Endogenous glucocorticoids in inflammation: contributions of systemic and local responses
DOI: https://doi.org/10.4414/smw.2012.13650
Rowan S.
Hardy, Karim
Raza, Mark S.
Cooper
Summary
The anti-inflammatory actions of therapeutic glucocorticoids are well established and these drugs are widely used to treat a variety of inflammatory conditions. It is also clear that endogenously synthesised glucocorticoids have an important role in regulating inflammatory responses. Traditionally, our understanding of the effects of endogenous glucocorticoids has been based on the levels of glucocorticoids within the circulation. These levels are controlled by the hypothalamic-pituitary-adrenal axis. However, more recently it has been established that the local level of glucocorticoids is of potential importance. Situations where the local level of glucocorticoids may differ from the level in the circulation are illustrated in this review. In addition, the mechanisms regulating local glucocorticoid levels and actions are identified. Increasingly, it will be important to understand how the levels of glucocorticoids within the circulation and within the tissues are regulated in a coordinated manner.
Introduction
Since their initial discovery by Kendall, Reichstein and Hench, glucocorticoids (GCs) have proven to be a highly effective tool in the treatment of all manner of inflammatory conditions. Consequently, our understanding of the adrenal synthesis of the endogenously active GC ‘cortisol’ and its regulation by the hypothalamic-pituitary-adrenal (HPA) axis has grown significantly, as has our knowledge of the complex metabolic pathways that regulate local GC bioavailability. Unfortunately, it rapidly became apparent that prolonged exposure to elevated therapeutic GCs also results in a range of detrimental side effects, collectively termed ‘Cushings syndrome’. Consequently, a variety of modified GC analogues were developed, with varying side effect profiles and altered metabolic properties that have proven effective as therapeutic agents. Inflammation has also been shown to be an effective inducer of circulating endogenous cortisol, through its modulating effects on the HPA axis, however, the actions of inflammation on local GC metabolism have proven equally as complex. In this review we report the latest finding on the mechanisms regulating local and systemic levels of endogenous and therapeutic GCs during inflammatory disease. Increasingly, to balance the positive and negative properties of GCs, it will be important to understand how their levels within the circulation and within tissues are regulated in a coordinated manner.
Glucocorticoids
Glucocorticoids are a class of steroid hormones named for their affects on glucose metabolism. Cortisol, the active physiological GC in humans (referred to as hydrocortisone when given as a pharmaceutical), is synthesised primarily in the zona fasciculata of the adrenal gland, and elicits a diverse array of homeostatic effects. These include modifying carbohydrate, protein and fat metabolism as well as complex immune suppression and modulator actions. The isolation and first use of cortisone (the metabolic precursor to the active GC cortisol) was first achieved by Kendall, Reichstein and Hench, for which they received a Nobel prize following its successful use in the treatment of rheumatoid arthritis (RA). Since then, therapeutic GC use in the treatment of inflammatory diseases has expanded dramatically, although their diverse biological actions continue to cause significant complications collectively referred to as Cushing’s syndrome [1].
The systemic metabolic actions of GCs include a general decrease in the uptake and utilisation of glucose in conjunction with an increase in hepatic gluconeogenesis, favouring increased circulating glucose levels. This is accompanied by increased muscle proteolysis and lipolysis. Other notable roles of GCs include maintenance of the circulation, influencing vascular tone and increasing salt and water retention. Both endogenous and exogenous GCs also have diverse anti-inflammatory and immune modulatory actions. These effects are mediated through a number of distinct mechanisms. These include actions on leukocyte populations, interfering with their trafficking, chemotaxis, phagocytosis and inflammatory activation as well as reducing the synthesis and release of many secreted mediators of inflammation [2]. Many of the direct clinical signs of inflammation, such as pain, warmth and swelling are directly opposed through increased annexin-1 expression and interference with pro-inflammatory prostaglandin and leukotriene production in conjunction with interference with intracellular pro-inflammatory pathways such as AP-1, MAPK and NF-κB [3].
The molecular mechanisms of GC signalling have been extensively characterised and reviewed [3, 4]. Classically, GCs elicit their actions through their binding to cytosolic glucocorticoid receptors (GRs). Circulating free cortisol, being lipophilic, passes directly across the cell membrane into the cytoplasm. In the absence of ligand, the GR exists within the cytoplasm bound to various heat shock proteins (HSP). Upon ligand binding, the GR detaches from these HSPs and translocates to the nucleus where it dimerises with another ligand bound GR. The resulting dimer binds to sequences of GR target genes termed glucocorticoid response elements. Consequently, the GR dimer can recruit multiple co-activators or repressors to either up or down-regulate gene expression. Further diversity is introduced through multiple GR isotypes. Of these GRα, the dominant GR isoform, is the primary mediator of GC effects [5]. In contrast GRβ appears to possess partial agonist activity, and acts as a competitive negative regulator of GR signalling [6–8]. Many of the anti-inflammatory actions of GCs occur through non-genomic mechanisms. In particular, the ligand bound GR can interfere with both AP-1 and NF-κB pro-inflammatory signalling [3]. There may also be a role for membrane bound GR in these responses [9].
The HPA axis
Systemically, circulating cortisol levels are regulated by the HPA axis (fig. 1). The hypothalamus produces corticotropin releasing hormone (CRH) in response to many distinct circadian, neurosensory, blood-borne and limbic signals [10]. CRH in turn acts on the pituitary gland resulting in the secretion of adrenocorticotrophic hormone (ACTH) into the circulation where it acts on its target organ, the adrenal gland, resulting in the synthesis and release of cortisol. Within the circulation cortisol is primarily bound to corticosteroid binding protein (CBG), with only a small fraction (~10%) existing as free, biologically active, cortisol. Circulating cortisol negatively regulates both CRH and ACTH release at the hypothalamus and pituitary respectively, creating a classical negative feedback loop.
