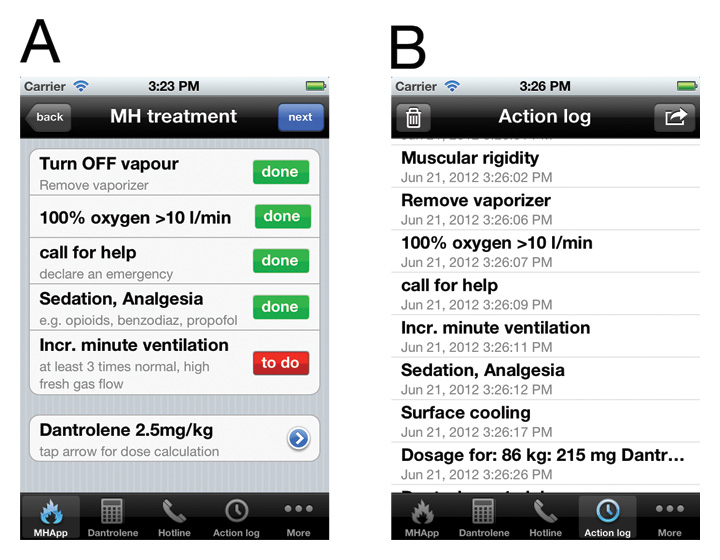
Figure 1
Two screenshots of the MHApp. Panel A shows the first five treatment steps, which all have to be confirmed before proceeding. Panel B shows the automatically generated protocol. This protocol can be forwarded by email.
DOI: https://doi.org/10.4414/smw.2012.13652
Peri-operative fatalities associated with hyperthermia have been reported since the introduction of general anaesthesia in the 19th century [1]. It was not until 1960, however, when Denborough and Lovell described a peri-operative hyperthermia disorder in a family with an autosomal dominant mode of inheritance, that the true nature of these fatalities became clearer [2]. Denborough and Lovell named the disorder ‘malignant hyperthermia’. In the 1970s, mortality from malignant hyperthermia (MH) crises was still over 80% [3]. Dedicated research has since unveiled the pathogenesis of this potentially fatal anaesthesia complication (i.e., being a pharmacogenetic disorder of skeletal muscle calcium regulation [4]). Furthermore, a specific and effective medical treatment with dantrolene has been discovered. Nowadays, if the disorder is recognised expeditiously and an adequate treatment is initiated, an MH episode represents a very dangerous yet treatable state; and mortality has decreased to less than 5% [5]. Furthermore, preventive (pre-symptomatic) diagnostic measures including in vitrocontracture testing and molecular genetic analysis have substantially increased peri-operative patient safety. Nevertheless and even more tragic at this stage of knowledge, fatal events linked to MH continue to appear in the literature [6]. This underlines the importance of continued vigilance towards this subclinical myopathy. If not, MH remains as fatal as it was a century ago [6]. This review summarises the essential and important facts about MH for the clinician and presents new developments in the field.
MH is defined as a disturbance of the skeletal muscle calcium homeostasis, triggered by volatile anaesthetics and depolarising muscle relaxants (i.e., all halogenated inhalative anaesthetics such as halothane, isoflurane, sevoflurane, desflurane and the depolarising muscle relaxant succinylcholine). In humans, exposure to stresses like heat or vigorous exercise has been infrequently implicated in the disorder. In the absence of triggering agents, the affected individuals appear normal without any pathologic signs, as MH represents (in most cases) a subclinical myopathy. However, some known characteristic muscle diseases with a clinical phenotype (e.g., central core disease, multi-minicore disease and King-Denborough syndrome) are associated with MH episodes [7, 8], whereas other muscle diseases (e.g., Thomsen’s disease and Becker type myotonia) most probably are not [9]. Once a vulnerable individual is exposed to a triggering agent, a pathologic hypermetabolic response featuring a rapid increase in oxygen consumption and expired carbon dioxide, muscle rigidity, hyperthermia, acidosis and hyperkalemia ensues.
The prevalence of MH episodes is estimated to range from 1:10,000 to 1:220,000 [10, 11]. In a recent report from Japan evaluating the actual nationwide prevalence of MH events based on a large-scale cross-sectional data analysis, the prevalence was calculated to be 1:73,000 in the years 2006-2008 [12]. Men have repeatedly been shown to be significantly more prone compared to women [12, 13], and more than 50% of all MH reactions involve the paediatric population [5]. All ethnic groups are affected [5]. In a prevalence and clinical outcome study from New York State from 2001 to 2005, most of the MH events (71%) originated from emergency/urgent admissions [13].
An MH episode may develop upon first exposure to a triggering agent. However, on average, affected individuals undergo three anaesthesias before a first MH crisis occurs [5], and cases are known where even more uneventful inhalational anaesthesias were performed before an MH crisis. Therefore, the prevalence of the genetic predisposition for MH can neither be estimated nor determined. In a French study of 104 MH families, six individuals carried two MH mutations [14]. These authors estimated MH mutations to be as frequent as 1 in 2000.
