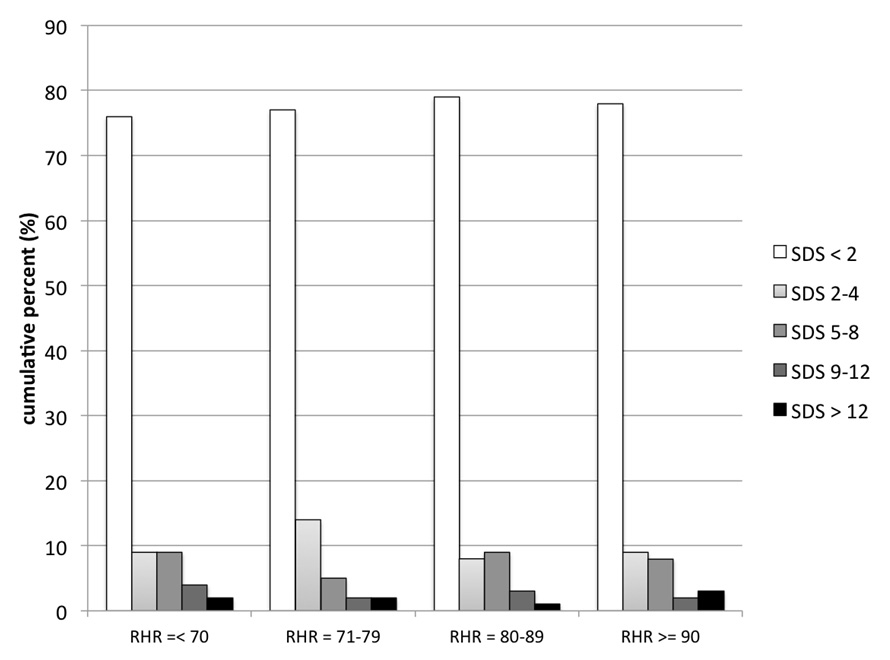
Figure 1
Graphics illustrating the interrelation between the quartiles of resting heart rate (RHR in bpm) and the severity of myocardial ischaemia (SDS),
DOI: https://doi.org/10.4414/smw.2012.13660
There is a growing body of evidence to suggest that sustained elevation of resting heart rate (RHR) is a cardiovascular risk factor [1, 2]. Numerous studies have shown a relationship between increased RHR and cardiovascular mortality in the general population [3, 4], and among patients with various cardiovascular diseases including hypertension [5, 6], coronary artery disease (CAD) [7, 8] and heart failure [9–11].
The pathophysiological link between increased RHR and the cardiovascular outcome, however, is not fully understood. Even if suggestive of a causative relation, the association between increased RHR and cardiovascular outcome does not by itself prove causality. Increased RHR may in fact represent a surrogate marker of the global burden of risk factors or the presence of more advanced cardiac end-organ damage [12, 13]. However, an incremental and independent impact of RHR on prognosis (and sudden cardiac death) was shown by multiple studies [3, 7], and recent randomised trials provided the evidence that heart rate reduction with ivabradine, a selective negative chronotropic drug, reduced cardiovascular endpoints in patients with CAD [14] and heart failure [15], suggesting some independent mechanism through which elevated RHR may affect cardiovascular risk. In this setting, increased RHR has been shown to accelerate coronary atherosclerosis in animals and humans [16, 17], to increase risk of rupture of pre-existing coronary plaques [18], and to trigger myocardial ischaemia in CAD patients [19, 20].
Based on this, the question is whether increased RHR is also associated with the presence and the severity of CAD itself. We therefore sought to assess the association between RHR and extent of CAD in a population of consecutive patients with clinically suspect but previously unknown CAD referred for myocardial perfusion SPECT (MPS).
All consecutive patients referred for exercise or vasodilator stress MPS for CAD evaluation at University Hospital Basel between 2001 and 2005 were considered for this study (n = 6,719). Patients with known CAD (n = 3,332), atrial fibrillation or pacemaker rhythm (n = 372), and medical treatment with negative chonotropic drugs, such as beta-blockers (also in the wash-out phase) and negative chonotropic calcium channel blockers (n = 1,550), were excluded. The final population consisted of 1,465 patients. Certainly this is a selected and lower risk population than the average population referred for MPS. Detailed medical history was collected in all patients.