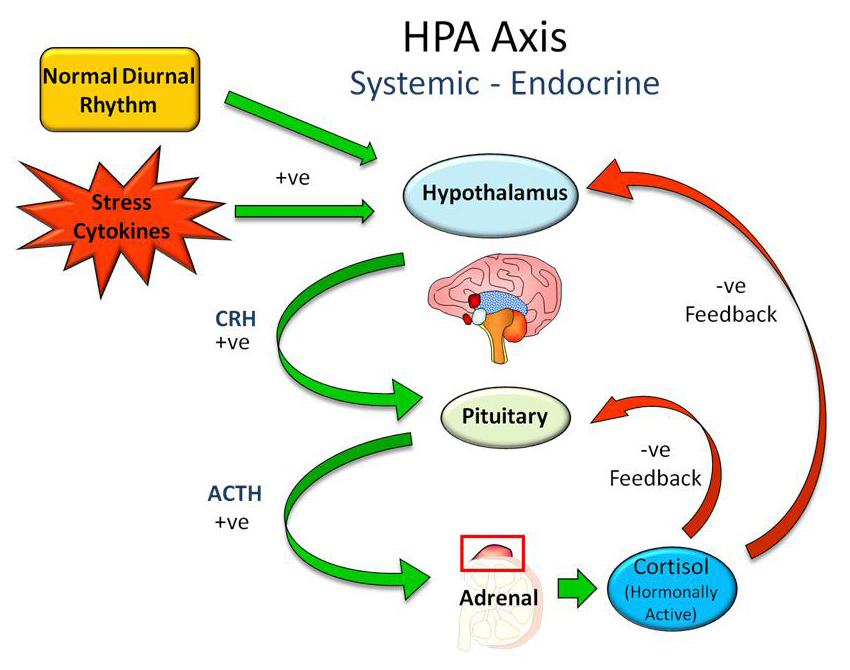
Figure 1
Systemic regulation of circulating cortisol levels by the hypothalamic pituitary adrenal (HPA) axis. Signalling to the hypothalamus by the normal circadian rhythm, stress, and pro-inflammatory cytokines increase release of corticotropin releasing hormone (CRH) from the hypothalamus. This acts on the pituitary to increase release of adrenocorticotropic hormone (ACTH) that acts to increase cortisol synthesis and secretion from the adrenal gland. Cortisol suppresses both CRF and ACTH at the pituitary and hypothalamus and a negative feedback loop.
In additional to the normal homeostatic regulation of cortisol secretion, a wide range of stressors are potent stimulators of CRH release and lead to an increase in the level of circulating cortisol. This can be clearly observed in patients who have undergone surgery or in patients with severe illness, who present with significantly higher circulating levels of cortisol [11, 12]. This normal response of the HPA axis to stress and inflammation has been well documented and characterised. During the early onset of infection, tissue damage and autoimmune disease, many cytokines produced as part of the innate immune response increase cortisol output via activation of the HPA axis. These include TNFα, IL-1β, IL-6 and the type I interferons (IFN-α/β) which are produced by macrophages, vascular endothelial cells, fibroblasts, and neurons [13]. Similarly, cytokines produced as part of the adaptive immune response such as IL-2 and IFN-γ are also effective in increasing HPA cortisol output [14]. In humans, these cytokines act not only on the hypothalamus to increase CRH release, but on the pituitary and adrenals resulting in marked increases in circulating ACTH and cortisol. Their respective mechanisms of action and effects on ACTH and cortisol are reviewed extensively by Turnbull et al. [15]. The resulting increase in circulating GCs has several important anti-inflammatory actions. Firstly the increased GC levels are able to minimise some of the detrimental effects of excessive inflammation, protecting the body from excessive inflammatory tissue damage and autoimmune shock [16]. Secondly they are able to modify the immune response, favouring a switch to a humoral Th2 rather than cell mediated Th1 response [17].
Abnormal HPA axis regulation in disease
The normal HPA axis response to inflammation appears to be important and beneficial. Abnormal HPA axis responses are implicated in a range of acute and chronic diseases. Structural abnormalities that reduce the function of the HPA axis are associated with an impaired acute response to stress and a greatly increased risk of death. This is seen in patients with untreated pituitary disease (hypopituitarism) or Addison’s disease [11]. The prolonged use of the anaesthetic agent etomidate in the ITU setting was associated with a dramatic increase in mortality, an effect that was subsequently determined to be due to this drug’s powerful inhibition of adrenal cortisol synthesis [18]. An area of intense debate in the critical care community is whether some patients develop functional abnormalities of the HPA axis during severe illness. This has been termed Critical Illness Related Corticosteroid Insufficiency (CIRCI) but it remains unclear how this condition should best be defined or treated. A complicating factor in the interpretation of HPA axis responses during acute illness is the alteration in the binding of cortisol to CBG. This is partly through a decrease in CBG synthesis as part of the acute phase response and partly due to peripheral degradation of CBG at sites of inflammation [11, 19]. The result is that the ratio of ‘free’ to total cortisol within the circulation and within tissues changes during inflammatory illness with an increase in the relative amount of free cortisol.