The characteristic hypermetabolism during an MH crisis produces increased carbon dioxide and despite increases in minute ventilation, carbon dioxide levels continue to rise [15]. Rising end-tidal carbon dioxide can be an early warning sign of an impending MH crisis especially when succinylcholine is used as the relaxing agent, in which case an abrupt rise in CO2 can be observed. In the absence of succinylcholine, however, the rise in CO2 is more gradual and can be missed [16]. Unexplained tachycardia, muscle rigidity, hyperthermia, diffuse sweating, acidosis and hyperkalemia are further key signs of an MH episode [5]. Internationally, MH experts have developed a clinical grading scale to assess the likelihood of an intra-operative event representing an MH episode [17]. According to this scale, differential weighting is given to the various manifestations of the syndrome (i.e., muscle rigidity, muscle breakdown, respiratory acidosis, metabolic acidosis, temperature increase, cardiac involvement and family history) to facilitate the clinical diagnosis of MH [15, 17]. Of note, increase in temperature generally appears later. Therefore, one should not wait for this sign to appear before making the diagnosis. On the other hand, there are numerous conditions (e.g., surgical stress, inadequate anaesthetic depth, sepsis, thyreotoxic crisis, etc.) that present unspecific signs such as tachycardia and hypercapnia, which can mimic MH [1, 15]. So, the clinical diagnosis of MH can at times be hard to make. However, prompt recognition and treatment of an MH episode is absolutely crucial; any delay in diagnosis and treatment increases mortality [15]. A further clinical sign of potential MH is succinylcholine-induced masseter muscle rigidity (MMR). Often, after the administration of succinylcholine, an increase in masseter muscle tone can be observed. This must be distinguished from a true MMR event, the “jaws of steel”, which is defined to last for at least 2 min after succinylcholine administration. MMR occurs in about 1 in 100 children when anaesthesia is induced with halothane and succinylcholine [18]. The same incidence is probably true for the combination sevoflurane and succinylcholine. However, if one starts anaesthesia intravenously with the combination of thiopental-succinylcholine, the incidence of MMR is much less [19]. Importantly, propofol was shown to have even greater muscle relaxant properties compared to thiopental when combined with succinylcholine [20]. Half the patients exhibiting MMR were diagnosed as MH susceptible [21]. The clinical incidence of MH defined by arterial blood gas analysis, however, is only about 15% in patients experiencing MMR. This shows that MMR can be an early sign of MH, but in most cases it merely exposes an underlying myopathy, not necessarily representing MH. There are many variables influencing pharmacosensitivity of masseter muscle (e.g., serum electrolyte levels, size of motor units, expression of ectopic nicotinic acetylcholine receptors). So, MMR is very unspecific and has to be considered as an unreliable sign of MH. Furthermore, this symptom cannot be quantified by measurements (in daily clinical practice) and may therefore be vulnerable to subjective error, most notably in a stressful clinical situation. Still, it may be prudent to discontinue anaesthesia following a MMR event. In case of an emergency, one should ensure a ‘trigger-free’ anaesthesia and be especially alert for signs of hypermetabolism. Post-operatively, these individuals should be monitored for hypermetabolism and signs of muscle damage (i.e. temperature, creatine kinase (CK) and blood gases). If there is any second suspicion (such as inadequate CK elevation or CO2 increase), these patients with a typical MMR episode should best be referred to an MH testing centre [5].
Figure 1
Two screenshots of the MHApp. Panel A shows the first five treatment steps, which all have to be confirmed before proceeding. Panel B shows the automatically generated protocol. This protocol can be forwarded by email.
In case an MH episode is suspected, all triggering agents should be discontinued immediately and anaesthesia should be changed to propofol [22]. Help should be called as early as possible. Smart phone applications, such as ‘MHApp’ for the iPhone, can help to manage the complex and time-consuming therapy (fig. 1). Hyperventilation with a high fresh gas flow (>10 l/min) should be started and treatment with a bolus of dantrolene at 2.5 mg/kg initiated to limit MH. Dantrolene is the primary drug used to treat MH, and, as a specific ryanodine receptor antagonist, it inhibits the pathologically increased calcium release from the sarcoplasmic reticulum in the affected individuals [23]. The initial bolus of dantrolene (2.5 mg/kg) should be repeated until clinical signs and acidosis subside (i.e., titrate the treatment to tachycardia and hypercarbia) [1]. The recommended upper limit of 10 mg/kg dantrolene may have to be surpassed, as guided by the clinical and laboratory signs of the individual patient. Cooling measures need to be started (e.g., placing ice packs on the groin, axillae and neck) and maintained until the temperature of the patient is below 38.5°C. Blood should be regularly drawn to evaluate acid-base status, blood cell count, coagulation profile, electrolytes, creatinine, CK and myoglobin. Urine should be secured for myoglobin measurement. This extended monitoring helps to guide the management of the MH episode [15]. Any increase in potassium or worsening acidosis should be followed by a dantrolene bolus. Coagulopathy, such as disseminated intravascular coagulation can develop and may need to be treated. In case of increased myoglobin in blood or urine samples due to the muscular breakdown and the risk of kidney damage, more aggressive hydration has to be started, together with urine alkalinisation and the administration of diuretics to help clear tubular myoglobin; urine output of at least 2 ml/kg/h should be targeted [24]. Regular CK measurements, every 6-8 h, should be conducted until the values start to fall. Rhabdomyolysis can be associated with accentuated derangements in the serum electrolytes; hyperkalemia has to be especially diagnosed and treated, as severe life-threatening cardiac arrhythmias can develop. Patients experiencing an MH episode have to be followed up in an intensive care unit for at least 36-48 hours. Dantrolene at a dose of 1 mg/kg should be continued every 4-8 hours as guided by clinical and laboratory parameters (fig. 2). Importantly, about 20% of patients will experience a recrudescence of the syndrome (even under dantrolene treatment) [25]. Of note, according to an analysis by Burkman et al., the mean time from initial reaction to recrudescence was 13 h, well beyond the immediate peri-operative period [25].