All patients underwent routine rest/stress (bicycle ergometry or adenosine vasodilator stress) dual isotope (Tl-201 rest/Tc-99m sestamibi) MPS protocol as previously described [21, 22]. Rest-single photon emission computed tomography (SPECT) was obtained after administration of 111 MBq Tl-201. Tl-201 SPECT was performed 10 min after. Stress-SPECT was performed after injection of a 740 MBq dose of Tc-99m sestamibi was injected. Post-stress gated SPECT was acquired on average 95–100 min post-stress. SPECT imaging was performed following standard protocols on triple head cameras (Picker Prism 3000, Philips IRIX). No attenuation or scatter correction was used. SPECT images were acquired and processed as previously described [22], with a circular 180° acquisition (head to head 90°). During imaging, two energy windows were used for Tl-201, including a 30% window centred on the 70-keV peak and a 20% window centred on the 167-keV peak. For Tc-99m sestamibi SPECT, a 15% window centred on the 140-keV peak was used. Semiquantitative visual interpretation was performed using a 20-segment model. Each segment was scored using a 5-point scoring system: 0 = normal, 1 = equivocal, 2 = moderate, 3 = severe reduction of radioisotope uptake, and 4 = apparent absence of detectable tracer uptake in a segment. A summed stress score (SSS) was calculated by adding the scores of the 20 segments of the stress images, and a summed rest score (SRS) by adding the scores of the 20 segments of the rest images. To assess defect reversibility, a summed difference score (SDS) was calculated by subtracting SRS from SSS, reflecting the severity and the extent of ischaemia. MPS was considered abnormal in case of scar (SRS ≥4) or ischaemia (SDS ≥2). For the degree of ischaemia, an SDS of <2 was considered nonischaemic, SDS 2–4 mildly, SDS 5–8 moderately, SDS 9-12 severely and SDS >12 extensively ischaemic. For the definition of myocardial scar, an SRS <4 was considered normal, SRS 4–10 moderately and >10 severely abnormal [23].
Whenever possible an exercise stress protocol was performed (n = 1,071). RHR, blood pressure and 12-lead ECG were recorded before exercise. A standardised, sitting, stepwise and symptom limited bicycle exercise test was performed to the end points as defined in the exercise testing guidelines [24]. At near-maximal exercise a 740 MBq dose of Tc-99m sestamibi was injected and exercise was continued for at least an additional minute after injection. A 12-lead ECG was continuously recorded during exercise and recovery. Blood pressure was recorded every minute during exercise and recovery.
In patients who were unable to perform an exercise stress test a pharmacological vasodilator stress protocol was performed (n = 394). RHR, blood pressure and 12-lead ECG were recorded before pharmacological stress testing. Intravenous adenosine (140 µg/kg/min.) was infused for 6 minutes, and a 740 MBq dose of 99mTc-sestamibi was injected at the end of the third minute of adenosine infusion [22]. A 12-lead ECG was continuously recorded during exercise and recovery. Blood pressure was recorded every minute during exercise and recovery. Patients were instructed to refrain from caffeine, and medications such as aminophylline for 24 hours before MPS.
The RHR was defined on the basis of the 12-lead ECG recorded at rest before exercise or vasodilator stress testing in the sitting position.
Numerical data are presented as mean ± standard deviation or median and interquartile range as appropriate. Categorical data are presented as numbers and percentages. A dichotomous division of the population was done on the basis of the RHR which was below or above to the median value. Comparison between groups was done with Fisher’s test for categorical variables, Student’s t-test for numerical variables with equal distribution and Mann-Whitney-U test for numerical variables with non-equal distribution. Variables associated with RHR identified by comparison (p ≤0.1) were included in a multivariate logistic regression model to identify independent predictors of increased RHR.
Figure 1
Graphics illustrating the interrelation between the quartiles of resting heart rate (RHR in bpm) and the severity of myocardial ischaemia (SDS),
The association between quartiles of the RHR and the severity of myocardial ischaemia (SDS) and scar (SRS) by MPS was further assessed by Kruskal-Wallis test.
The comparison between patients with and without CAD findings at MPS was done by Fisher’s test for categorical variables and Student’s t-test for numerical variables. Factors associated with CAD findings at MPS identified by comparison (p ≤0.1) were included in a multivariable logistic regression model to identify independent predictors of CAD findings.