Abnormal HPA axis responses have also been implicated in the pathogenesis or persistence of chronic inflammatory disorders such as RA and inflammatory bowel disease (IBD). The reasoning behind this was partly that patients with these conditions respond very well symptomatically to therapeutic steroids suggesting there might be a deficiency of endogenous GCs through a failure of the HPA to respond to inflammation [20]. This view also arose from animal experiments which demonstrated that related histocompatible strains of rats had markedly different sensitivities to the induction of chronic experimental arthritis. The susceptibility in the arthritis prone animals was found to be due to failure of the HPA axis to increase GC levels in response to inflammation [21]. Administration of RU486, a GC receptor antagonist, made arthritis resistant rats more susceptible to experimental arthritis and the administration of the synthetic GC dexamethasone to arthritis prone animals rendered them arthritis resistant. These findings suggested that a similar defect might be present in humans who develop chronic inflammation. However, it has proven difficult to find such a defect. Patients with established RA who have never received therapeutic GCs have no consistent clinical or biochemical evidence of adrenal insufficiency when undergoing standard endocrine testing [22]. More recent research has examined the hypothesis that in patients with inflammatory diseases such as RA and IBD there might be relative corticosteroid insufficiency, with ‘normal’ levels of cortisol in the circulation actually being inappropriately low for the degree of inflammatory stress [23]. In some patients with RA there does appear to be an inappropriately low level of serum ACTH for the level of inflammation. The impairment of ACTH production appears related to chronic exposure to either inflammation or TNFα since treatment with anti-TNF therapy increases ACTH secretion and cortisol levels in a subset of these individuals. In IBD, abnormalities in the normal relationship between measures of inflammation and cortisol and adrenal androgen levels have been reported [24]. Both cortisol and adrenal androgens are regulated by ACTH suggested an alteration of the signaling pathways between the hypothalamus and the adrenal gland.
The major endpoint of these experiments was generally the level of cortisol in the systemic circulation. However, it is now clear that the HPA axis extends into the tissues and the axis itself can be influenced by changes occurring at the tissue level. This raises the possibility that the underlying defect in the HPA axis could be at a level beyond the circulation. The mechanism most likely to be important in the action of GCs at a tissue level is local GC metabolism.
Local glucocorticoid metabolism
In addition to the systemic regulation of GCs by the HPA axis, local GC bioavailability can be further modified through their pre-receptor metabolism. The most important enzymes in this regard are the 11β-hydroxysteroid dehydrogenases (11β-HSDs). These intracellular enzymes regulate the metabolism of the active GC cortisol with its inactive precursor cortisone (fig. 2) and thus alter the exposure of GC receptors to their ligands. There are two 11β-HSD enzymes termed 11β-HSD1 and 11β-HSD2. The first is a bidirectional enzyme that under physiological conditions favours the activation of cortisol from cortisone, increasing local GC concentrations greatly above circulating levels [25]. This enzyme is expressed in a range of tissues, including the liver, adipose, and bone, as well as showing marked expression within inflamed tissues such as the synovium [26]. Although widely used as an oral steroid, cortisone actually has no direct ability to bind to the GR. Instead, its action depends on its first pass metabolism to cortisol by 11β-HSD1 activity within the liver. In contrast, 11β-HSD2 solely inactivates GCs, converting active cortisol to inactive cortisone. Expressed within the kidney, pancreas and other mineralocorticoid sensitive tissues, 11β-HSD2 protects the mineralocorticoid receptor (MR) from occupation by cortisol, which circulates at a much higher concentration than aldosterone. 11β-HSD2 thus confers specificity on the MR. In addition to metabolism of endogenous GCs, these enzymes also metabolise synthetic therapeutic GCs and their 11-keto derivatives. Of note, prednisolone and its inactive metabolite prednisone share a similar metabolism to that of cortisol and cortisone [27]. In contrast, dexamethasone, as a result of a substitution of a fluoride molecule at the 9α position of the glucocorticoid structure, undergoes very different metabolism by the 11β-HSD enzymes. For example, 11β-HSD2 is less effective at inactivating dexamethasone and interestingly, whereas 11β-HSD2 solely inactivates endogenous GCs, it is bidirectional for dexamethasone, favouring steroid activation [28].
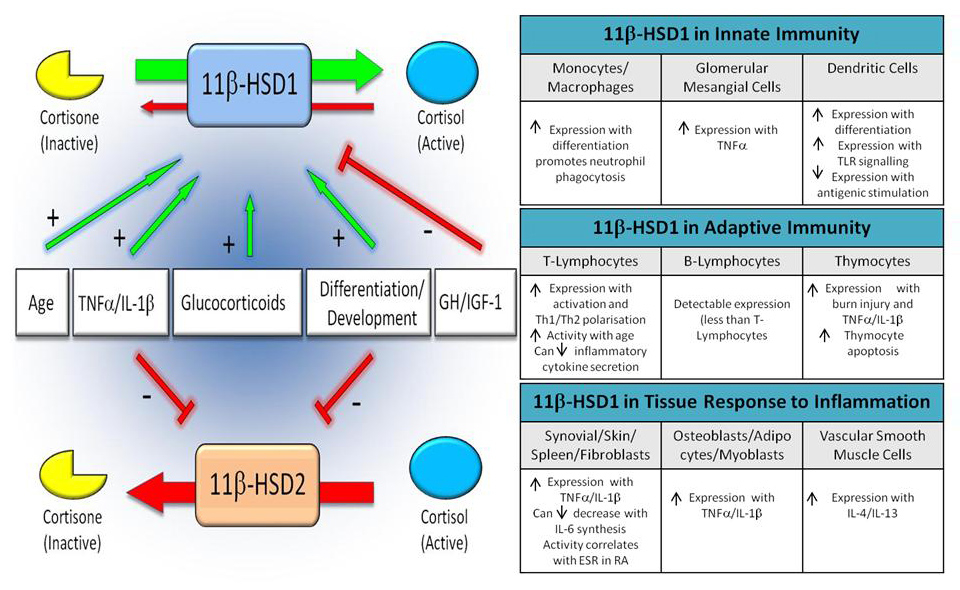
Figure 2
Overview of the cortisol activating enzyme 11β-HSD1, and the cortisol inactivating enzyme 11β-HSD2 and their regulation within different leukocyte subsets.