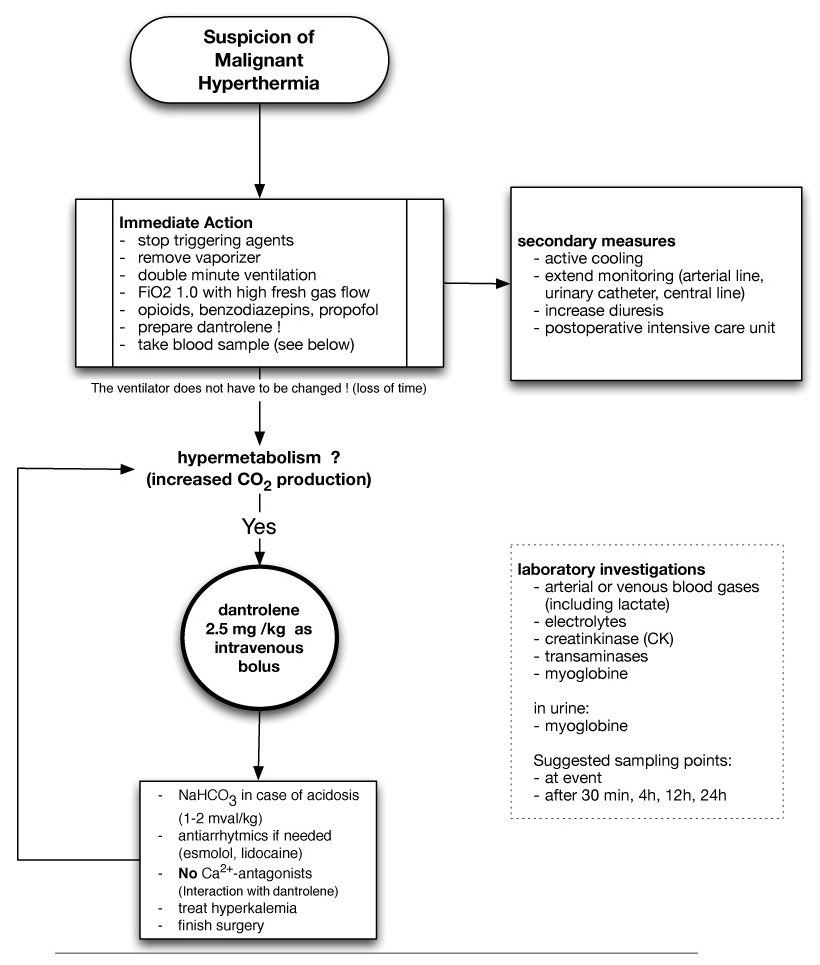
Figure 2
Treatment algorithm for malignant hyperthermia according to the Swiss MH investigation unit.
In skeletal muscle specimens from MH-susceptible swine, Iaizzo and co-authors showed that exposure to halothane led to higher myoplasmic calcium concentrations as compared to specimens from control animals [26], and that these higher myoplasmic calcium concentrations were associated with contractures of the muscle fibres tested (fig. 3). Further experimental evidence from various sources has indicated the same underlying mechanism as causative for MH: uncontrolled release of calcium from the sarcoplasmic reticulum [5]. This exaggerated intracellular calcium release in MH-susceptible skeletal muscle leads to large increases in aerobic and anaerobic metabolism as the muscle cells attempt to re-establish homeostasis by sequestering unbound calcium [15]. However, in MH-susceptible muscle, the rise in calcium caused by the triggering agents overwhelms the cellular capacity to re-establish homeostasis. The pathologically enhanced intracellular calcium rise eventually reaches the threshold levels for myofibrillar contraction and muscular rigidity begins. This leads to increased oxygen consumption and increased carbon dioxide production. Heat is generated with rising lactic acid levels. Adenosine triphosphate stores become depleted, which progressively endangers the membrane integrity of the skeletal muscle cells. Rhabdomyolysis ensues with leakage of muscle cell contents (including electrolytes, myoglobin and various other sarcoplasmic proteins, like CK) into the circulation, with potential consequences for renal function and integrity [5, 15, 24].