A p-value of ≤0.05 was considered statistically significant. Analyses were performed using the commercially available statistical package (SPSS version 16.0. Inc., Chicago, IL, USA).
The baseline characteristics of the study population are shown in table 1.
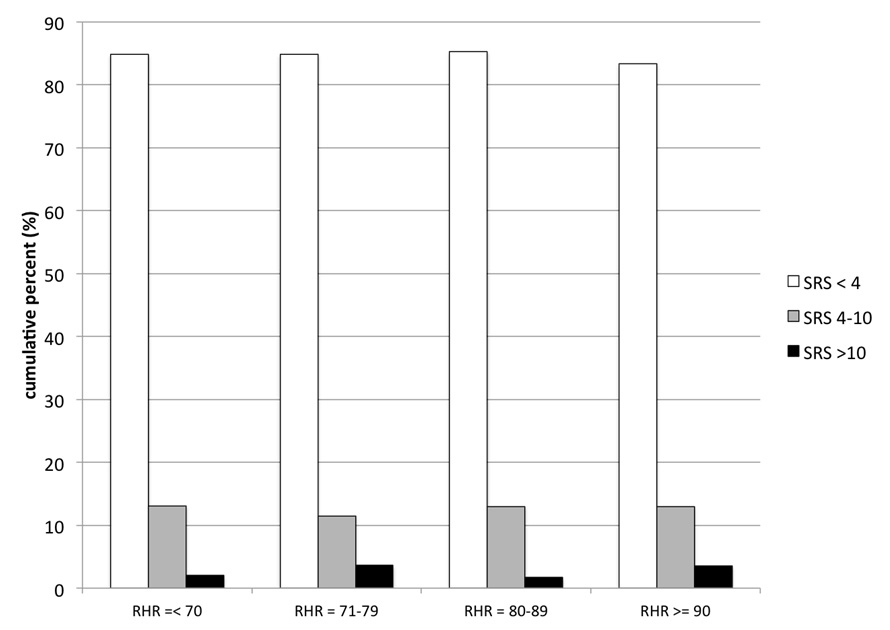
Figure 2
Graphics illustrating the interrelationship between the quartiles of resting heart rate (RHR in bpm) and the extent of myocardial scar (SRS) as assessed by MPS. Note that RHR was related neither to severity of myocardial ischaemia (p = 0.798), nor to the extent of myocardial scar (p = 0.869).
Approximately half of the patients presented with angina and the global burden of cardiovascular risk factors was high, as half of the patients had at least 2 cardiovascular risk factors. The average and median RHR was of 80 bpm. and 79 bpm. respectively.
As shown in (table 1), patients with an RHR above the median value of 79 bpm. were younger, more likely to be female, non-smokers, more frequently had diabetes and hypercholesterolaemia and were more likely to be on statins. Of note, that RHR was significantly higher in diabetic patients than their non-diabetic counterparts, 84 ±14 and 80 ±13 respectively (p <0.001). In female patients the heart rate was higher than in male patients, 81 ±13 and 80 ±14 respectively (p = 0.02).
In the multivariate analysis younger age, female gender, hypercholesterolaemia and diabetes were identified as independent predictors of elevated RHR (table 3). Another finding of note was that presence of CAD was not a predictor of elevated heart rate.
As shown in (table 2), more than ¼ of this patient population without a prior history of CAD did have MPS findings of CAD, including myocardial ischaemia (n = 332; 23%) or myocardial scar (n = 225; 15%). Patients with an RHR above the median value of 79 bpm did not have a higher prevalence of CAD findings as compared to patients with lower RHR. Moreover, increased RHR rate was not associated with the extent of myocardial ischaemia (SDS) or myocardial scar (SRS), nor with the global burden of CAD as assessed with the summed stress score (SSS). As shown in figs. 1 and 2, a quartile dependent analysis confirmed that elevated RHR was not associated with the severity of myocardial ischaemia (p = 0.798) or scar (p = 0.869) at MPS. On the other hand, patients presenting with angina (35% vs 22%; p <0.001), dyspnoea (40% vs 26%; p <0.001), or patients with established cardiovascular risk factors such as diabetes (34% vs 27%; p = 0.013), active smoking (32% vs 26%; p = 0.029), male gender (36% vs 18%; p <0.001) and higher age (65 ± 11 vs 60 ± 11; p <0.001) were more likely to have CAD findings at MPS. In a multivariate analysis angina, dyspnea, diabetes, male gender, and higher age turned out to be independent predictors of CAD findings at MPS but not RHR (table 4).