Tissue expression of 11β-HSD1 enzyme
The 11β-HSD1 enzyme has an extensive distribution throughout human tissues including liver, adipose tissue, skin, muscle, bone, gonads, eye, adrenal, pituitary and lymphoid tissues [29]. Of these, the highest 11β-HSD1 expression has been reported within tissues that are known to be highly GC sensitive such as liver, gonads, adipose and brain. 11β-HSD1 has diverse roles, including the regulation of development, adiposity, glucose metabolism, bone turnover and neurological development [30, 31]. More recently 11β-HSD1 has been implicated in the regulation of the local immune responses through its expression in stromal cells and in leukocytes at sites of inflammation.
Stromal expression of 11β-HSD1 enzymes
11β-HSD1 is expressed in a wide range of mesenchymal derived stromal cells, including adipocytes, osteoblasts, myoblasts, fibroblasts, and vascular smooth muscle cells [29]. Its expression has been explored most extensively in fat tissue (where it might play a role in visceral obesity) and bone (where it might regulate bone formation relative to resorption [32]). 11β-HSD1 has been examined in multiple fibroblast populations, including those isolated from thymus, bone marrow, skin and synovium [33, 34]. These fibroblasts possess variable basal expression of 11β-HSD1 that is highly site specific and retained over prolonged culture. Despite these differences, the level of enzyme activity in all of these cells was strongly upregulated by pro-inflammatory cytokines such as TNFα and IL-1β. Interestingly this upregulation of enzyme activity occurred in the presence of GCs and, in many of these cells, the combination of proinflammatory cytokines and GCs dramatically increased enzyme expression further still [35].
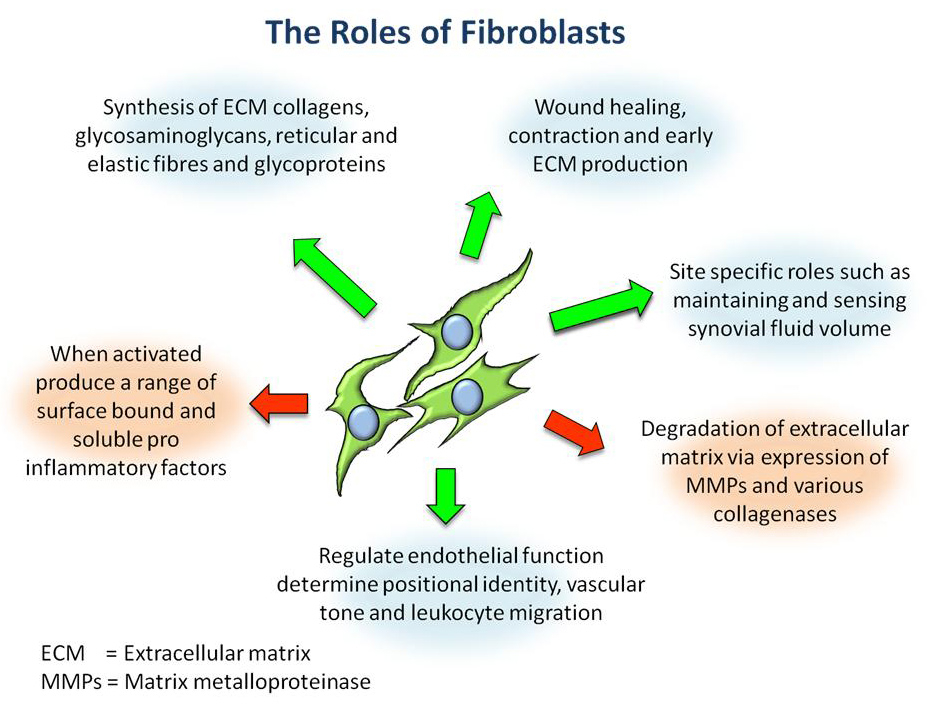
Figure 3
Overview of the diverse roles of stromal fibroblasts within connective tissues.