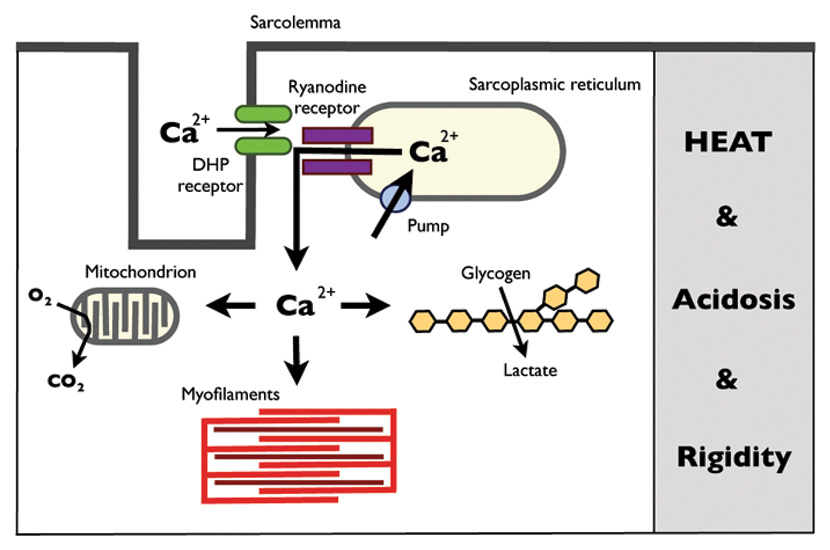
Figure 3
A schematic representation of the excitation-contraction coupling mechanism and the pathophysiological key elements of malignant hyperthermia (adapted from Lehmann-Horn et al. [36]). The muscle action potential is propagated along the sarcolemmal membrane into the transverse tubule, where the dihydropyridine receptors (DHP receptor) sense the action potential voltage change and open up. The conformational change of the opening DHP receptors then activates the ryanodine receptors, which release calcium from the sarcoplasmic reticulum. In malignant hyperthermia, an uncontrolled calcium release from the sarcoplasmic reticulum overwhelms the compensatory mechanisms within the skeletal muscle cell: increased muscle contractile activity and metabolic stimulation ensues.
The cause of this abnormal regulation and release of calcium in affected skeletal muscle lies, in most cases, in a defective calcium release channel in the skeletal muscle sarcoplasmic reticular membrane, named the ryanodine receptor (RYR). In close contact to the RYR is the dihydropyridine-sensitive voltage-gated calcium channel (DHPR). DHPR are located in the transverse tubule membranes and function as voltage sensors. Every time a muscle action potential is propagated along the sarcolemmal membrane into the transverse tubule, the voltage change is sensed by DHPR and the channel opens up. The conformational change of the opening DHPR is directly transmitted to the RYR, which themselves respond by opening. The resultant calcium release then leads to muscular contraction. This is the fundamental excitation-contraction coupling (ECC) needed for normal skeletal muscle function. Aberrations in these two receptors, as well as in other proteins involved in the myoplasmic calcium regulation, are responsible for the functional changes seen in calcium regulation in MH. In particular, mutations in the gene for the ryanodine receptor (RYR1 gene on chromosome 19) were linked to MH susceptibility [27, 28]. Approximately 70% of MH families carry mutations in this specific gene [29, 30]. However, not all MH families exhibit linkage to this gene. Other potential genetic loci have been identified, such as mutations of adult muscle sodium channel alpha-subunit (SCN4A) [31] or of the main subunit of DHPR (CACNA1S) [32], although MH mutations have so far only been identified in CACNA1S.
