This change served to definitely find a conventional definition for acute changes of renal function. Then, it has been underlined that the amount of such markers change during AKI is variable: as a continuum of kidney damage, renal dysfunction ranges from a low level (or “risk” of being injured), to an actual “injury” (with reduced but still present renal function), to a definitive “failure” (severely reduced or lost renal physiology). This idea brought Acute Dialysis Quality Initiative (ADQI), back in 2004, to the consensus definition of the RIFLE criteria [26]: such acronym conventionally described 3 stages of progressively higher creatinine levels or decreased urine output flows (Risk, Injury, Failure) and 2 outcomes (Loss of function and End stage kidney disease) (table 1). Subsequently the Acute Kidney Injury Network (AKIN) introduced small though important modifications to this classification (table 1) [27]: a number of comparative studies could not show significant differences in their diagnostic/prognostic performance [28]. The ease of the classification stages achievement at the bedside, even if at a first glance superficial and completely questionable (as every consensus definition is), originally aimed to increase clinicians awareness on the fact that even small increases (0.3 mg/dl) in serum creatinine should be looked carefully because that patient already has AKI (or, as recently defined, a “kidney attack” [2]). Furthermore, after years without a common renal failure definition with scientific literature providing dissimilar epidemiological studies, RIFLE and AKIN finally created a common epidemiological framework of reference. In this light, today it has been definitely confirmed (in disparate clinical settings) that AKI is independently associated with greater risk of death and that this risk is linearly correlated to AKI stages [29]. It has to be remarked that the concept of “definition” should not be confounded with the concept of “aetiological diagnosis”. RIFLE/AKIN criteria do not intend to identify the cause of AKI (especially because, as stated above, it is blurred and difficult to establish): they are only useful to mark the pathology and to identify it. The next step to be verified by prospective studies on RIFLE/AKIN classifications will be the possibility of applying different therapeutic strategies at different AKI stages: timing of different therapies might be standardised or reproduced in different centres and trials. This idea most recently brought a group of Belgian researchers to utilise a “real-time electronic alert system”, based on the RIFLE classification criteria, in order to verify if clinicians, once proactively and timely alerted that their patients AKI class was worsening, did improve AKI management. Interestingly, they showed that, during the “alert period”, the number and timeliness of AKI therapeutic interventions was significantly increased and short-term renal outcome in the Risk patients was improved [30] with respect to a control period. Two provocative observational studies on the possibility of deciding RRT timing on a risk-injury-failure basis seemed instead to substantially reject this hypothesis, as far as dialysis is concerned [31–32]. The authors showed that many Failure patients not treated with RRT achieved a similar outcome (if not better) to Failure patients undergoing RRT [31]. They also claimed that RRT-treated Risk and Injury patients showed worse outcomes than Failure RRT-treated patients [32]. However, as also remarked below, it is possible that the most effective intervention on AKI management is (medical) prevention of patient progression to a worse AKI stage. Then, since RRT is clearly applied to patients with the most severe clinical presentation irrespective of creatinine levels, AKI classifications may poorly perform in a cohort of dialysed critically ill patients.