Of particular relevance to the local immune response in inflammatory arthritis are synovial fibroblasts that play a diverse role within connective tissues (fig. 3). Early studies by Gay et al. identified that when activated during inflammatory joint disease, synovial fibroblasts take on an aggressive, invasive phenotype, breaking down articular cartilage through the action of matrix metalloproteinases (MMPs) [36]. This was supported by further discoveries that their production of secreted factors such as RANKL increases osteoclast differentiation, survival and activity, thus contributing to bone erosion, dysregulation of bone metabolism and juxta articular osteoporosis [37]. Synovial fibroblasts are also actively involved in maintaining the inflammatory process, activating leukocyte populations to regulate their migration, survival and retention. In their basal state, primary synovial fibroblasts possess significant 11β-HSD1 expression and activity [33]. This has been shown for synovial fibroblasts isolated from patients with inflammatory arthritis but also for patients with non-inflammatory joint conditions. The 11β-HSD1 activity in synovial fibroblasts is greater than that seen in matched fibroblasts from skin or bone marrow obtained from the same donor. Enzyme activity also increases dramatically in response to pro-inflammatory cytokines [33]. Enzyme activity appears functionally important since synovial fibroblasts isolated from inflamed tissues are able to attenuate IL-6 and CCL-2 expression in response to endogenously derived GCs, following stimulation with TNFα and IL-1β [33]. As mentioned above, endogenously derived GCs can synergise with pro-inflammatory cytokines in stromal fibroblasts, significantly increasing 11β-HSD1 by an autocrine feed-forward mechanism [35]. Expression of 11β-HSD1 in synovial fibroblasts has also been demonstrated in synovial tissue in vivo. Schmidt et al. demonstrated that 11β-HSD1 was expressed in synovial tissue and that the level of expression increased in relation to the extent of inflammation [34]. Hardy et al. also demonstrated 11β-HSD1 expression in synovial tissue fibroblasts using immunohistochemisty [26]. Enzyme activity assays in synovial tissue also demonstrated the ability of synovial tissue to interconvert cortisone and cortisol including the ability to regenerate cortisol – an exclusive property of 11β-HSD1. Despite this, Schmidt et al. went on to show that 11β-HSD1 activity varied between different disease states, with rheumatoid arthritis synovium having less cortisol activation relative to OA [34]. One implication of this is that persistent inflammatory disease may arise as a result of an attenuated 11β-HSD1 response in susceptible individuals.
Immune system; - monocytes, lymphocytes
Many cells of both the innate and adaptive immunity also possess 11β-HSD1 activity. These include macrophages, dendritic cells, mast cells and T cells [33, 38, 39]. Whilst circulating peripheral blood mononuclear cells have no identifiable 11β-HSD1 expression, it is significantly up-regulated during macrophage differentiation [38, 40]. In type 1 macrophages (anti-microbial, phagocytic, lymphokine producing), elevated expression of 11β-HSD1 increases their sensitivity to endogenous GCs. This in turn down regulates their pro-inflammatory behaviour and reduces their survival. In contrast endogenously derived GCs boost the capacity to clear apoptotic neutrophils and cell debris in type 2 macrophages (wound healing, repair, anti-inflammatory) [41]. The role of GC signalling in macrophages is further complicated by the discovery of 11β-HSD2 within the inflamed synovium and tissue macrophages specifically [26, 34, 42]. This could be particularly important as macrophages expressing this enzyme may be rendered insensitive to local endogenous cortisol, as well as certain therapeutic GCs such as prednisolone. As a consequence these cells may be protected from the normal pro-resolution actions of GCs. Schmidt et al. in particular identified that 11β-HSD2 was elevated to a greater extent than 11β-HSD1 in RA, suggesting that this may account for relative GC insensitivity in these tissues, although this was not observed in following studies [26, 34]. Identifying and characterising the expression of 11βHSD2 in different macrophage subtypes remains to be achieved. Detectable levels of 11β-HSD1 have been identified within CD4+ T cells, CD8+ T cells, mast cells, B220+ and CD11+ dendritic cells of lymph nodes and spleen [39, 43]. Closer examination of CD4+ cells confirmed that they up-regulated this expression upon activation and polarisation into Th1 and Th2 cells. It is believed that the presence of this enzyme in these cells will perform multiple functions, supporting resolution of inflammation but also influencing function, survival and differentiation of T cells.
Evidence for a role of 11β-HSD1 in regulating inflammation in vivo
In addition to regulation of GCs via the HPA axis, there is a growing body of work demonstrating the importance of 11β-HSD1 in regulating local GC levels during inflammation in vivo [43]. At the tissue level in patients with colitis or inflammatory bowel disease, 11β-HSD1 is increased, whilst 11β-HSD2 expression is decreased compared to normal controls [44, 45]. Similarly, when looking at inflamed synovium isolated from patients with either RA or osteoarthritis, there is a marked increase in both 11β-HSD1 expression and its associated steroid activating activity [26]. Furthermore, within the synovium of RA patients it was shown that the capacity for the tissue to generate cortisol correlated with disease activity. This was mirrored in systemic measures of 11β-HSD1 activity in these individuals (based on urinary corticosteroid metabolite analysis), with the greatest activity being seen in patients with the highest levels of CRP and ESR.
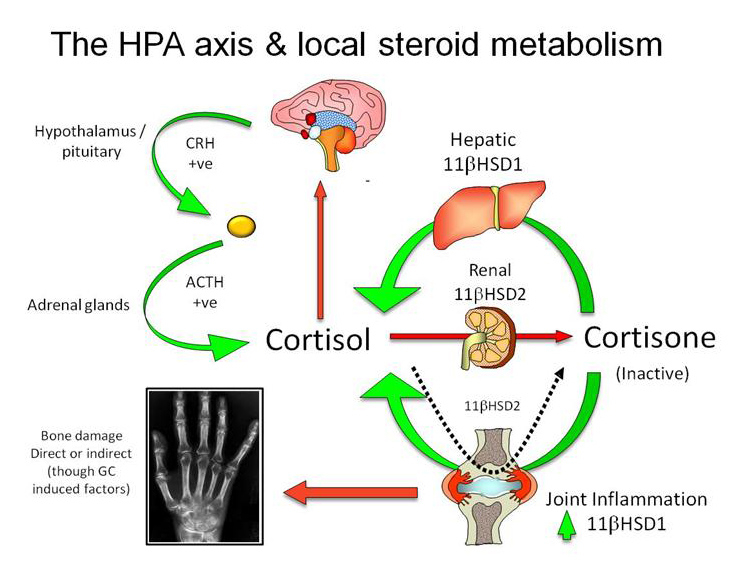
Figure 4
The integration of the local metabolism of cortisol within the liver, kidneys and inflamed tissues, with systemic regulation of circulating cortisol by the HPA axis. Hepatic 11β-HSD1 reactivates circulating cortisone, whilst renal 11β-HSD2 expression inactivates cortisol. During inflammation, elevated 11β-HSD1 expression within inflamed tissues amplifies the local cortisol concentration, which can result in detrimental side effects such as bone loss in inflammatory disease. Persistent 11β-HSD2 expression within specific leukocyte subsets during inflammation will confer steroid resistance.