As MH is a subclinical myopathy, special testing is needed to confirm or exclude MH susceptibility. Testing for MH susceptibility is invasive and, therefore, not applicable as a screening method. Indication for MH testing is given for individuals who have survived a suspected MH episode or for close relatives of known MH susceptible individuals. The gold standard for such pre-symptomatic testing is the in vitro muscle contracture testing (IVCT). An invasive, open muscle biopsy is conducted under regional anaesthesia, and the muscle strips are mounted and immersed in tissue baths for measurement of isometric contractions [33]. These muscle fibres are exposed to increasing concentrations of halothane and caffeine according to the standardised protocol of the European Malignant Hyperthermia Group (EMHG) [33]. Following the test, one of the following conclusions regarding MH susceptibility can be reached: MH susceptible (MHS), i.e., significant contracture of at least one muscle bundle both at ≤ 2 mM caffeine and ≤ 2% halothane; MH negative (MHN), i.e., significant contracture of all the tested bundles only at ≥ 3 mM caffeine and > 2% halothane; MH equivocal (MHE), i.e., all other test results are deemed equivocal (MHEh if reacting to halothane only or MHEc if reacting to caffeine only). Clinically, MHE patients are regarded as MH susceptible. This specific EMHG protocol reaches a sensitivity of 99% and a specificity of 94% [34]. Although still regarded as the gold standard for the diagnosis of MH, IVCT testing is invasive, costly and confined to specialised centres in Europe. Therefore, DNA analysis and molecular genetic testing is an attractive alternative. Only a small volume of blood is required, which can then be sent to an accredited diagnostic laboratory for further testing. Although, molecular genetic testing in patients from MH families with known causative mutations has been introduced relatively recently, today it represents the routine testing for such patients [27, 35]. Despite the fact that more than 200 RYR1 variants have been identified, only 30 of these have been investigated for their functional effect and have, therefore, been approved as ‘diagnostic’ mutations by the EMHG (http://www.emhg.org). It is of utmost importance to realise that absence of RYR1 mutations does not exclude MH. Patients, in whom the familial RYR1 mutation was not identified, must undergo an IVCT in order to confirm or exclude MH susceptibility [35]. Ongoing investigations will uncover further causative mutations and thereby improve the usefulness and efficacy of genetic testing in future.
As described earlier, it is thought that most patients with susceptibility to MH, except for those with congenital myopathies associated with RYR1 mutations, have no clinical manifestations of muscle disease except when exposed to the typical triggering agents (i.e., only during anaesthesia). However, over the years sporadic reports of confirmed fulminant and fatal non-anaesthetic “awake” MH episodes have appeared in the literature [29, 36]. It is thought that a subset of MHS individuals may be prone to MH-like symptoms if exposed to non-anaesthetic triggers (e.g., environmental heat stress, exercise or even emotional stress). In porcine MH, such an association between environmental stressors and MH crisis is well established and is even known as the porcine stress syndrome [36, 37]. MHS (i.e., homozygous) pigs are sensitive to environmental stress, and MH episodes can be triggered in awake animals by exposure to exercise, heating or excitement [38]. However, MH is different among species. In humans, MH has an autosomal dominant mode of inheritance, while pigs have to be homozygous to be susceptible. In 1980, Gronert and co-authors reported the case of a 42-year-old patient with episodic hyperthermia [39]. This patient suffered from recurrent episodes of aching joints, malaise and fever of 40°C or more if exposed to extreme physical or emotional stress. As his episodes had similarities to MH, a muscle biopsy and contracture testing was part of the diagnostic work-up. Pathologic muscle contracture responses confirmed this patient to be MH susceptible.
In 2001, a case of non-anaesthetic, stress-induced hyperpyrexic death in an individual with a history of MH was reported [40], and most recently similar non-anaesthetic MH-like events have been described in two unrelated children triggered by either exposure to environmental heat or infection [29, 36, 41]. Both children had the same RYR1 variant (p.R3983C) on one allele. While the boy also had bilateral ptosis and muscle hypotonia (suggestive of additional myopathy), the girl had a second mutation (p.D4505H) on the other allele. Maybe this specific RYR1 variant is associated with a distinct phenotype and predisposes patients to massive myoplasmic calcium release (in response to heat, fever and infection) [6, 36]. This discussion is ongoing [6], and further research is needed.
We would like to cite recent clinical recommendations and guidelines for advice to MH patients in this regard [42]. The vast majority of MH patients are not vulnerable and will not experience an MH-like episode in response to stress, heat or infection and, therefore, need not limit environmental or stress situations. However, a subgroup of MHS patients with a personal or family history of intolerance to heat or other stressful conditions should be cautious and should avoid over-exertion. In case of early symptoms, stressful activity should be reduced to limit the likelihood of an “awake” MH episode. Symptomatic treatment with cooling should be initiated.
A potential association of MH with idiopathic hyperCKemia, defined as a persistently elevated serum concentration of CK (i.e., at least 3 serum CK levels more than twice normal over at least 3 months) with no evidence of neuromuscular disease, has been a matter of debate for years. Recently, a case of a healthy 18-year-old man was reported, known only for asymptomatic (familial) CK elevation, who exhibited an MH reaction intra-operatively [43]. Further diagnostics revealed a mutation in the RYR1 gene of this patient. Patients with asymptomatic hyperCKemia have long been suspected to have a susceptibility to MH. While in a recent investigation of patients with unexplained hyperCKemia the in vitro contracture testing detected one out of 37 individuals to be MH susceptible (2.7%) [44], the incidence of positive contracture tests in patients with unexplained hyperCKemia in another study was much higher (49%) [45]. This may be explained by varying selection of the cases selected for testing [43]. Indeed, while various older studies have concluded increased asymptomatic CK levels to be, at best, a weak predictor of MH [46–48], unexplained persistently elevated serum CK levels in an otherwise healthy individual should alert the clinician for a thorough neurological examination and history taking [49]. In the context of selection bias of patients it might be helpful to remember that thyroid dysfunction is a very common cause of hyperCKemia. Still, in case of anaesthesiological complications within a family, referral of the patient to an MH investigation centre seems advisable [44, 50].