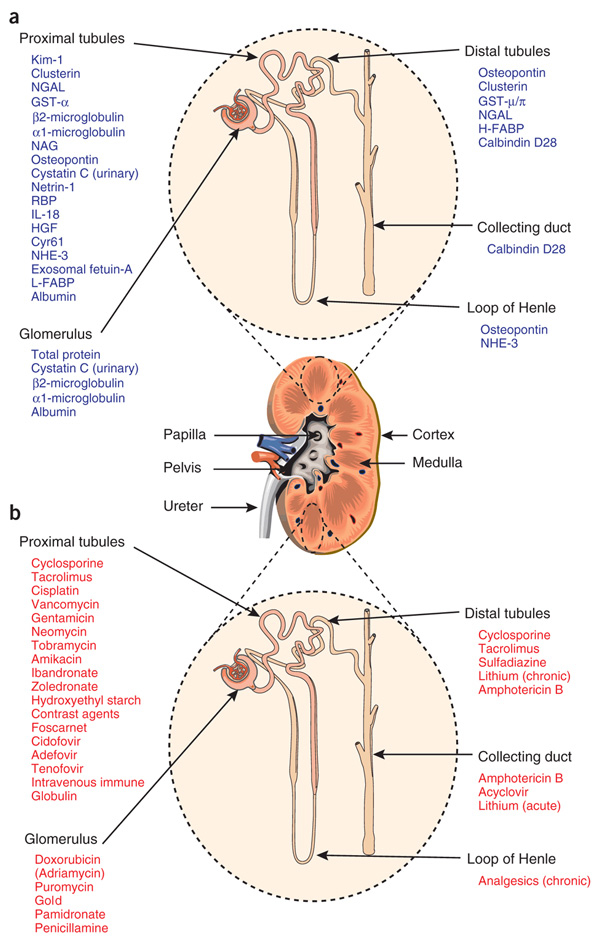
Figure 1
The utility of biomarkers to detect injury to specific nephron segments affected by various nephrotoxins.
a Nephron segment-specific biomarkers of kidney injury.
b Drugs that elicit site-specific toxicity in the kidney.
(Source: Bonventre JV, Vaidya VS, Schmouder R, Feig P, Dieterle F. Next-generation biomarkers for detecting kidney toxicity. Nat Biotechnol. 2010;28:436–40. © Macmillan publishers Ltd, 2012. Reprinted with permission).
Finally, the recent enthusiasm of basic research on AKI diagnosis highlighted the fact that renal dysfunction begins long before sufficient loss of excretory kidney function can be detected with standard laboratory tests. This happens because urea and creatinine are insensitive markers of glomerular filtration rate (GFR) and are modified by nutrition, the use of steroids, the presence of gastrointestinal blood, muscle mass, age, gender, or muscle injury [4]. Furthermore, creatinine starts increasing only when more than 50% of GFR is lost, cannot reflect GFR trends and may be diluted after aggressive fluid resuscitation [33]. New techniques based on proteomics have identified several novel biomarkers of AKI: cystatin C (CysC), neutrophil gelatinase associated lipocalin (NGAL), interleukin 18 (IL18), kidney injury molecule 1 (KIM1), and many others (fig. 1) [34]. Current state of the art does not allow declaring which of these biomarkers is the ideal one: however, a biochemical pattern can be drawn basing on the different features of each of them. In many pivotal studies biomarkers showed to change earlier than changes in serum creatinine [35]. Others appeared to reflect different aetiologies of renal injury [36]. Then, they seemed to dynamically change with treatment or recovery, which suggests that they can be used to monitor interventions [37]. In recent studies subpopulations of patients who did not have AKI according to creatinine-based criteria, but actually had a degree of subclinical renal damage, were identified by biomarkers and associated with worse outcomes [38]. Finally, by identifying possible mechanisms of injury, these biomarkers might increase our understanding of the pathogenesis of AKI and/or help in prognosis/triaging of AKI syndrome [39]. Although urinary NGAL and CysC are probably the most studied renal biomarkers, multiple other molecules are currently under investigation. Interestingly enough, such “biomarkers wave” allowed also to evaluate organ cross talk, as in the case of cardiorenal syndrome: brain natriuretic peptide (BNP) commonly used to screen or follow up patients with heart failure, showed to be associated with post-operative AKI in a cohort cardiac surgery patients [40]. No study so far, however, attempted to show a positive association of biomarkers use (and relative cost) with hard clinical outcomes.