The possible functional role of 11β-HSD1 in the immune response has been examined in vivo in studies using chemical inhibitors of 11β-HSD1 and in transgenic animals with global deletion of 11β-HSD1. A major limitation of these studies is that it is difficult to examine the relative contributions of 11β-HSD1 within different cell populations to the immune response. A particular limitation is the inability to separate out the contribution that stromal 11β-HSD1 makes relative to enzyme expression within leukocytes. The earliest reports examining the effect of 11β-HSD inhibition were performed before the widespread appreciation that there are two distinct 11β-HSD enzymes and that 11β-HSD1 predominantly mediates a steroid activating reaction. Systemic inhibition of 11β-HSD activity using glycyrrhetinic acid resulted in reduced resistance to infection with Listeria monocytogenese [46]. Experiments in 11β-HSD1 knockout mice have shown defects in macrophage phagocytosis of apoptotic neutrophils during peritoneal inflammation [41]. This was attributed to failure of upregulation of macrophage 11β-HSD1 since glucocorticoids are known to stimulate the clearance of apoptotic neutrophils by macrophages. 11β-HSD1 knockout mice also display enhanced sensitivity in response to LPS injection [47]. This effect was attributed to enhanced cytokine production from macrophages, an effect associated with their abnormally rapid differentiation after reduction in cortisol levels (in the absence of 11β-HSD1).
A wider range of immune responses were recently examined in global 11β-HSD1 knockout mice [48]. These included models of joint inflammation, peritonitis and lung inflammation. Inflammation was greater in these models and took longer to resolve. A few studies have gone further to examine the possible role of inflammation associated 11β-HSD1 in humans in vivo. As discussed above, 11β-HSD1 activity measured at a tissue or systemic level, increases substantially with inflammation in patients with RA [26]. It seems likely that the consequent increase in cortisol production within the joint limits inflammation locally. In keeping with this, administration of metyrapone, an inhibitor of 11β-HSD1 activity, worsened synovial inflammation in patients with RA [49]. There is also evidence that IBD is associated with increases in 11β-HSD1 activity. This has been demonstrated at mRNA and protein levels in biopsies of colonic tissue obtained from patients with or without colitis [45]. Patients with IBD with acute exacerbations of their condition show a significant increase in systemic 11β-HSD1 activity, most likely originating from the inflamed bowel [50]. Interestingly, patients with IBD who are in clinical remission also have high levels of systemic 11β-HSD1 activity compared to healthy volunteers. This suggests local GC production within inflamed tissues might be sufficient to suppress the clinical features of inflammation. Overall these findings suggest that 11β-HSD1 is probably an important regulator of the immune response and limits the degree of acute inflammation. This is probably through increased 11β-HSD1 activity in inflamed tissues (and the consequent increase in tissue GC levels) and through effects of altered 11β-HSD1 activity in immune cells (fig. 4).
What determines whether responses are systemic or local?
It appears that a rise in tissue GC levels is a common feature of the inflammatory response. However, it remains unclear the extent to which the increased level of GC within the tissue is due to activation of the HPA axis or to local tissue responses. This is further complicated by the finding that two potent stimulators of the HPA axis (TNFα and IL-1β) are also potent stimulators of 11β-HSD1 expression in many tissues [35]. In the most simplistic model, low levels of inflammation within a tissue might be countered by a rise in local GC production that might be sufficient (along with other components of the immune response) to contain inflammation and lead to its resolution. With greater degrees of inflammation the local production of GC might be insufficient to control inflammation locally so a systemic response might be required. However, in chronic inflammatory disease such as RA and IBD neither local nor systemic steroid responses, nor their combination, appear sufficient to control inflammation. Further studies will be required to clarify whether differences in GC response at a local or a systemic level are linked to the phenotypic manifestations of inflammatory conditions or to whether inflammation resolves appropriately or inappropriately.
References
1 Hardy RS, Cooper MS. Glucocorticoid-induced osteoporosis – a disorder of mesenchymal stromal cells? Front Endocrin. 2011;2(24 ).
2 McKay LI, Cidlowski JA. Molecular control of immune/inflammatory responses: interactions between nuclear factor-kappa B and steroid receptor-signaling pathways. Endocr Rev. 1999;20(4):435–59.
3 Schoneveld OJ, Gaemers IC, Lamers WH. Mechanisms of glucocorticoid signalling. Biochim Biophys Acta. 2004;1680(2):114–28.
4 Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids - new mechanisms for old drugs. N Engl J Med. 2005;353(16):1711–23.
5 Hollenberg SM, Weinberger C, Ong ES, Cerelli G, Oro A, Lebo R, et al. Primary structure and expression of a functional human glucocorticoid receptor Cdna. Nature. 1985;318(6047):635–41.