Exertional rhabdomyolysis has been linked to MH susceptibility (with positive contracture testing) in a small number of case reports. Recently, a case was reported of a 30-year-old patient on statin therapy who first presented with exertional rhabdomyolysis after a 2.5 mile walk and had to be taken to the operating room because of bilateral calf compartment syndrome [51]. During one of the subsequent operations, the patient presented with a fulminant MH reaction. In vitro contracture testing confirmed the MH diagnosis. In a previous study, Wappler and co-authors found ten out of twelve patients with exertional rhabdomyolysis (and with no personal or family history of MH) to be MHS by contracture testing; three of the MHS individuals had causative RYR1 mutations [52].
Muscular side effects due to statin therapy are frequent, and rhabdomyolysis can occur [6, 51]. Three cases of statin-associated myalgia have been reported with special relevance, as these patients underwent muscle biopsy and in vitro contracture testing. Thereafter, one patient had associated myoglobinuria and the other two presented with persistently elevated CK levels despite discontinuation of statin therapy. All three were found to be MHS by IVCT [6]. In a case series, nine patients with increased CK levels and myalgias after statin treatment were evaluated by IVCT: 7 out of 9 patients exhibited abnormal IVCT test results [53]. Although these abnormal results have to be interpreted with caution, considering the lack of specificity of the IVCT in the presence of muscle damage, statin therapy may well be able to unmask underlying muscle pathology such as MH. Nevertheless, given the various important beneficial effects, statin therapy should not be withheld from MHS patients [6]. Recently, Hopkins recommended starting statin treatment cautiously in MHS patients. Specifically, a pre-treatment CK level (with further follow-up) should be taken and the patient be instructed to report muscle symptoms or dark urine [6].
Lastly, we would like to add that RYR1 mutations have recently been found in patients suffering from neuroleptic malignant syndrome, which beforehand has been seen as an entirely separate entity [54]. There might be a causal relationship or perhaps this finding is just a coincidence.
The genetic prevalence of MH cannot be adequately estimated, because clinical penetrance of MH is highly variable and the majority of MHS individuals lack clinical symptoms in absence of triggering agents. Furthermore, there is no screening method to test for MH, as current tests are invasive (open muscle biopsy) or restricted to MH families with known MH-associated mutations (molecular genetic testing). Although prevalence is historically thought to be at approximately 1 in 50,000 or less, molecular genetic investigations have suggested the prevalence to be as high as 1 in 2000. Such a high frequency is further supported by the fact, that MH mutations have been identified in patients with exertional heat stroke and statin-induced rhabdomyolysis. Although MH is still a condition mainly of importance to anaesthesiologists, non-anaesthetic – and non-pharmacological – triggering of MH have substantially increased the number of people interested in MH. For further information and advice we refer the reader to the following popular weblinks on the topic: http://www.emhg.org and http://www.mhaus.org .
1 Girard T, Ginz H, Urwyler A. Maligne Hyperthermie. Schweiz Med Forum.2004:1192–7.
2 Denborough M, Lovell R. Anaesthetic deaths in a family. Lancet.1960;2:45.
3 Litman RS, Rosenberg H. Malignant hyperthermia: update on susceptibility testing. JAMA.2005;293:2918–24.
4 Denborough M. Malignant hyperthermia. Lancet. 1998;352:1131–6.
5 Rosenberg H, Davis M, James D, Pollock N, Stowell K. Malignant hyperthermia. Orphanet J Rare Dis.2007;2:21.
6 Hopkins PM. Malignant hyperthermia: pharmacology of triggering. Br J Anaesth. 2011;107:48–56.
7 Klingler W, Rueffert H, Lehmann-Horn F, Girard T, Hopkins PM. Core myopathies and risk of malignant hyperthermia. Anesth Analg.2009;109:1167–73.
8 Kinder Ross A. Muscular dystrophy versus mitochondrial myopathy: the dilemma of the undiagnosed hypotonic child. Paediatr Anaesth.2007;17:1–6.
9 Parness J, Bandschapp O, Girard T. The myotonias and susceptibility to malignant hyperthermia. Anesth Analg.2009;109:1054–64.
10 Ording H. Incidence of malignant hyperthermia in Denmark. Anesth Analg. 1985;64:700–704.
11 Urwyler A, Hartung E. Die Maligne Hyperthermie. Anaesthesist.1994;43:557–69.
12 Sumitani M, Uchida K, Yasunaga H, Horiguchi H, Kusakabe Y, Matsuda S, et al. Prevalence of malignant hyperthermia and relationship with anesthetics in Japan: data from the diagnosis procedure combination database. Anesthesiology.2011;114:84–90.