It must finally be remembered that apart from biomarkers expectations, the operators never have to forget AKI causes and physiopathology: the current effort to standardise AKI and to diagnose it with magic tools before it can be clinically manifest will never be able to track AKI before renal damage has even occurred. Furthermore, any diagnostic approach to the cause or trigger of AKI must take into account the local context and epidemiology. In this light, each critically ill patient should be closely observed, AKI occurrence never overlooked and physiopathology/prevention of renal dysfunction always taken in consideration.
|
Table 1: Classification/staging system for acute kidney injury. |
|
RIFLE
|
|
Class
|
Serum creatinine criteria
|
Urine output criteria
|
| R |
Serum creatinine increase to 1.5-fold or GFR decrease >25% from baseline |
<0.5 ml/kg/hour for 6 hours |
| I |
Serum creatinine increase to 2-fold or GFR decrease >50% from baseline |
<0.5 ml/kg/hour for 12 hours |
| F |
Serum creatinine to 3-fold, GFR decrease >75% from baseline
or serum creatinine ≥4 mg/dl
(≥354 μmol/l) with an acute increase of at least 0.5 mg/dl (44 μmol/l) |
Anuria for 12 hours |
|
AKIN
|
|
Stage
|
Serum creatinine criteria
|
Urine output criteria
|
| 1 |
Serum creatinine increase ≥0.3 mg/dl (≥26.4 μmol/l) or increase to 1.5- to 2-fold from baseline |
<0.5 ml/kg/hour for 6 hours |
| 2 |
Serum creatinine increase >2- to 3-fold from baseline |
<0.5 ml/kg/hour for 12 hours |
| 3 |
Serum creatinine increase >3-fold from baseline or serum creatinine ≥4.0 mg/dl (≥354 μmol/l) with an acute increase of at least 0.5 mg/dl (44 μmol/l) |
Less than 0.3 ml/kg per hour for 24 hours or anuria for 12 hours |
|
|
Need for renal replacement therapy |
| Synopsis of RIFLE and AKIN criteria for AKI classification/staging. Small but significant changes can be identified between the two definitions. A time constraint of 48 hours for diagnosis (creatinine or urine output modifications) is required in AKIN criteria. Glomerular Filtration Rate (GFR) decreases for diagnosis are reported only in RIFLE. In both cases, only one criterion (creatinine or urine output) has to be fulfilled to qualify for a class/stage. Classes L (loss of function) and E (end stage kidney disease) of RIFLE criteria are not reported in AKIN. Given wide variation in indications and timing of initiation of renal replacement therapy (RRT), individuals who receive RRT are considered to have met the criteria for AKIN stage 3 irrespective of the stage they are in at the time of RRT. |
Treatment
Prevention of AKI
Avoiding AKI occurrence is probably the best way to improve outcomes of critically ill patients with renal dysfunction. Hence, when possible, patients should not receive potentially nephrotoxic drugs such as vancomycin, aminoglycosides, contrast agents and acetyl salicylic acid, especially in patients with additive risk factors for AKI development (elderly, diabetic, congestive heart disease or chronic renal failure patients) [41] (fig. 1). Furthermore, management of “pre-renal” causes is mandatory from the initial phases of the shock state: typically a colloid/crystalloid 10–40 ml/kg intravenous fluid challenge is considered the first line therapy to target a mean arterial pressure of 65 mm Hg and a central venous pressure of 12–15 mm Hg (in case of mechanical ventilation) [42]. An on-going debate has not fully elucidated so far if colloids or crystalloids should be preferably infused and if they can be safely used in septic patients, especially in the light of their potential nephrotoxic role [43]. Unfortunately, today, no striking evidence from human data, contradictory animal studies and no understanding of the physiological effects of fluid bolus resuscitation in critically ill patients exist [44]. If restrictive fluid management of haemodynamically unstable patients is considered, an early and aggressive use of vasopressors should be recommended in order to restore adequate perfusion pressure after an initial moderate fluid administration [44]. In this light, norepinephrine seems to be the drug of choice and its beneficial effects on renal perfusion have been clearly shown in an interesting experimental model [45]. Vasopressin (antidiuretic hormone) is another powerful agent for rapid and effective increase of systemic resistances and mean arterial pressure. It is important to highlight that vasopressors, when carefully titrated to the minimal dose able to restore mean arterial pressure in critically ill patients, are not harmful for kidney function and may be potentially beneficial [46].