6 Bamberger CM, Bamberger AM, Decastro M, Chrousos GP. Glucocorticoid receptor-beta, a potential endogenous inhibitor of glucocorticoid action in humans. J Clin Invest. 1995, 95(6):2435–41.
7 Yudt MR, Jewell CM, Bienstock RJ, Cidlowski JA. Molecular origins for the dominant negative function of human glucocorticoid receptor beta. Mol Cell Biol. 2003;23(12):4319–30.
8 Oakley RH, Jewell CM, Yudt MR, Bofetiado DM, Cidlowski JA. The dominant negative activity of the human glucocorticoid receptor beta isoform. Specificity and mechanisms of action. J Biol Chem. 1999;274(39):27857–66.
9 Strehl C, Gaber T, Lowenberg M, Hommes DW, Verhaar AP, Schellmann S, et al. Origin and functional activity of the membrane-bound glucocorticoid receptor. Arthritis Rheum. 2011;63(12):3779-–88.
10 Chrousos GP. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N Engl J Med. 1995;332(20):1351–62.
11 Cooper MS, Stewart PM. Corticosteroid insufficiency in acutely ill patients. N Engl J Med. 2003;348(8):727–34.
12 Rothwell PM, Udwadia ZF, Lawler PG. Cortisol response to corticotropin and survival in septic shock. Lancet. 1991;337(8741):582–3.
13 Besedovsky HO, DelRey A. Immune-neuro-endocrine interactions: facts and hypotheses. Endocr Rev. 1996;17(1):64–102.
14 Naito Y, Fukata J, Tominaga T, Masui Y, Hirai Y, Murakami N, et al. Adrenocorticotropic hormone-releasing activities of interleukins in a homologous invivo system. Biochem Bioph Res Co. 1989;164(3):1262–7.
15 Turnbull AV, Rivier CL. Regulation of the hypothalamic-pituitary-adrenal axis by cytokines: actions and mechanisms of action. Physiol Rev. 1999;79(1):1–71.
16 Silverman MN, Pearce BD, Biron CA, Miller AH. Immune modulation of the hypothalamic-pituitary-adrenal (HPA) axis during viral infection. Viral Immunol. 2005;8(1):41–78.
17 Chrousos GP. The stress response and immune function: clinical implications. The 1999 Novera H. Spector Lecture. Ann N Y Acad Sci. 2000;917:38–67.
18 Allolio B, Stuttmann R, Fischer H, Leonhard W, Winkelmann W. Long-term etomidate and adrenocortical suppression. Lancet. 1983;2(8350):626.
19 Cooper MS, Stewart PM. Adrenal insufficiency in critical illness. J Intensive Care Med. 2007;22(6):348–62.
20 Panayi GS. Hormonal control of rheumatoid inflammation. Br Med Bull. 1995;51(2):462–71.
21 Sternberg EM, Hill JM, Chrousos GP, Kamilaris T, Listwak SJ, Gold PW, et al. Inflammatory mediator-induced hypothalamic-pituitary-adrenal axis activation is defective in streptococcal cell wall arthritis-susceptible Lewis rats. Proc Natl Acad Sci U S A. 1989;86(7):2374–8.
22 Jessop DS, Harbuz MS. A defect in cortisol production in rheumatoid arthritis: why are we still looking? Rheumatology. 2005;44(9):1097–100.
23 Straub RH, Paimela L, Peltomaa R, Scholmerich J, Leirisalo-Repo M. Inadequately low serum levels of steroid hormones in relation to interleukin-6 and tumor necrosis factor in untreated patients with early rheumatoid arthritis and reactive arthritis. Arthritis Rheum. 2002;46(3):654–62.
24 Straub RH, Vogl D, Gross V, Lang B, Scholmerich J, Andus T. Association of humoral markers of inflammation and dehydroepiandrosterone sulfate or cortisol serum levels in patients with chronic inflammatory bowel disease. Am J Gastroenterol. 1998;93(11):2197–202.
25 Cooper MS, Stewart PM. 11Beta-hydroxysteroid dehydrogenase type 1 and its role in the hypothalamus-pituitary-adrenal axis, metabolic syndrome, and inflammation. J Clin Endocrinol Metab. 2009;94(12):4645–54.
26 Hardy R, Rabbitt EH, Filer A, Emery P, Hewison M, Stewart PM, et al. Local and systemic glucocorticoid metabolism in inflammatory arthritis. Ann Rheum Dis. 2008;67(9):1204–10.
27 Cooper MS, Bujalska I, Rabbitt E, Walker EA, Bland R, Sheppard MC, et al.Modulation of 11 beta-hydroxysteroid dehydrogenase isozymes by proinflammatory cytokines in osteoblasts: an autocrine switch from glucocorticoid inactivation to activation. J Bone Miner Res. 2001;16(6):1037–44.
28 Patel P, Hardy R, Vaiyapuri S, Bartle G, Kindblom LG, Grimer R, et al. Expression of 11beta-hydroxysteroid dehydrogenase enzymes in human osteosarcoma: potential role in pathogenesis and as targets for treatments. Endocr Relat Cancer. 2012.
29 Tomlinson JW, Walker EA, Bujalska IJ, Draper N, Lavery GG, et al. 11beta-hydroxysteroid dehydrogenase type 1: a tissue-specific regulator of glucocorticoid response. Endocr Rev. 2004;25(5):831–66.
30 Lakshmi V, Nath N, Muneyyirci-Delale O. Characterization of 11 beta-hydroxysteroid dehydrogenase of human placenta: evidence for the existence of two species of 11 beta-hydroxysteroid dehydrogenase. J Steroid Biochem Mol Biol. 1993;45(5):391–7.