| Table 1: Baseline characteristics and univariable predictors of increased RHR. | ||||
| Overall (n = 1,465) | Resting HR <79 bpm (n = 736) | Resting HR ≥79 bpm (n = 729) | p-value | |
| Age | 61.8 ± 11.4 | 62.7 ± 11.2 | 60.9 ± 11.5 | 0.002 |
| Male gender | 845 (58) | 449 (61) | 396 (54) | 0.011 |
| BMI, kg/m2 | 27.2 ± 4.9 | 27.0 ± 4.6 | 27.4 ± 5.2 | 0.117 |
| Heart rate, bpm | 80±14 | 69 ± 7 | 91 ± 9 | <0.001 |
| Symptoms | ||||
| Angina | 689 (47) | 353 (48) | 336 (46) | 0.496 |
| Dyspnoea | 191 (13) | 91 (12) | 100 (14) | 0.485 |
| Risk factors | ||||
| Hypertension | 743 (51) | 358 (48) | 385 (54) | 0.117 |
| Diabetes | 250 (17) | 97 (13) | 153 (21) | <0.001 |
| Active smoking | 421 (29) | 231 (31) | 190 (26) | 0.028 |
| Hypercholesterolaemia | 555 (38) | 251 (34) | 302 (42) | 0.003 |
| Family history | 393 (27) | 188 (26) | 205 (28) | 0.288 |
| ≥2 risk factors | 733 (50) | 347 (47) | 386 (53) | 0.028 |
| Medication | ||||
| Aspirin | 624 (43) | 320 (44) | 304 (42) | 0.493 |
| ACE-inhibitors | 255 (17) | 119 (16) | 136 (19) | 0.215 |
| Angiotensin receptor blockers | 266 (18) | 121 (17) | 145 (20) | 0.091 |
| Statins | 318 (22) | 144 (20) | 174 (24) | 0.049 |
| Calcium channel blockers* | 212 (15) | 113 (15) | 99 (14) | 0.335 |
| Diuretics | 324 (22) | 148 (20) | 176 (24) | 0.068 |
| Data of continuous variables are given as mean ± standard deviation. Data of categorical variables are given as numbers (percentage). BMI = body mass index; ACE = angiotensin converting enzyme. * Nifedipin or amlodipine type calcium channel blockers. | ||||
| Table 2: Results of the SPECT study. | ||||
| Overall (n = 1,465) | Resting HR <79 bpm (n = 736) | Resting HR >79 bpm (n = 729) | p-value | |
| Physical stress testing | 1,071 (73) | 493 (70) | 578 (79) | 0.02 |
| LVEF | 58 ± 11 | 58 ± 11 | 58 ± 12 | 0.238 |
| SRS | 0 (0–2) | 0 (0–2) | 0 (0–2) | 0.752 |
| SSS | 0 (0–4) | 0 (0–4) | 0 (0–4) | 0.486 |
| SDS | 0 (0–0) | 0 (0–1) | 0 (0–0) | 0.278 |
| Ischaemia | 332 (23) | 174 (24) | 158 (22) | 0.383 |
| Scar | 225 (15) | 111 (15) | 114 (16) | 0.772 |
| CAD findings | 409 (28) | 205 (28) | 204 (28) | 1.000 |
| Data of continuous variables are given as mean ± standard deviation or median (interquartile ranges) as appropriate. Data of categorical variables are gives as numbers (percentage). LVEF = left ventricular ejection fraction; SRS = summed rest score; SSS = summed stress score; SDS = summed difference score. Ischaemia: SDS ≥2; Scar: SRS ≥4; CAD findings: presence of scar or ischemia at MPS. | ||||
| Table 3: Independent predictors of elevated RHR. | |||
| OR | 95%-CI | p-value | |
| Age, per year | 0.982 | 0.972–0.991 | <0.001 |
| Female gender | 1.369 | 1.102–1.702 | 0.005 |
| Hypercholesterolaemia | 1.307 | 1.010–1.702 | 0.042 |
| Diabetes | 1.718 | 1.287–2.298 | <0.001 |
| OR = odds ratio; CI = confidence interval. | |||
| Table 4: Independent clinical predictors of CAD findings at MPS. | |||
| OR | 95%-CI | p-value | |
| Angina | 2.075 | 1.616–2.659 | <0.001 |
| Dyspnoea | 1.678 | 1.192–2.358 | 0.003 |
| Diabetes | 1.379 | 1.012–1.879 | 0.042 |
| Male gender | 3.311 | 2.518–4.376 | <0.001 |
| Age, per year | 1.043 | 1.031–1.055 | <0.001 |
| RHR, per beat | 1.007 | 0.998–1.016 | 0.14 |
| OR = odds ratio; CI = confidence interval. | |||
Sustained elevation of RHR is thought to play an important role in the pathogenesis of atherosclerosis, and heart rate dependent acceleration of CAD development has been hypothesised as a possible pathomechanism through which elevated RHR raises cardiovascular morbidity and mortality [25]. To test the hypothesis that elevated RHR correlates with the presence and the extent of CAD in patients without a prior history of CAD, we analysed the association between RHR and MPS finding of CAD in consecutive patients with suspected CAD evaluated by MPS at our institution.