13 Brady JE, Sun LS, Rosenberg H, Li G. Prevalence of malignant hyperthermia due to anesthesia in New York State, 2001–2005. Anesth Analg.2009;109:1162-–6.
14 Monnier N, Krivosic-Horber R, Payen JF, Kozak-Ribbens G, Nivoche Y, Adnet P, et al. Presence of two different genetic traits in malignant hyperthermia families: implication for genetic analysis, diagnosis, and incidence of malignant hyperthermia susceptibility. Anesthesiology.2002;97:1067–74.
15 Tautz TJ, Urwyler A, Antognini JF, Riou B. Case scenario: Increased end-tidal carbon dioxide: a diagnostic dilemma. Anesthesiology.2010;112:440–6.
16 Karan SM, Crowl F, Muldoon SM. Malignant hyperthermia masked by capnographic monitoring. Anesth Analg.1994;78:590–2.
17 Larach MG, Localio AR, Allen GC, Denborough MA, Ellis FR, Gronert GA, et al. A clinical grading scale to predict malignant hyperthermia susceptibility. Anesthesiology. 1994;80:771–9.
18 Schwartz L, Rockoff MA, Koka BV. Masseter spasm with anesthesia: incidence and implications. Anesthesiology.1984;61:772–5.
19 Lazzell VA, Carr AS, Lerman J, Burrows FA, Creighton RE. The incidence of masseter muscle rigidity after succinylcholine in infants and children. Can J Anaesth.1994;41:475–9.
20 Ummenhofer WC, Kindler C, Tschaler G, Hampl KF, Drewe J, Urwyler A. Propofol reduces succinylcholine induced increase of masseter muscle tone. Can J Anaesth. 1998;45:417–23.
21 O'Flynn RP, Shutack JG, Rosenberg H, Fletcher JE. Masseter muscle rigidity and malignant hyperthermia susceptibility in pediatric patients. An update on management and diagnosis. Anesthesiology.1994;80:1228–33.
22 Glahn KP, Ellis FR, Halsall PJ, Muller CR, Snoeck MM, Urwyler A, et al. Recognizing and managing a malignant hyperthermia crisis: guidelines from the European Malignant Hyperthermia Group. Br J Anaesth.2010;105:417–20.
23 Inan S, Wei H. The cytoprotective effects of dantrolene: a ryanodine receptor antagonist. Anesth Analg.2010;111:1400–10.
24 Bosch X, Poch E, Grau JM. Rhabdomyolysis and acute kidney injury. N Engl J Med.2009;361:62–72.
25 Burkman JM, Posner KL, Domino KB. Analysis of the clinical variables associated with recrudescence after malignant hyperthermia reactions. Anesthesiology.2007;106:901–6.
26 Iaizzo PA, Klein W, Lehmann-Horn F. Fura-2 detected myoplasmic calcium and its correlation with contracture force in skeletal muscle from normal and malignant hyperthermia susceptible pigs. Pflugers Arch.1988;411:648–53.
27 Girard T, Johr M, Schaefer C, Urwyler A. Perinatal diagnosis of malignant hyperthermia susceptibility. Anesthesiology.2006;104:1353–4.
28 McCarthy TV, Quane KA, Lynch PJ. Ryanodine receptor mutations in malignant hyperthermia and central core disease. Hum Mutat.2000;15:410–7.
29 Groom L, Muldoon SM, Tang ZZ, Brandom BW, Bayarsaikhan M, Bina S, et al. Identical de novo mutation in the type 1 ryanodine receptor gene associated with fatal, stress-induced malignant hyperthermia in two unrelated families. Anesthesiology.2011;115:938–45.
30 Robinson R, Carpenter D, Shaw MA, Halsall J, Hopkins P. Mutations in RYR1 in malignant hyperthermia and central core disease. Hum Mutat.2006;27:977–89.
31 Vita GM, Olckers A, Jedlicka AE, George AL, Heiman-Patterson T, Rosenberg H, et al. Masseter muscle rigidity associated with glycine1306-to-alanine mutation in the adult muscle sodium channel alpha-subunit gene. Anesthesiology.1995;82:1097–103.
32 Monnier N, Procaccio V, Stieglitz P, Lunardi J. Malignant-hyperthermia susceptibility is associated with a mutation of the alpha 1-subunit of the human dihydropyridine-sensitive l-type voltage-dependent calcium-channel receptor in skeletal muscle. Am J Hum Genet.1997;60:1316–25.
33 A protocol for the investigation of malignant hyperpyrexia (MH) susceptibility. The European Malignant Hyperpyrexia Group. Br J Anaesth.1984;56:1267–9.
34 Ording H, Brancadoro V, Cozzolino S, Ellis FR, Glauber V, Gonano EF, et al. In vitro contracture test for diagnosis of malignant hyperthermia following the protocol of the European MH Group: results of testing patients surviving fulminant MH and unrelated low-risk subjects. The European Malignant Hyperthermia Group. Acta anaesthesiol Scand. 1997;41:955–66.