A completely different approach to AKI prophylaxis has been attempted with the use of fenoldopam: in a randomised controlled clinical trial, this agent, a selective agonist of renal dopaminergic receptors (vasodilation of glomerular afferent arterioles), showed to significantly reduce the incidence of AKI in a cohort of patients with sepsis and normal baseline renal function [47]. This study is one of the few that attempted a specific preventive medical nephroprotective strategy in critically ill patients with sepsis. As a matter of fact, one reason for the lack of an effective therapy of septic AKI might rely on the delay of clinical signs and symptoms of sepsis with respect of renal injury that presumably occur in the earliest phases of shock. In this light, the use of novel early diagnostic biomarkers and AKI classifications might encourage a timely preventive nephroprotective intervention [36–37].
Medical therapy of AKI
Diuretics, in particular loop diuretics, exemplify the typical symptomatic medication of critically ill patients with oliguric AKI. Loop diuretics act on the medullary thick ascending loop of Henle to inhibit the Na+/K+/2Cl– pump on the luminal cell membrane surface and reduce oxygen demand. Diuretics main role is to manage volume overload and optimise acid–base and electrolyte homeostasis. Currently, however, controversy exists as to whether or not diuretics can actually reduce or delay RRT use and eventually improve clinical outcomes in sepsis and septic AKI [48]. Thus, although a strong rationale for their use seems to exist, understanding of how and when diuretics should be used is limited. Most interestingly, however, a recent post hoc analysis [49], from the Fluid and Catheter Treatment Trial (FACTT) study, which evaluated a conservative versus liberal fluid management strategy in patients with acute lung injury, showed that a positive fluid balance after AKI was associated with 60-day mortality, that the risk of death in patients with acute lung injury and AKI was approximately 1.6-fold higher per liter/day of fluid accumulated and, Importantly, that diuretic use after AKI was associated with decreased mortality, with the protective effect on 60-day mortality being limited after adjustment for fluid balance. This analysis suggests that the benefit of furosemide in critically ill patients was derived from the resultant optimisation of fluid overload.
Several specific medications for AKI have been attempted in the last ten years but their use in the clinical field has hopelessly failed, even after promising experimental studies. On the clinical field, an isolated positive investigation evaluated the administration of bovine alkaline phosphatase (AP) to sepsis patients with AKI [50]. AP is an endogenous enzyme that acts through dephosphorylation of endotoxins and pro-inflammatory extracellular ATP with a specific local action on renal tubules. In this trial, AP significantly improved renal function, indicated by the authors with a composite endpoint including endogenous creatinine clearance, RRT requirement and duration of dialysis. The findings of this study were further reinforced by a significant reduction in some of the evaluated circulating inflammatory markers (C-reactive protein, Interleukin-6, LPS-binding protein) and urinary markers of tubular injury (Kidney Injury Molecule-1 and Interleukin-18) in AP-treated patients compared to placebo.
In conclusion, specific medical therapy of renal dysfunction is not currently available for routine clinical practice. Critically ill patients primarily benefit from preventive measures, aggressive haemodynamic optimisation, proper management of fluid balance and avoidance of overload. Medical (supportive) therapy for severe AKI, so far, can be considered a strategic method to gain time before extracorporeal renal support is decided.
Renal replacement therapy
Artificial blood purification and renal function substitution is indicated when kidney dysfunction is severe enough to cause acute harm to the patient. Given this consideration, it is clear that the best time to start RRT is controversial (except in the honestly rare case of rapidly increasing potassium levels) and difficult to establish. Several observational studies evaluating timing described “early” RRT as beneficial [51]: as it happened for AKI diagnosis, currently no standard definition of such earliness exists and, above all, it has not been clarified with respect of what we should define “early timing” (ICU admission? AKI diagnosis? Severe AKI diagnosis? Sepsis occurrence? Fluid balance?).