The results of this study suggest that there is no association between elevated RHR and (presence and extent of) CAD in patients without a prior history of CAD as assessed by MPS. On the other hand, the presence of other established cardiovascular risk factors, such as diabetes, male gender and age were predictors of CAD as assessed by MPS.
Such findings are somewhat in contradiction with the idea that sustained elevation of RHR may have a detrimental effect on initiation and progression of coronary atherosclerosis. This may in part be because of the different patient population we used for the analysis. A lot of the data in the literature has been acquired in animal studies or in congestive heart failure patients [8, 14].
Multiple animal studies showed in fact that increased RHR may cause dysfunction of the endothelial and vascular smooth muscle cells via heart rate dependent alterations of vascular shear stress [26–28]. Additionally, several experimental animal studies consistently showed that heart rate reduction leads to improvement of the endothelial function [29, 30]. However, while animal studies provide growing evidence about the pathophysiologic link between elevated RHR and atherosclerosis, data on humans are limited and still inconclusive. While Perski et al. reported that RHR rate was associated with progression of CAD among survivors of acute myocardial infarction [31] and Sipahi et al. showed that beta-blocker treatment was associated with reduced progression of CAD [32], Bangalore et al. reported that treatment with beta-blockers was associated with increased cardiovascular morbidity and mortality in hypertensive patients [33]. This finding may be explained by the haemodynamic consequences of beta-blocker therapy on the central circulation, where an augmentation of the central aortic systolic blood pressure may be observed [34, 35]. On the other hand, the interpretation of such analyses in humans is complicated by the fact that increased RHR in CAD and hypertensive patients may result from inadequate adherence to the medical regimen, since patients with higher RHR may have discontinued the intake of beta-blockers and other cardiovascular drugs. Therefore, adherence to the medical regimen, which is known to be a frequent problem in cardiovascular patients [36, 37], may represent an important confounder in the above-mentioned studies.
Considering that the association between RHR and the extent of CAD has not previously been well established in humans, the results of the current study provide important new insight in this context. Of course, as opposed to diagnostic techniques such as coronary angiography and intravascular ultrasonography, MPS does not provide information about the anatomy, morphology and function of the coronary vessels. However, MPS findings of ischaemia are known to correlate with the morphological extent of CAD and, even more, the prognostic value of MPS is superior to that provided by coronary angiography [38].
Hence the results of the current study may question the concept that initiation and progression of atherosclerosis and CAD is responsible for the increased cardiovascular and non-cardiovascular mortality related to sustained elevation of RHR in humans. In this context, other mechanisms such as reduced myocardial blood supply in patients with haemodynamically significant coronary stenosis related to increased heart rate and consequent shortening of the diastolic phase [19, 20], triggering of cardiac arrhythmias [39, 40], altered myocardial energetics [41], or consequences of autonomic neuropathy, especially in diabetics [42]. It is worthy of note that a higher RHR per se is not proof of autonomic neuropathy.
However, autonomic neuropathy may dominate the pathophysiological scenario linking sustained elevation of RHR and cardiovascular outcomes. Of note, cardiac autonomic neuropathy is linked with increased mortality in diabetic patients [43].