31 Cooper MS, Hewison M, Stewart PM. Glucocorticoid activity, inactivity and the osteoblast. J Endocrinol. 1999;163(2):159–64.
32 Cooper MS, Bujalska I, Rabbitt E, Walker EA, Bland R, Sheppard MC, et al. Modulation of 11beta-hydroxysteroid dehydrogenase isozymes by proinflammatory cytokines in osteoblasts: an autocrine switch from glucocorticoid inactivation to activation. J Bone Miner Res. 2001;16(6):1037–44.
33 Hardy RS, Filer A, Cooper MS, Parsonage G, Raza K, Hardie DL, et al. Differential expression, function and response to inflammatory stimuli of 11beta-hydroxysteroid dehydrogenase type 1 in human fibroblasts: a mechanism for tissue-specific regulation of inflammation. Arthritis Res Ther. 2006;8(4):R108.
34 Schmidt M, Weidler C, Naumann H, Anders S, Scholmerich J, Straub RH. Reduced capacity for the reactivation of glucocorticoids in rheumatoid arthritis synovial cells: possible role of the sympathetic nervous system? Arthritis Rheum. 2005;52(6):1711–20.
35 Kaur K, Hardy R, Ahasan MM, Eijken M, van Leeuwen JP, Filer A, et al. Synergistic induction of local glucocorticoid generation by inflammatory cytokines and glucocorticoids: implications for inflammation associated bone loss. Ann Rheum Dis. 2010;69(6):1185–90.
36 MullerLadner U, Kriegsmann J, Franklin BN, Matsumoto S, Geiler T, Gay RE, et al. Synovial fibroblasts of patients with rheumatoid arthritis attach to and invade normal human cartilage when engrafted into SCID mice. Am J Pathol. 1996;149(5):1607–15.
37 Shigeyama Y, Pap T, Kunzler P, Simmen BR, Gay RE, Gay S. Expression of osteoclast differentiation factor in rheumatoid arthritis. Arthritis Rheum. 2000;43(11):2523–30.
38 Thieringer R, Le Grand CB, Carbin L, Cai TQ, Wong B, Wright SD, et al. 11 Beta-hydroxysteroid dehydrogenase type 1 is induced in human monocytes upon differentiation to macrophages. J Immunol. 2001;167(1):30–5.
39 Zhang TY, Ding X, Daynes RA. The expression of 11 beta-hydroxysteroid dehydrogenase type I by lymphocytes provides a novel means for intracrine regulation of glucocorticoid activities. J Immunol. 2005;174(2):879–89.
40 Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol. 2006;177(10):7303–11.
41 Gilmour JS, Coutinho AE, Cailhier JF, Man TY, Clay M, Thomas G, et al. Local amplification of glucocorticoids by 11 beta-hydroxysteroid dehydrogenase type 1 promotes macrophage phagocytosis of apoptotic leukocytes. J Immunol. 2006;176(12):7605–11.
42 Haas CS, Creighton CJ, Pi X, Maine I, Koch AE, Haines GK, Ling S, et al. Identification of genes modulated in rheumatoid arthritis using complementary DNA microarray analysis of lymphoblastoid B cell lines from disease-discordant monozygotic twins. Arthritis Rheum. 2006;54(7):2047–60.
43 Chapman KE, Coutinho AE, Gray M, Gilmour JS, Savill JS, Seckl JR. The role and regulation of 11beta-hydroxysteroid dehydrogenase type 1 in the inflammatory response. Mol Cell Endocrinol. 2009;301(1-2):123–31.
44 Zbankova S, Bryndova J, Leden P, Kment M, Svec A, Pacha J. 11beta-hydroxysteroid dehydrogenase 1 and 2 expression in colon from patients with ulcerative colitis. J Gastroenterol Hepatol. 2007;22(7):1019–23.
45 Stegk JP, Ebert B, Martin HJ, Maser E. Expression profiles of human 11beta-hydroxysteroid dehydrogenases type 1 and type 2 in inflammatory bowel diseases. Mol Cell Endocrinol. 2009;301(1-2):104–8.
46 Hennebold JD, Mu HH, Poynter ME, Chen XP, Daynes RA. Active catabolism of glucocorticoids by 11 beta-hydroxysteroid dehydrogenase in vivo is a necessary requirement for natural resistance to infection with Listeria monocytogenes. Int Immunol. 1997;9(1):105–15.
47 Zhang TY, Daynes RA. Macrophages from 11beta-hydroxysteroid dehydrogenase type 1-deficient mice exhibit an increased sensitivity to lipopolysaccharide stimulation due to TGF-beta-mediated up-regulation of SHIP1 expression. J Immunol. 2007;179(9):6325–35.
48 Coutinho AE, Gray M, Brownstein DG, Salter DM, Sawatzky DA, Clay S, et al. 11beta-hydroxysteroid dehydrogenase type 1, but not type 2, deficiency worsens acute inflammation and experimental arthritis in mice. Endocrinology. 2012;153(1):234–40.
49 Saldanha C, Tougas G, Grace E. Evidence for anti-inflammatory effect of normal circulating plasma cortisol. Clin Exp Rheumatol. 1986;4(4):365–6.
50 Cooper MS, Kriel H, Sayers A, Fraser WD, Williams AM, Stewart PM, et al. Can 11beta-hydroxysteroid dehydrogenase activity predict the sensitivity of bone to therapeutic glucocorticoids in inflammatory bowel disease? Calcif Tissue Int. 2011;89(3):246–51.