Once RRT is indicated, a prescription on dialysis dose has to be made and the treatment carefully delivered. Intensity of a dialytic treatment could be roughly indicated by the amount of dialysis/hemofiltration flow delivered to the patient: after the milestone trial by the group of Vicenza back in 2000 [52], such flow has been indexed to patient’s body weight, in order to highlight that this variable is of straightforward importance in AKI patients. Two large multi-centre randomised controlled studies published in 2009 (the randomised evaluation of normal versus augmented level [RENAL] replacement therapy study [53] and the VA/NIH Acute Renal Failure Trial Network [ATN] study [54]) finally clarified the concept of optimal dialysis dose. The RENAL and ATN studies were designed to compare “normal” or “less intensive” renal support to an “augmented” or “intensive” therapy: in particular, the RENAL study compared 25 ml/kg per h continuous veno-venous haemodiafiltration (CVVHDF) to 40 ml/kg per h; the ATN study compared 20 ml/kg per h CVVHDF or thrice weekly intermittent dialysis to 35 ml/kg/h CVVHDF or daily intermittent dialysis. Surprisingly, both studies showed that increases in intensity of RRT dose did not improve patient outcomes, essentially confuting a large body of evidence coming from previous smaller trials. As a consequence, the definition of “normal dose” is now recommended in a range of 20–30 ml/kg per h for continuous therapies and/or thrice weekly intermittent haemodialysis.
No randomised controlled trials have addressed the issue of RRT stop. Observational studies have suggested that urine output during RRT can be used to predict successful weaning. A spontaneous urine output >500 ml/day seems to have sufficient discrimination to be used for the purpose of considering a trial of RRT cessation [55]. Furthermore fluid balance has recently been claimed as a major outcome determinant of critically ill patients with AKI: it is possible that a negative fluid balance should be targeted in CRRT patients, once haemodynamic stability has been warranted by initial resuscitation efforts [56]. However, it is currently impossible to recommend a priori the net UF rate, that should instead be tailored on patients’ needs. The authors of the RENAL trial recently retrospectively analysed the association between daily fluid balance and several hard clinical outcomes on the cohort of more than 1400 patients previously enrolled in RENAL study [56]. Interestingly, not only they found that a negative mean daily fluid balance during the study treatment was independently associated with a decreased risk of death at 90 days, increased survival time and increased renal replacement-free days, intensive care unit-free days and hospital-free days but also they showed how this association remained after adjustment for propensity and all available markers of illness severity at randomisation. Such a robust association between a positive fluid balance and unfavourable outcomes suggests the need to decrease, as soon as clinically possible, fluid administration in patients with AKI or to specifically target negative fluid balance during RRT.
Conclusion
The mortality of AKI remains high (about 50%). It is possible that even a short episode of AKI may contribute to long-term organ and patient morbidity and mortality. Thus, this complex syndrome should be prevented, aggressively treated and never overlooked, especially the milder forms. When RRT becomes necessary, it is possible that the (critically ill) patient has a clinical picture of multiple organ failure: in case, unfortunately, probability of surviving is still dramatically low: a correct dialysis prescription and delivery, avoidance of dialysis underdosing and prevention of harmful complications (such as hypotension and bleeding) are currently recommended to target “standard of care”. AKI survivors have to be followed up being progression to forms of chronic renal failure currently increasing.
References
1 Singbartl K, Kellum JA. AKI in the ICU: definition, epidemiology, risk stratification, and outcomes. Kidney Int. 2012; 81:819–25.
2 Kellum JA, Bellomo R, Ronco C. Kidney Attack. JAMA. 2012 May 9. [Epub ahead of print]
3 Piccinni P, Cruz DN, Gramaticopolo S, Garzotto F, Dal Santo M, Aneloni G, et al; NEFROINT investigators. Prospective multicenter study on epidemiology of acute kidney injury in the ICU: a critical care nephrology Italian collaborative effort (NEFROINT). Minerva Anestesiol. 2011;77:1072–83.