The results of the current study only apply to this “selected” population of patients without a prior history of CAD referred for MPS at the discretion of their treating physician. Here too, patients with atrial fibrillation, pacemaker rhythm, and intake of drugs with negative chronotropic effects were excluded. Hence some selection bias may have affected the results of this study. Importantly, patients with known CAD were excluded because the majority were on beta-blocker treatment and, even if anti-anginal drugs were discontinued whenever possible, a sustained effect on RHR has to be assumed.
Coronary angiography is known to be more sensitive than MPS in detecting an early manifestation of CAD such as coronary plaques without haemodynamically relevant stenosis. Thus we cannot exclude that the results would be different if coronary angiography were used to define the presence and extent of CAD.
RHR was not measured according to a pre-defined standard, as recommended by the European Society of Hypertension, meaning that the RHR was not measured after a resting period of 5 minutes and in sitting position in all patients [44, 45]. However, RHR was assessed by 12-lead ECG, thus ensuring a very precise measurement. In general a higher RHR than those measured according to the above-mentioned guidelines has to be expected, since RHR measurements are affected by a number of confounders, including psychic stimuli, body position, and environmental factors [45]. We feel that the method with which RHR was assessed in the current study does not influence the paper’s conclusions because we compared patient groups with lower versus higher heart rates as dichotomised by the median RHR of the population.
Elevated RHR is not associated with the presence and extent of CAD in patients evaluated for suspected but previously unknown CAD. This observation suggests that the impact of a higher RHR on mortality may be linked with other factors than only CAD itself.
1 Cook S, Togni M, Schaub MC, Wenaweser P, Hess OM. High heart rate: a cardiovascular risk factor? Eur Heart J. 2006;27:2387–93.
2 Fox K, Borer JS, Camm AJ, Danchin N, Ferrari R, Lopez Sendon JL, et al. Resting heart rate in cardiovascular disease. J Am Coll Cardiol. 2007;50:823–30.
3 Benetos A, Rudnichi A, Thomas F, Safar M, Guize L. Influence of heart rate on mortality in a French population: role of age, gender, and blood pressure. Hypertension. 1999;33:44–52.
4 Greenland P, Daviglus ML, Dyer AR, Liu K, Huang CF, Goldberger JJ, et al. Resting heart rate is a risk factor for cardiovascular and noncardiovascular mortality: the Chicago Heart Association Detection Project in Industry. Am J Epidemiol. 1999;149:853–62.
5 King DE, Everett CJ, Mainous AG, 3rd, Liszka HA. Long-term prognostic value of resting heart rate in subjects with prehypertension. Am J Hypertens. 2006;19:796–800.
6 Kolloch R, Legler UF, Champion A, Cooper-Dehoff RM, Handberg E, Zhou Q, et al. Impact of resting heart rate on outcomes in hypertensive patients with coronary artery disease: findings from the INternational VErapamil-SR/trandolapril STudy (INVEST). Eur Heart J. 2008;29:1327–34.
7 Diaz A, Bourassa MG, Guertin MC, Tardif JC. Long-term prognostic value of resting heart rate in patients with suspected or proven coronary artery disease. Eur Heart J. 2005;26:967–74.
8 Fox K, Ford I, Steg PG, Tendera M, Robertson M, Ferrari R. Heart rate as a prognostic risk factor in patients with coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): a subgroup analysis of a randomised controlled trial. Lancet. 2008;372:817–21.
9 Lechat P, Hulot JS, Escolano S, Mallet A, Leizorovicz A, Werhlen-Grandjean M, et al. Heart rate and cardiac rhythm relationships with bisoprolol benefit in chronic heart failure in CIBIS II Trial. Circulation. 2001;103:1428–33.
10 McAlister FA, Wiebe N, Ezekowitz JA, Leung AA, Armstrong PW. Meta-analysis: beta-blocker dose, heart rate reduction, and death in patients with heart failure. Ann Intern Med. 2009;150:784–94.
11 Pocock SJ, Wang D, Pfeffer MA, Yusuf S, McMurray JJ, Swedberg KB, et al. Predictors of mortality and morbidity in patients with chronic heart failure. Eur Heart J. 2006;27:65–75.