35 Urwyler A, Deufel T, McCarthy T, West S. Guidelines for molecular genetic detection of susceptibility to malignant hyperthermia. Br J Anaesth.2001;86:283–7.
36 Lehmann-Horn F, Klingler W, Jurkat-Rott K. Nonanesthetic malignant hyperthermia. Anesthesiology. 2011;115:915–7.
37 Fujii J, Otsu K, Zorzato F, de Leon S, Khanna VK, Weiler JE, et al. Identification of a mutation in porcine ryanodine receptor associated with malignant hyperthermia. Science. 1991;253:448–51.
38 Wappler F, Fiege M, Schulte am Esch J. Pathophysiological role of the serotonin system in malignant hyperthermia. Br J Anaesth.2001;87:794–8.
39 Gronert GA, Thompson RL, Onofrio BM. Human malignant hyperthermia: awake episodes and correction by dantrolene. Anesth Analg.1980;59:377–8.
40 Tobin JR, Jason DR, Challa VR, Nelson TE, Sambuughin N. Malignant hyperthermia and apparent heat stroke. JAMA2001;286:168-–9.
41 Gronert GA, Tobin JR, Muldoon S. Malignant hyperthermia – human stress triggering. Biochim Biophys Acta. 2011;1813:2191–2.
42 Maclennan DH, Zvaritch E. Response to "malignant hyperthermia - human stress triggering" in reference to original article "mechanistic models for muscle diseases and disorders originating in the sarcoplasmic reticulum". Biochim Biophys Acta. 2011;1813:2193–4.
43 Kasi PM. Malignant hyperthermia and idiopathic hyperckemia. Case Reports in Medicine. 2011;2011:3.
44 Malandrini A, Orrico A, Gaudiano C, Gambelli S, Galli L, Berti G, et al. Muscle biopsy and in vitro contracture test in subjects with idiopathic hyperCKemia. Anesthesiology. 2008;109:625–8.
45 Weglinski MR, Wedel DJ, Engel AG. Malignant hyperthermia testing in patients with persistently increased serum creatine kinase levels. Anesth Analg.1997;84:1038–41.
46 Ellis FR, Clarke IM, Modgill M, Currie S, Harriman DG. Evaluation of creatinine phosphokinase in screening patients for malignant hyperpyrexia. Br Med J. 1975;3:511–3.
47 Amaranath L, Lavin TJ, Trusso RA, Boutros AR. Evaluation of creatinine phosphokinase screening as a predictor of malignant hyperthermia. A prospective study. Br J Anaesth.1983;55:531–3.
48 Paasuke RT, Brownell AK. Serum creatine kinase level as a screening test for susceptibility to malignant hyperthermia. JAMA.1986;255:769–71.
49 Lingaraju N, Rosenberg H. Unexplained increases in serum creatine kinase levels: its relation to malignant hyperthermia susceptibility. Anesth Analg.1991;72:702–5.
50 Monnier N, Kozak-Ribbens G, Krivosic-Horber R, Nivoche Y, Qi D, Kraev N, et al. Correlations between genotype and pharmacological, histological, functional, and clinical phenotypes in malignant hyperthermia susceptibility. Hum Mutat2005;26:413–25.
51 Capacchione JF, Sambuughin N, Bina S, Mulligan LP, Lawson TD, Muldoon SM. Exertional rhabdomyolysis and malignant hyperthermia in a patient with ryanodine receptor type 1 gene, l-type calcium channel alpha-1 subunit gene, and calsequestrin-1 gene polymorphisms. Anesthesiology.2010;112:239–44.
52 Wappler F, Fiege M, Steinfath M, Agarwal K, Scholz J, Singh S, et al. Evidence for susceptibility to malignant hyperthermia in patients with exercise-induced rhabdomyolysis. Anesthesiology.2001;94:95–100.
53 Guis S, Figarella-Branger D, Mattei JP, Nicoli F, Le Fur Y, Kozak-Ribbens G, et al. In vivo and in vitro characterization of skeletal muscle metabolism in patients with statin-induced adverse effects. Arthritis Rheum.2006;55:551–7.
54 Sato T, Nishio H, Iwata M, Kentotsuboi, Tamura A, Miyazaki T, et al. Postmortem molecular screening for mutations in ryanodine receptor type 1 (ryr1) gene in psychiatric patients suspected of having died of neuroleptic malignant syndrome. Forensic Sci Int.2010;194:77–9.
Acknowledgment: We thank Allison Dwileski for editorial assistance.
Funding / potential competing interests: Support was provided from institutional and/or departmental sources. No potential conflict of interest was relevant to this article. T. Girard is the author of the iPhone app "MHApp". All revenue go to the Eruopean MH Group (EMHG) and the MH Association of the United States (MHAUS) by a 50/50 share. He has no personal financial interest.