4 Bellomo R, Kellum JA, Ronco C. Acute kidney injury. Lancet. 2012 May 18. [Epub ahead of print]
5 Augusto JF, Lassalle V, Fillatre P, Perrotin D, Meziani F, Schenck-Dhif M, et al. Safety and diagnostic yield of renal biopsy in the intensive care unit. Intensive Care Med. 2012 Jul 10. [Epub ahead of print]
6 Thuillier R, Favreau F, Celhay O, Macchi L, Milin S, Hauet T. Thrombin inhibition during kidney ischemia-reperfusion reduces chronic graft inflammation and tubular atrophy. Transplantation 2010;90:612–21.
7 Versteilen AM, Blaauw N, DiMaggio F, et al. Rho-kinase inhibition reduces early microvascular leukocyte accumulation in the rat kidney following ischemia-reperfusion injury: roles of nitric oxide and blood flow. Nephron Exp Nephrol. 2011;118:e79–e86.
8 Kwon O, Hong SM, Ramesh G. Diminished NO generation by injured endothelium and loss of macula densa nNOS may contribute to sustained acute kidney injury after ischemia-reperfusion. Am J Physiol Renal Physiol. 2009;296:F25–33.
9 Kato N, Yuzawa Y, Kosugi T, et al. The E-selectin ligand basigin/CD147 is responsible for neutrophil recruitment in renal ischemia-reperfusion. J Am Soc Nephrol. 2009;20:1565–76.
10 Thurman JM. Triggers of inflammation after renal ischemia/reperfusion. Clin Immunol. 2007;123:7–13.
11 Pulsken WP, Teske GJ, Butter LM, et al. Toll-like receptor-4 coordinates the innate immune response of the kidney to renal ischemia/reperfusion injury. PLoS One 2008;3:e3596.
12 Conger DJ. Vascular alterations in acute renal failure: roles of initiation and maintenance. In: Molitoris BA, Finn WF, eds. Acute renal failure: a companion to Brenner and Rector’s the kidney, 1st edn. Philadelphia: WB Saunders 2001; 13–29.
13 Saikumar P, Venkatachalam AM. Role of apoptosis in hypoxic/ischemic damage in the kidney. Semin Nephrol. 2003;23:511–21.
14 Zuk A, Bonventre JV, Brown D, Matilin KS. Polarity, integrin, and extracellular matrix dynamics in the post-ischemic rat kidney. Am J Physiol. 1998;275:C711–31.
15 Abuelo JG. Normotensive ischemic acute renal failure. N Engl J Med. 2007;23;357:797–805.
16 Lipcsey M, Bellomo R. Septic acute kidney injury: hemodynamic syndrome, inflammatory disorder, or both? Crit Care. 2011;15:1008.
17 Langenberg C, Bellomo R, May C, Wan L, Egi M, Morgera S: Renal blood flow in sepsis. Crit Care 2005;9:R363–R374.
18 Prowle JR, Molan MP, Hornsey E, Bellomo R. Measurement of renal blood flow by phase-contrast magnetic resonance imaging during septic acute kidney injury: A pilot investigation. Crit Care Med. 2012;40:1768–76.
19 Ramchandra R, Wan L, Hood SG, Frithiof R, Bellomo R, May CN. Septic shock induces distinct changes in sympathetic nerve activity to the heart and kidney in conscious sheep. Am J Physiol Regul Integr Comp Physiol. 2009;297:R1247–53.
20 Schrier RW, Wang W. Acute renal failure and sepsis. N Engl J Med. 2004;351:159–69.
21 Loutzenhiser R, Griffin K, Williamson G, Bidani A. Renal autoregulation: new perspectives regarding the protective and regulatory roles of the underlying mechanisms. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1153–67.
22 Ishikawa K, Bellomo R, May CN. The impact of intrarenal nitric oxide synthase inhibition on renal blood flow and function in mild and severe hyperdynamic sepsis. Crit Care Med. 2011;39:770–6.