12 Inoue T, Oshiro S, Iseki K, Tozawa M, Touma T, Ikemiya Y, et al. High heart rate relates to clustering of cardiovascular risk factors in a screened cohort. Jpn Circ J. 2001;65:969–73.
13 Palatini P, Julius S. The physiological determinants and risk correlations of elevated heart rate. Am J Hypertens. 1999;12:3S–8S.
14 Fox K, Ford I, Steg PG, Tendera M, Ferrari R. Ivabradine for patients with stable coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): a randomised, double-blind, placebo-controlled trial. Lancet. 2008;372:807–16.
15 Swedberg K, Komajda M, Bohm M, Borer JS, Ford I, Dubost-Brama A, et al. Ivabradine and outcomes in chronic heart failure (SHIFT): a randomised placebo-controlled study. Lancet. 2010;376:875–85.
16 Huikuri HV, Jokinen V, Syvanne M, Nieminen MS, Airaksinen KE, Ikaheimo MJ, et al. Heart rate variability and progression of coronary atherosclerosis. Arterioscler Thromb Vasc Biol. 1999;19:1979–85.
17 Kaplan JR, Manuck SB, Clarkson TB. The influence of heart rate on coronary artery atherosclerosis. J Cardiovasc Pharmacol. 1987;10Suppl 2:S100–2; discussion S3.
18 Heidland UE, Strauer BE. Left ventricular muscle mass and elevated heart rate are associated with coronary plaque disruption. Circulation. 2001;104:1477–82.
19 Andrews TC, Fenton T, Toyosaki N, Glasser SP, Young PM, MacCallum G, et al. Subsets of ambulatory myocardial ischemia based on heart rate activity. Circadian distribution and response to anti-ischemic medication. The Angina and Silent Ischemia Study Group (ASIS). Circulation. 1993;88:92–100.
20 Panza JA, Diodati JG, Callahan TS, Epstein SE, Quyyumi AA. Role of increases in heart rate in determining the occurrence and frequency of myocardial ischemia during daily life in patients with stable coronary artery disease. J Am Coll Cardiol. 1992;20:1092–8.
21 Berman DS, Kiat H, Friedman JD, Wang FP, van Train K, Matzer L, et al. Separate acquisition rest thallium-201/stress technetium-99m sestamibi dual-isotope myocardial perfusion single-photon emission computed tomography: a clinical validation study. J Am Coll Cardiol. 1993;22:1455–64.
22 Zellweger MJ, Kaiser C, Brunner-La Rocca HP, Buser PT, Osswald S, Weiss P, et al. Value and limitations of target-vessel ischemia in predicting late clinical events after drug-eluting stent implantation. J Nucl Med. 2008;49:550–6.
23 Muzzarelli S, Pfisterer ME, Muller-Brand J, Zellweger MJ. Interrelation of ST-segment depression during bicycle ergometry and extent of myocardial ischaemia by myocardial perfusion SPECT. Eur J Nucl Med Mol Imaging. 2009;36:1842–50.
24 Gibbons RJ, Balady GJ, Bricker JT, Chaitman BR, Fletcher GF, Froelicher VF, et al. ACC/AHA 2002 guideline update for exercise testing: summary article. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Update the 1997 Exercise Testing Guidelines). J Am Coll Cardiol. 2002;40:1531–40.
25 Custodis F, Schirmer SH, Baumhakel M, Heusch G, Bohm M, Laufs U. Vascular pathophysiology in response to increased heart rate. J Am Coll Cardiol. 2010;56:1973–83.
26 Chatzizisis YS, Coskun AU, Jonas M, Edelman ER, Feldman CL, Stone PH. Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling: molecular, cellular, and vascular behavior. J Am Coll Cardiol. 2007;49:2379–93.
27 Cucherat M. Quantitative relationship between resting heart rate reduction and magnitude of clinical benefits in post-myocardial infarction: a meta-regression of randomized clinical trials. Eur Heart J. 2007;28:3012–9.
28 Williams B. Mechanical influences on vascular smooth muscle cell function. J Hypertens. 1998;16:1921–9.
29 Custodis F, Baumhakel M, Schlimmer N, List F, Gensch C, Bohm M, et al. Heart rate reduction by ivabradine reduces oxidative stress, improves endothelial function, and prevents atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2008;117:2377–87.
30 Drouin A, Gendron ME, Thorin E, Gillis MA, Mahlberg-Gaudin F, Tardif JC. Chronic heart rate reduction by ivabradine prevents endothelial dysfunction in dyslipidaemic mice. Br J Pharmacol. 2008;154:749–57.
31 Perski A, Olsson G, Landou C, de Faire U, Theorell T, Hamsten A. Minimum heart rate and coronary atherosclerosis: independent relations to global severity and rate of progression of angiographic lesions in men with myocardial infarction at a young age. Am Heart J. 1992;123:609–16.
32 Sipahi I, Tuzcu EM, Wolski KE, Nicholls SJ, Schoenhagen P, Hu B, et al. Beta-blockers and progression of coronary atherosclerosis: pooled analysis of 4 intravascular ultrasonography trials. Ann Intern Med. 2007;147:10–8.
33 Bangalore S, Sawhney S, Messerli FH. Relation of beta-blocker-induced heart rate lowering and cardioprotection in hypertension. J Am Coll Cardiol. 2008;52:1482–9.
34 Williams B, Lacy PS. Impact of heart rate on central aortic pressures and hemodynamics: analysis from the CAFE (Conduit Artery Function Evaluation) study: CAFE-Heart Rate. J Am Coll Cardiol. 2009;54:705–13.
35 Williams B, Lacy PS, Thom SM, Cruickshank K, Stanton A, Collier D, et al. Differential impact of blood pressure-lowering drugs on central aortic pressure and clinical outcomes: principal results of the Conduit Artery Function Evaluation (CAFE) study. Circulation. 2006;113:1213–25.
36 Granger BB, Swedberg K, Ekman I, Granger CB, Olofsson B, McMurray JJ, et al. Adherence to candesartan and placebo and outcomes in chronic heart failure in the CHARM programme: double-blind, randomised, controlled clinical trial. Lancet. 2005;366:2005–11.
37 Muzzarelli S, Brunner-La Rocca H, Pfister O, Foglia P, Moschovitis G, Mombelli G, et al. Adherence to the medical regime in patients with heart failure. Eur J Heart Fail. 2010;12:389–96.
38 Marie PY, Danchin N, Durand JF, Feldmann L, Grentzinger A, Olivier P, et al. Long-term prediction of major ischemic events by exercise thallium-201 single-photon emission computed tomography. Incremental prognostic value compared with clinical, exercise testing, catheterization and radionuclide angiographic data. J Am Coll Cardiol. 1995;26:879–86.
39 Bauer A, Kantelhardt JW, Barthel P, Schneider R, Makikallio T, Ulm K, et al. Deceleration capacity of heart rate as a predictor of mortality after myocardial infarction: cohort study. Lancet. 2006;367:1674–81.
40 La Rovere MT, Pinna GD, Hohnloser SH, Marcus FI, Mortara A, Nohara R, et al. Baroreflex sensitivity and heart rate variability in the identification of patients at risk for life-threatening arrhythmias: implications for clinical trials. Circulation. 2001;103:2072–7.
41 Ceconi C, Cargnoni A, Francolini G, Parinello G, Ferrari R. Heart rate reduction with ivabradine improves energy metabolism and mechanical function of isolated ischaemic rabbit heart. Cardiovasc Res. 2009;84:72–82.
42 Maser RE, Mitchell BD, Vinik AI, Freeman R. The association between cardiovascular autonomic neuropathy and mortality in individuals with diabetes: a meta-analysis. Diabetes Care. 2003;26:1895–901.
43 Vinik AI, Maser RE, Mitchell BD, Freeman R. Diabetic autonomic neuropathy. Diabetes Care. 2003;26:1553–79.
44 Cook S, Delacretaz E, Hess OM. Is resting heart rate a cardiovascular risk factor?. Praxis. 1994;2008;97:601–11.
45 Palatini P, Benetos A, Grassi G, Julius S, Kjeldsen SE, Mancia G et al. Identification and management of the hypertensive patient with elevated heart rate: statement of a European Society of Hypertension Consensus Meeting. J Hypertens. 2006;24:603–10.
Funding / potential competing interests: No financial support and no other potential conflict of interest relevant to this article was reported.
Authors’ contribution: A. Kohler and S. Muzzarelli share the first authorship.