23 Ronco C, House AA, Haapio M. Cardiorenal and renocardiac syndromes: the need for a comprehensive classification and consensus. Nat Clin Pract Nephrol. 2008;4:310–1.
24 Damman K, Navis G, Smilde TD,et al. Decreased cardiac output, venous congestion and the association with renal impairment in patients with cardiac dysfunction. Eur J Heart Fail. 2007;9:872–8.
25 Singbartl K. Renal-pulmonary crosstalk. Contrib Nephrol. 2011;174:65–70.
26 Bellomo R, Ronco C, Kellum JA, Mehta RL, Palevsky P, Acute Dialysis Quality Initiative workgroup. Acute renal failure – definition, outcome measures, animal models, fluid therapy and information technology needs: the Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit Care. 2004;8:R204–12.
27 Mehta RL, Kellum JA, Shah SV, Molitoris BA, Ronco C, Warnock DG, Levin A; Acute Kidney Injury Network. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care. 2007;11:R31.
28 Cruz DN, Ricci Z, Ronco C. Clinical review: RIFLE and AKIN – time for reappraisal. Crit Care. 2009;13:211.
29 Ricci Z, Cruz D, Ronco C. The RIFLE criteria and mortality in acute kidney injury: A systematic review. Kidney Int. 2008;73:538–46.
30 Colpaert K, Hoste EA, Steurbaut K, Benoit D, Van Hoecke S, De Turck F, Decruyenaere J. Impact of real-time electronic alerting of acute kidney injury on therapeutic intervention and progression of RIFLE class. Crit Care Med. 2012;40:1164–70.
31 Schneider AG, Uchino S, Bellomo R. Severe acute kidney injury not treated with renal replacement therapy: characteristics and outcome. Nephrol Dial Transplant. 2012;27:947–52.
32 Schneider AG, Eastwood GM, Seevanayagam S, Matalanis G, Bellomo R. A risk, injury, failure, loss, and end-stage renal failure score-based trigger for renal replacement therapy and survival after cardiac surgery. J Crit Care 2012 Apr 3. [Epub ahead of print]
33 Macedo E, Bouchard J, Soroko SH, Chertow GM, Himmelfarb J, Ikizler TA, Paganini EP, Mehta RL. Program to Improve Care in Acute Renal Disease Study. Fluid accumulation, recognition and staging of acute kidney injury in critically-ill patients. Crit Care. 2010;14(3):R82.
34 Devarajan P, Krawczeski CD, Nguyen MT, Kathman T, Wang Z, Parikh CR. Proteomic identification of early biomarkers of acute kidney injury after cardiac surgery in children. Am J Kidney Dis. 2010;56:632–42.
35 Mishra J, Dent C, Tarabishi R, Mitsnefes MM, Ma Q, Kelly C, et al. Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker for acute renal injury after cardiac surgery. Lancet. 2005;365:1231–8.
36 Ricci Z, Cruz DN, Ronco C. Classification and staging of acute kidney injury: beyond the RIFLE and AKIN criteria. Nat Rev Nephrol. 2011;7:201–8.
37 Ricci Z, Luciano R, Favia I, Garisto C, Muraca M, Morelli S, Di Chiara L, Cogo P, Picardo S. High-dose fenoldopam reduces postoperative neutrophil gelatinase-associated lipocaline and cystatin C levels in pediatric cardiac surgery. Crit Care. 29; 15:R160.
38 Haase M, Devarajan P, Haase-Fielitz A, Bellomo R, Cruz DN, Wagener G, et al. The outcome of neutrophil gelatinase-associated lipocalin-positive subclinical acute kidney injury: a multicenter pooled analysis of prospective studies. J Am Coll Cardiol. 2011;57:1752–61.
39 Kümpers P, Hafer C, Lukasz A, Lichtinghagen R, Brand K, Fliser D, Faulhaber-Walter R, Kielstein JT. Serum neutrophil gelatinase-associated lipocalin at inception of renal replacement therapy predicts survival in critically ill patients with acute kidney injury. Crit Care. 2010;14:R9.
40 Patel UD, Garg AX, Krumholz HM, Shlipak MG, Coca SG, Sint K, et al. Translational Research Investigating Biomarker Endpoints in Acute Kidney Injury (TRIBE-AKI) Consortium. Preoperative serum brain natriuretic peptide and risk of acute kidney injury after cardiac surgery. Circulation. 2012;125:1347–55.
41 Liangos O. Drugs and AKI. Minerva Urol Nefrol. 2012;64:51–62.
42 Dellinger RP, Levy MM, Carlet JM, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med. 2008;36:296–327.
43 Perel P, Roberts I. Colloids versus crystalloids for fluid resuscitation in critically ill patients. Cochrane Database Syst Rev 2012 Jun 13;6:CD000567.
44 Hilton AK, Bellomo R. A critique of fluid bolus resuscitation in severe sepsis. Crit Care. 2012;16:302.
45 Di Giantomasso D, May CN, Bellomo R. Norepinephrine and vital organ blood flow during experimental hyperdynamic sepsis. Intensive Care Med. 2003;29:1774–81.
46 Gordon AC, Russell JA, Walley KR, et al. The effects of vasopressin on acute kidney injury in septic shock. Intensive Care Med. 2010;36:83–91.
47 Morelli A, Ricci Z, Bellomo R, et al. Prophylactic fenoldopam for renal protection in sepsis: a randomized, double-blind, placebo-controlled pilot trial. Crit Care Med. 2005;33:2451–6.
48 Bagshaw SM, Gibney RT, McAlister FA, Bellomo R. The SPARK Study: a phase II randomized blinded controlled trial of the effect of furosemide in critically ill patients with early acute kidney injury. Trials. 2010;11;11:50.
49 Grams ME, Estrella MM, Coresh J, Brower RG, Liu KD; National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome Network. Fluid balance, diuretic use, and mortality in acute kidney injury. Clin J Am Soc Nephrol. 2011;6:966–73.
50 Pickkers P, Heemskerk S, Schouten J, et al. Alkaline phosphatase for treatment of sepsis-induced acute kidney injury: a prospective randomized double-blind placebo-controlled trial. Crit Care. 2012;16:R14.
51 Karvellas CJ, Farhat MR, Sajjad I, Mogensen SS, Leung AA, Wald R, et al. A comparison of early versus late initiation of renal replacement therapy in critically ill patients with acute kidney injury: a systematic review and meta-analysis. Crit Care. 2011;15:R72.
52 Ronco C, Bellomo R, Homel P, Brendolan A, Dan M, Piccinni P, et al. Effects of different doses in continuous veno-venous haemofiltration on outcomes of acute renal failure: a prospective randomised trial. Lancet. 2000;356:26–30.
53 RENAL Replacement Therapy Study Investigators, Bellomo R, Cass A, Cole L, Finfer S, Gallagher M, Lo S, McArthur C, et al. Intensity of continuous renal-replacement therapy in critically ill patients. N Engl J Med. 2009;361:1627–38.
54 VA/NIH Acute Renal Failure Trial Network, Palevsky PM, Zhang JH, O’Connor TZ, Chertow GM, Crowley ST, Choudhury D, et al. Intensity of renal support in critically ill patients with acute kidney injury. N Engl J Med. 2008;359:7–20.
55 Uchino S, Bellomo R, Morimatsu H, et al. Discontinuation of continuous renal replacement therapy: a post hoc analysis of a prospective multicenter observational study. Crit Care Med. 2009;37:2576–82.
56 The RENAL Replacement Therapy Study Investigators, Bellomo R, Cass A, Cole L, Finfer S, Gallagher M, Lee J, et al. An observational study fluid balance and patient outcomes in the randomized evaluation of normal vs augmented level of replacement therapy trial. Crit Care Med. 2012;40:1753–60.